

Influenza D virus (IDV), a new genus of Orthomyxoviridae family, presents a large host range and a worldwide circulation. The pathogenicity of this virus has been studied in the calf model. The mouse model is frequently used to enable a first assessment of a pathogen’s fitness, replication, and pathogenesis for influenza A and B viruses. We showed that DBA/2 mice are a relevant in vivo model for the study of IDV replication. This model will allow for rapid IDV fitness and replication evaluation and will enable phenotypic comparisons between isolated viruses. It will also allow for a better understanding of the immune response induced after IDV infection.
KEYWORDS: influenza D virus, mouse model, replication
ABSTRACT
A novel genus within the Orthomyxoviridae family was identified in the United States and named influenza D virus (IDV). Bovines have been proposed to be the primary host, and three main viral lineages (D/OK-like, D/660-like, and D/Japan-like) have been described. Experimental infections had previously been performed in swine, ferrets, calves, and guinea pigs in order to study IDV pathogenesis. We developed a murine experimental model to facilitate the study of IDV pathogenesis and the immune response. DBA/2 mice were inoculated with 105 50% tissue culture infective dose (TCID50) of D/bovine/France/5920/2014 (D/OK-like). No clinical signs or weight loss were observed. Viral replication was observed mainly in the upper respiratory tract (nasal turbinates) but also in the lower respiratory tract of infected mice, with a peak at 4 days postinfection. Moreover, the virus was also detected in the intestines. All infected mice seroconverted by 14 days postinfection. Transcriptomic analyses demonstrated that IDV induced the activation of proinflammatory genes, such as gamma interferon (IFN-γ) and CCL2. Inoculation of NF-κB-luciferase and Ifnar1−/− mice demonstrated that IDV induced mild inflammation and that a type I interferon response was not necessary in IDV clearance. Adaptation of IDV by serial passages in mice was not sufficient to induce disease or increased pathogenesis. Taken together, present data and comparisons with the calf model show that our mouse model allows for the study of IDV replication and fitness (before selected viruses may be inoculated on calves) and also of the immune response.
IMPORTANCE Influenza D virus (IDV), a new genus of Orthomyxoviridae family, presents a large host range and a worldwide circulation. The pathogenicity of this virus has been studied in the calf model. The mouse model is frequently used to enable a first assessment of a pathogen’s fitness, replication, and pathogenesis for influenza A and B viruses. We showed that DBA/2 mice are a relevant in vivo model for the study of IDV replication. This model will allow for rapid IDV fitness and replication evaluation and will enable phenotypic comparisons between isolated viruses. It will also allow for a better understanding of the immune response induced after IDV infection.
INTRODUCTION
In 2011, a new influenza virus was isolated from a pig with influenza-like symptoms in Oklahoma. Electronic microscopy and real-time reverse transcription-PCR (RT-PCR) revealed that it was neither an influenza A virus (IAV) nor an influenza B virus (IBV). Next-generation sequencing (NGS) analyses allowed for the identification of 7 Orthomyxovirus-like RNA segments, but this virus presented only 50% overall identity to human influenza C virus (ICV). Furthermore, serological analyses demonstrated that antibodies against this new virus failed to cross-react with IAV, IBV, or ICV (1). All of these results suggested that it was a new genus of Orthomyxoviridae, temporarily named C/swine/Oklahoma/1334/2011 (C/swine/OK) and then influenza D virus (IDV) (1, 2).
IDV is widely distributed in the world: so far, it has been detected in North and Central America (1, 3–6), Asia (7–11), Europe (12–18), and Africa (19, 20). Two clades of IDV that are antigenically and phylogenetically different were identified in the United States, D/OK-like and D/660-like. Both were shown to frequently reassort in the United States (21). More recently, a third clade, circulating specifically in Japan, was identified (D/Japan-like) (11, 22, 23).
Several studies detected IDV in cattle, with a higher prevalence in sick than in healthy cattle (2, 10, 21) and a higher prevalence in cattle than in swine, suggesting that bovines could be a main host of IDV (1, 3). IDV was also detected in small ruminants, horses, and camels (19, 22, 23).
The zoonotic potential of IDV is still not clear, but serological and virological studies suggested that the virus might infect humans, especially those exposed to cattle (1, 24–26). An experimental study in ferrets was conducted in order to understand IDV pathogenesis and zoonotic potential (1). The ferret is a good model for studying human influenza virus. Indeed, these animals express the same pattern of viral receptors and present clinical signs similar to those observed in humans (27). Despite the absence of clinical signs, the virus replicated in the upper respiratory tract of the ferrets. Moreover, direct contact transmission between ferrets was observed. Taken together, these results suggested that humans could be susceptible to IDV, but studies are still needed to confirm this hypothesis.
IDV is involved in bovine respiratory disease complex (BRDC). BRDC causes a major economic and public health problems in young calves worldwide. The causes are multifactorial, represented by the following: (i) the presence of one or several pathogens (virus and/or bacteria), (ii) a compromised immune system of bovines, and (iii) environmental factors. Recently, three metagenomics studies identified IDV among viruses associated with BRDC. IDV was mainly associated with BRDC, alone or in combination with bovine adenovirus-3, and bovine rhinitis A in U.S. cattle (6) or with bovine rhinitis A and B viruses, bovine coronavirus, or bovine respiratory syncytial virus in Canadian cattle (5). The role of IDV in BRDC remains unclear, but the high prevalence in cattle and the mild clinical signs by experimental infections suggest that it could be an initiating pathogen (28, 29).
Little is known about IDV pathogenesis, transmission, or the associated immune response. It is therefore necessary to develop a small-animal model in order to have a better understanding of IDV’s biology. Four experimental infection models had been developed in swine, ferrets, guinea pigs, and calves (1, 28, 30, 31). Swine and ferrets did not present clinical signs, and IDV was only detected in the upper respiratory tract (1). In guinea pigs, IDV replicated with high titers in the upper and lower respiratory tracts but was not associated with clinical signs (30). Ferguson et al. (28) and Salem et al. (29) also studied the pathogenesis and transmission of the virus in calves, the main host known so far. They observed mild clinical signs, and IDV was detected in both upper and low respiratory tracts. Transmission by direct contact and aerosols was observed. Salem et al. (29) studied the immune response of calves post-IDV inoculation. The calves presented an innate immune response involving proinflammatory cytokines and chemokines, such as CCL2, CCL3, and CCL2. Surprisingly, the type I interferon mRNAs were not overexpressed. A mixed Th1 and Th2 response was also observed, with IDV-specific IgG1 production starting from 10 days postinfection.
Mice have so far not been used for studying the pathogenesis of IDV. Mice are, however, the most used animal model for studying influenza viruses (32). The mouse model is a very convenient model for studying viral replication (33), tissue tropism (34), and the immune response (35) but also for testing vaccines or antiviral molecules (36, 37). It has clear practical advantages such as a low cost, small size, ease of husbandry, and good availability of reagents for immunology testing (32). The main disadvantages are the inefficient influenza virus transmission between mice, the rare clinical signs, and the requirement of adaptation for some influenza virus strains. It was shown that the mouse genetic background has also an impact on their susceptibility to IAV (38).
The mouse model was especially used to study the pathogenicity of IAV, namely, tissue tropism, fitness of replication, and the immune response. The most visible clinical signs post-IAV infection are weight loss and changes in behavior. IAV was shown to replicate mainly in the upper and lower respiratory tract of mice, especially in epithelial cells, endothelial cells, and type I pneumocytes. A viral peak has often been observed around 3 to 4 days postinfection (dpi). Systemic replication was observed with a highly pathogenic H5N1 virus and was associated with severe disease (38, 39).
Here, we developed a murine experimental model to study the pathogenesis and immune response of IDV. We aimed to reproduce the clinical signs, viral replication, and tissue tropism as observed in the main host. We also used this model to better understand the immune response induced after infection of IDV in DBA/2, Ifnar1−/−, and NF-κB-luciferase transgenic mice.
RESULTS
Influenza D virus infects mice but does not cause clinical signs.
DBA/2 mice are known to be highly susceptible to IAV infection and were therefore selected for the present study. In order to determine if the mice are susceptible to IDV, we infected a total of 50 DBA/2 mice intranasally with 105 50% tissue culture infective dose (TCID50) of D/bovine/France/5920/2014 (D/5920; D/OK-like clade), and 25 mice served as negative controls (inoculated with phosphate-buffered saline [PBS]). This infection was carried out in two separate experiments.
We did not observe clinical signs or weight loss in the DBA/2-infected mice. No mortality was recorded either, suggesting that IDV could not induce disease in mice (Fig. 1).
FIG 1.
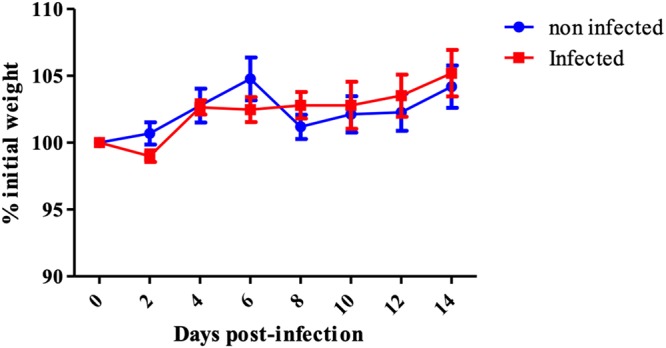
Absence of clinical signs during IDV infection in mice. A group of 36 mice was intranasally infected with 105 TCID50 of D/5920, and a group of 11 noninfected mice was used as a control. Clinical signs and mortality were recorded every 2 days for 14 days. There was no significant weight variation between noninfected and infected DBA/2 mice (P > 0.05). Weight changes expressed as the mean percentage of initial body weight ± standard deviation (SD).
The antibody response against IDV was measured at 14 days postinfection (dpi), using a hemagglutination inhibition (HI) assay. Two viral strains were used, D/5920 (inoculum) and D/bovine/Nebraska/9-5/2012 (D/660-like, heterologous strain). All of the infected mice seroconverted, suggesting that they had all been infected by the virus. The mice presented high antibody titers ranging from 15 to 240 against D/5920 (homologous strain), but the antibody titers against D/Neb were much lower (>20 but <40) (Table 1).
TABLE 1.
Seroconversion in infected DBA/2 mice 14 days postinfection with D/5920
| Strain | HI titer in mouse no.: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| D/5920a | 120 | 80 | 80 | 15 | 160 | 240 | 240 | 120 | 60 | 120 |
| D/Nebb | 20 | 20 | 20 | 20 | 20 | 40 | 40 | 40 | 20 | 20 |
D/5920, D/bovine/France/5920/2014.
D/Neb, D/bovine/Nebraska/9-5/2012. HI assays were performed with viral inoculum (D/5920) and a genetically and antigenically distinct strain of IDV (D/Neb). All noninfected mice did not seroconvert after IDV infection (data not shown).
IDV efficiently replicates in DBA/2 mice with a peak at 4 dpi.
To assess the viral replication and tissue tropism of the virus, eight mice were necropsied at 2, 4, 6, and 8 dpi, and different organs and samples (brain, nasal turbinates, trachea, lungs, spleen, liver, kidneys, intestines, and blood) were collected. IDV was titrated by TCID50 (Fig. 2). The virus was detected in nasal turbinates of all mice (n = 10) with high titers at 4 dpi (103.4 to 104.7 TCID50/g) but not at 2 or 6 to 8 dpi (Fig. 2B). The virus was detected in the trachea of three mice only at 4 dpi but with low titers (102 to 103 TCID50/g) compared to those in nasal turbinates (Fig. 2C). In the lungs, IDV was detected in two mice (n = 2/10) at 2 dpi (102.6 to 103 TCID50/g), in four mice (n = 4/10) at 4 dpi (101.9 to 102.8 TCID50/g), and in one mouse (n = 1/10) at 8 dpi (102.1 TCID50/g) (Fig. 2D).
FIG 2.

IDV mainly presented upper respiratory tropism. Mice were infected with 105 TCID50 of D/5920. (A to H) At 2, 4, 6, and 8 dpi, 8 mice per days were euthanized, and brain (A), nasal turbinates (B), trachea (C), lungs (D), spleen (E), liver (F), kidneys (G), and intestines (H) were collected. Virus titers were determined using the TCID50 method and are expressed in log10 of TCID50/g of tissue. Each dot represents one mouse. The dotted line represents the positivity threshold (1.5 log10 TCDI50/g of organ).
The virus was also sporadically detected in nonrespiratory organs (Fig. 2A, F, and G). Four mice at 2 dpi were positive for IDV in the liver (102.4 to 102.6 TCID50/g), suggesting viremia (Fig. 2E). However, blood samples taken at 2 and 6 dpi were negative for IDV (undetected titers). Finally, the virus was detected in the intestines of mice with titers ranging from 102.1 to 103.3 TCID50/g (6/8 mice virus positive at 2 dpi, 3/8 positive at 4 dpi, 2/8 positive at 6 dpi, and 3/5 positive at 8 dpi), suggesting that IDV could have an enteric tropism.
Histology analyses were performed for each organ at 2 and 6 dpi for negative-control mice and infected mice. At 2 dpi, no lesion was observed irrespective of the organ. At 6 dpi, mild inflammation was observed only in the nasal turbinates. In comparison with noninfected mice, there was an infiltration of lymphocytes, macrophages, and plasma cells, cell degeneration, and loss of cilia. Immunohistochemistry (IHC) staining confirmed the presence of virus in the nasal turbinates at 6 dpi (Fig. 3). No immunohistochemistry signal was detected in any other organs (data not shown).
FIG 3.

Microscopic lesions at 6 dpi and evidence of IDV replication in nasal turbinates of infected mice as done by hematoxylin and eosin staining (H&E) and immunohistochemical reaction (IHC). Magnification, ×1,000; scale bar = 10 μm. (A and B) Hematoxylin and eosin staining shown for the control (A) and infected mice, with infiltration of macrophages, lymphocytes, and plasma cells (long arrow), epithelial cell degeneration, and loss of cilia (short arrows) (B). (C and D) IHC is shown for the control (C) and infected mice, with anti-IDV cytoplasmic and nuclear immunohistochemical expression (D).
These results confirmed that the virus presented a respiratory tropism, especially for the upper respiratory tract, regarding the viral titers. Although there were no clinical signs, we observed mild inflammation at 6 dpi in the nasal turbinates (Fig. 3).
IDV induces a mild proinflammatory response.
To investigate the immune response against IDV, we evaluated the induction of genes involved in innate and adaptive responses in tissues by measuring the transcript levels of 36 associated genes. The immune response was assessed in the lungs (Fig. 4) to understand the local response, and in the spleen (Fig. 5) to understand the systemic response. Twofold changes between infected and noninfected control mice were considered significant.
FIG 4.

IDV induced a mild local proinflammatory response in DBA/2 mice. Mice were infected with 105 TCID50 of D/5920. At 2, 4, and 6 dpi, 3 infected and 3 noninfected mice were euthanized per day, and lungs were collected. Relative expressions for each gene were calculated by ΔΔCT analysis after normalization with the GAPDH housekeeping gene. The results are expressed as mRNA fold induction.
FIG 5.

IDV induced a mild systemic proinflammatory response in DBA/2 mice. Mice were infected with 105 TCID50 of D/5920. At 2, 4, and 6 dpi, 3 infected and 3 noninfected mice were euthanized per day, and spleens were collected. Relative expressions for each gene were calculated by ΔΔCT analysis after normalization with the GAPDH housekeeping gene. The results are expressed as mRNA fold induction.
In the lungs, we observed overexpression of several genes of the innate response. Looking at the sensors of the innate immunity, we observed a ≥2-fold overexpression of Toll-like receptor 4 (TLR4) and TLR7 at 6 dpi compared to at 2 to 4 dpi. For TLR3, we observed a higher expression at 4 dpi than at 2 and 6 dpi. Finally, we did not observe significant overexpression of TLR9 or retinoic acid-inducible gene I (RIG-I) transcripts (1.77- and 1.41-fold increases, respectively; Fig. 4). We also noticed an inflammatory response with a significant increase in interleukin 6 (IL-6) or IL-1β at 2 dpi. The highest fold changes were observed at 2 dpi for CCL5 and gamma interferon (IFN-γ) (5.3- and 6.9-fold changes, respectively; Fig. 4), with a decrease in their expression at 4 to 6 dpi (5-fold decrease, Fig. 4). These results suggested that IDV could induce a mild innate response in the two first days postinfection.
The gene coding for 2′,5′-oligoadenylate synthetase (2′,5′-OAS) was slightly overexpressed at 2 dpi, suggesting that type I interferon could be induced after IDV infection. Moreover, we observed an increase in interferon regulatory factor 3 (IRF3) mRNA levels at 6 dpi. Surprisingly, we did not observe overexpression of IFN-β or IFN-λ mRNA levels. Finally, we also observed a slight increase of Gata3 and T-bet at 6 dpi, suggesting the involvement of an adaptive and mixed Th1/Th2 response (Fig. 4).
In the spleen, the pattern of transcript expression was different. We observed overexpression of the RIG-I transcript at 4 dpi but not of the Toll-like receptors (TLR). Overexpression of NF-κBp65 at 4 and 6 dpi was also detected, suggesting inflammation in the spleen. However, no increase in IL-6 or IL-1β mRNA levels was observed. We noticed an increase in the anti-inflammatory cytokine transforming growth factor beta 1 (TGF-β1) at 4 dpi. The highest increase in the expression of proinflammatory effectors was observed for CCL2 at 4 dpi. An increase in IFN-γ, lower than that observed in the lungs, was also observed. At 6 dpi, slight overexpression of T-bet transcript was also observed but not of Gata3, in contrast to what was observed in the lungs (Fig. 5).
The NF-κB transcription factor is involved in the activation of genes coding for some proinflammatory cytokines. As we observed overexpression of NF-κB in the spleen of DBA/2 mice (3.3- and 4.4-fold changes at 4 and 6 dpi, respectively), we inoculated transgenic mice expressing a firefly luciferase gene under the control of the NF-κB promoter (NF-κB-luciferase mice) with D/5920 to determine the inflammation induced by IDV. The NF-κB-dependent inflammatory response was analyzed daily by luminescence monitoring in the whole bodies of the infected mice. We observed mild inflammation in mice starting from 2 dpi and lasting until 6 dpi (Fig. 6A). A two-phase response was noticed, with peaks of luminescence at 2 and at 5 dpi (Fig. 6B).
FIG 6.

IDV infection induced two-phase inflammation in mice. NF-κB-luciferase mice were infected with 105 TCID50 of D/5920 intranasally. (A) Bioluminescence was measured from 1 to 7 dpi by inoculation of luciferin intranasally (0.75 mg kg−1) and using the IVIS system. The scale on the right indicates the average radiance, calculated as the sum of the photons per second from each pixel inside the region of interest (ROI)/number of pixels (photons/s/cm2/sr). (B) Bioluminescence activity was quantified using the Living Image software and represented as a graph. fc, fold change; ctrl, control.
These results suggested that IDV could induce only a mild proinflammatory response with a limited increase in NF-κB transcript involved in cellular pathway for cytokine production.
The type I interferon response is not essential to resolve IDV infection.
Type I interferons are especially involved in innate immunity against influenza A virus (IAV), and they are induced after NF-κB, IRF3, and IRF7 activation. Here, we conducted an experiment on mice with a type I interferon receptor knockout (Ifnar1−/−) in order to understand the role of this pathway during IDV infection. Ten 12-week-old Ifnar1−/− mice were infected intranasally with 105 TCID50 of D/5920, and seven Ifnar1−/− mice were inoculated with PBS as the controls.
As observed for the DBA/2 mice, the Ifnar1−/− mice did not show clinical signs or weight loss (Fig. 7A). All of the mice seroconverted (80 ≤ HI titers ≤160), demonstrating that they had all been successfully infected by IDV. These observations suggested that type I interferon response is not essential for the protection against IDV.
FIG 7.

Pathogenicity of D/5920 in Ifnar1−/− mice. Ifnar1−/− mice were infected with 105 TCID50 of D/5920. Clinical signs were recorded daily during 14 days. (A) Weight was recorded every 2 days for noninfected and infected mice. (B) At 4 dpi, 4 infected mice were euthanized, and different organs were collected. Virus titers were determined using the TCID50 method and are expressed in log10 of TCID50/g of tissue. The dotted line represents the positivity threshold (1.5 log10 TCDI50/g of organ). KO, knockout.
To assess viral replication and tropism, we performed necropsies of four infected mice at 4 dpi. IDV presented almost the same tropism and viral titers in Ifnar1−/− and DBA/2 mice, where the virus was detected in nasal turbinates (103.7 to 104 TCID50/g), lungs (101.4 to 102.9 TCID50/g), and intestines (102.9 to 103.4 TCID50/g). Contrary to what was observed in DBA/2 mice, IDV was not detected in the trachea and liver of Ifnar1−/− mice (Fig. 7B).
Together, these results suggested that type I IFN (IFN-I) response could be induced during IDV infection in mice but without being critical in antiviral response against IDV, in contrast to what has been observed for influenza A virus (40).
D/bovine/France/5920/2014 does not undergo much adaptation after passages in DBA/2 mice.
In order to understand if D/5920 could adapt to mice after a few passages and then could induce clinical signs or higher viral replication, we performed 5 serial passages in DAB/2 mice. Briefly, three mice were infected with 105 TCID50 of D/5920 intranasally. At 4 dpi, DBA/2 mice were euthanized, and the respiratory organs (nasal turbinates, trachea, and lungs) were collected and homogenized. The homogenates were used to inoculate three new mice intranasally again. We performed 5 serial passages and then infected eight DBA/2 mice intranasally with the “mouse passage 5” virus (D/5920-M5). Each passage was titrated to confirm the presence of the virus.
The DBA/2 mice infected with the D/5920-M5 virus did not show clinical signs or weight loss, as observed with DBA/2 mice infected with nonadapted virus (Fig. 8A). All of the mice infected with the D/5920-M5 virus seroconverted, with antibody titers similar to those observed with mice infected with nonadapted virus. The viral replication at 4 dpi was assessed in the different organs. IDV tissue tropism was unchanged, as it replicated in respiratory organs and intestines. Moreover, the viral titers were similar between mice infected with nonadapted and mouse passage 5 virus in nasal turbinates (104.1 to 105 TCID50/g), trachea (103.6 to 104.3 TCID50/g), lungs (103.1 to 103.4 TCID50/g), and intestines (103.1 to 104.4 TCID50/g) compared with mice infected with nonadapted virus (P > 0.05) (Fig. 8B).
FIG 8.

Adaptation of D/5920 in DBA/2 mice. Three DBA/2 mice were infected with 105 TCID50 of D/5920. At 4 dpi, mice were euthanized, and nasal turbinates, trachea, and lungs were collected and homogenized. The pure homogenate was used to inoculate three naive DBA/2 mice. After five serial passages, 8 DBA/2 mice were infected intranasally with 30 μl of mouse passage 5 IDV (D/5920-M5). (A) Weight was measured every 2 days, and clinical signs were recorded daily. (B) At 4 dpi, 4 infected mice were euthanized, and different organs were collected. Virus titers were determined using the TCID50 method and are expressed in log10 of TCID50/g of tissue. The dotted line represents the positivity threshold (1.5 log10 TCDI50/g of organ).
We then performed whole-genome sequencing on the D/5920-M5 homogenate. No mutations were observed in the PB1, PB2, P3, NS, and M1 genes. A single substitution (nucleotide [nt] C842T, amino acid [aa] A281V) was observed on the HEF gene.
These results suggested that adaptation did not increase the pathogenicity of IDV (clinical signs or higher viral replication) or that the number of passages (5 passages) was not sufficient to adapt the virus.
DISCUSSION
This study aimed to develop a murine model in order to understand the pathogenesis of a recently identified virus, influenza D virus (IDV).
IDV and absence of clinical signs in mice.
In this study, DBA/2 mice infected with D/5920 did not present respiratory or general clinical signs or weight loss. IDV does not seem to induce disease in mice, as observed in guinea pigs, ferrets, and specific-pathogen-free (SPF) and feral swine models. This contrasts with IAV infection in mice, where mortality and/or morbidity is often observed with high doses of low-pathogenic virus, with mouse-adapted strains, or with highly pathogenic strains (32, 41). IDV was discovered recently, and so far, the pathogenicity of the viral strains remains unknown. We cannot exclude the possibility that the D/5920 strain is more or less pathogenic than are other IDV strains, but further analyses, such as the screening of different viral strains in vivo, could help answer this question. It is likely that the viral dose used for the infection was not responsible for the absence of clinical signs; indeed, we used 105 TCID50, which is a strong dose in mice. The minimal infectious dose for IDV in mice remains unknown, and an experiment with mice infected with different doses of IDV would help determine if the infective dose may play a role in the induction of clinical signs. In the calf model, a dose of 107 TCID50 of D/5920 was used, and only mild clinical signs were observed (29).
The absence of pathogenesis in mice has also been observed with other respiratory viruses, such as the highly prevalent bovine and human respiratory syncytial viruses (bRSV and hRSV, respectively). Several studies used BALB/c mice to reproduce the clinical signs, viral replication, and immune response of hRSV. Almeida et al. developed a murine BALB/c model of bRSV; however, the mice did not present clinical signs, and the virus was difficult to detect using PCR (42). In another study, BALB/c mice were infected with recombinant hRSV expressing the firefly luciferase protein. Despite replication in the mouse respiratory tract, the mice did not present any clinical signs or mortality (43).
IDV adaptation in mice.
Sreenivasan et al. suggested that viruses from the swine D/OK-like clade and from the bovine D/660-like clade may have different tropisms in guinea pigs (30). They hypothesized that the recognition of the HEF glycoprotein to its receptor may vary depending on the clade. A mutation in the binding site of the HEF glycoprotein was observed between isolates from swine D/OK-like (K212) and bovine D/660-like clades (R212); this mutation may modify the specificity and affinity of HEF for its cellular receptor and thus may modify tissue tropism between the two clades (21). So far, the potential differences of IDV strains have not been studied.
As mice are not the main host for IDV, viral adaptation may be required to induce disease. We attempted to adapt the IDV in DBA/2 mice in order to see if this would reveal clinical signs or an increase in viral replication. Many IAV strains indeed need adaptation to induce disease or even viral replication in mice (32, 44). Here, we performed five serial passages in mice to obtain a virus named D/5920-M5. DBA/2 mice infected with D/5920-M5 did not present clinical signs or a significant increase in viral replication at 4 dpi compared to DBA/2 mice infected with original virus. We observed very little genetic adaptation in mice; indeed, full-genome sequencing showed that adaptation only induced one mutation in the HEF gene (not located in the receptor binding site). In comparison, 6 amino acid mutations were observed on D/bovine/Oklahoma/660/2013 after one passage in the guinea pig model (30).
The number of serial passages was maybe not sufficient to adapt IDV to DBA/2 mice. For example, an adaptation of seasonal H1N1 (A/Brisbane/59/2007) virus in mice showed that mice presented clinical signs from the fifth passage and mortality from the sixth. A higher level of viral replication was also observed after the fifth passage in mice (45). For influenza B virus (IBV), clinical signs and an increase in viral replication were observed after 12 passages in mice (46). D/5920, however, did not lose fitness in mice after passages, as evident by similar titers and tissue tropisms. Another hypothesis is that IDV may not induce disease in mice no matter the number of passages. However, we noticed an important D/5920 adaptation in swine testis (ST) cells. Our inoculum virus, passaged 5 times on ST cells, was indeed different from the initially sequenced D/5920 virus, with 2 mutations in HEF (G628A and G684A) suggesting adaptation in cells (data not shown). It is unknown whether a wild-type, non-cell-culture-adapted virus would be virulent in mice.
IDV’s tropism in mice is respiratory but also enteric.
The limited literature available on IDV pathogenesis in vivo suggested a respiratory tropism of the pathogen. In ferrets and pigs, IDV replicated in nasal turbinates only (1). In calves and guinea pigs, virus replicated in both the upper and lower respiratory tract (28–30).
Our findings confirmed the respiratory tropism of D/5920 in mice. Here, D/5920 (D/OK-like clade) replicated mainly in the upper respiratory tract, with a peak at 4 dpi (nasal turbinates). We also observed replication to a lower extent in the middle (trachea) and lower respiratory tract, as observed in a guinea pig model (30). Contrary to this model, we observed lesions only in the nasal turbinates but not in the lungs of mice. We also noticed a time difference between virus detection and microscopic lesions for D/5920 infection (positive virus detection data at 4 dpi and microscopic lesions at 6 dpi). The immune response at 6 dpi may be sufficient to inhibit IDV replication and avoid more severe lesions. Interestingly, viral replication was similar to that observed in the main host, bovines, experimentally (29). In the calf model developed by Salem et al., the viral strain D/5920 replicated both in the upper and lower respiratory tract (29). In this model, the virus was detected at a higher titer in nasal swabs (5.6.108 RNA copies/ml) than in bronchoalveolar lavage fluid (1.4.105 RNA copies/ml), and also with similar titers in organs (lungs and nasal mucosa).
IDV was also detected with low titers in the intestines, but no lesions were observed. Previous studies of IAV infection showed high titers of H1N1 viruses in the respiratory tract and low titers in the intestines and feces of DBA/2 mice (47). Bao et al. also observed low titers of H7N9 in BALB/c intestines (48). No IDV (or with very low titers) has been detected in the enteric organs so far in other experimental models (intestines for ferrets [1] and rectal swabs for a calf model [28, 29]). The differences in physiology and receptor expression in the intestines could be responsible for these results. In a recent study, however, IDV was detected in rectal swabs from goats, suggesting that IDV could present an enteric tropism or could be orally transmitted in other susceptible species (9). Unfortunately, no rectal swabs were so far tested for cattle or swine in the field, which makes difficult to determine if IDV harbors an enteric tropism. IDV is able to bind cells through 9-O-acetylated sialic acids expressed on the cell surface (49). So far, the presence of 9-O-acetylated sialic acids has not been demonstrated in mouse intestines. However, a study demonstrated that rats express these sialic acids in the intestines, suggesting that it could be the same in mice (50). Here, we cannot exclude the possibility of oral contamination during infection.
Surprisingly, IDV was detected in mouse livers with low titers at 2 dpi, while blood samples were negative at the same time point. Transient viremia may occur early in the course of IDV infection. Blood samples collected in the first hours postinfection would help us understand if IDV indeed causes viremia. It was previously suggested that IAV could be viremic in mice. Indeed, virus was detected in the red blood cell fraction in the early step of infection, but it was virus strain dependent and required high PCR sensitivity and a high dose of inoculum (51). Recently, a study from China detected IDV in diseased dairy cattle, buffalo, and goat serum samples using PCR (9). Transient viremia was also observed in feral swine and calf experimental models. The virus was detected at 3 and 5 dpi in feral swine and 3 and 5 dpi in cattle, with around 3 log10 TCID50/ml in both studies, suggesting that IDV induces viremia (31, 52). We hypothesize that IDV could pass into the bloodstream using blood vessels in nasal turbinates and thus infect other tissues, such as those of the liver or the intestines. Further evidence, however, is warranted to really assess the putative viremia associated with IDV in mice. Our IHC findings confirm the presence of IDV in nasal turbinates but not in any other organs (Fig. 3). This suggests either that the sensitivity of the technique does not allow for the detection of low levels of virus or that the nonrespiratory tissues may carry virus without it replicating locally. Previously, IDV seemed to harbor a respiratory tropism in the field and in all animal models (1, 28, 30). Most IDV studies focused on the respiratory tract, except for a pathogenesis study in ferrets, where no IDV replication was observed outside the respiratory tract (1). We now have confirmed the respiratory tropism of IDV, but our findings also warrant further investigation of a putative viremia and/or intestinal tropism of the pathogen at least in mice.
Influenza viruses and the humoral immune response.
The antibody response likely plays an important role in controlling IDV infection, as it does for IAV infection. Previously, when several antigens were used to determine the titer of IDV antibodies in ruminant sera using a hemagglutination inhibition assay, 2-fold differences in titers were observed in average (19), suggesting a limited antigenic diversity between the 2 IDV clades (genotypes). In contrast, here, all infected mice seroconverted at 14 dpi, with antibody titers ranging from 1:15 to 1:240 against the homologous strain and from 1:20 to 1:40 against a heterologous strain (D/Neb or D/5920), suggesting a high antigenic difference between the two clades, as was initially observed in the United States by Collin et al. (21).
A mild proinflammatory response is sufficient to clear IDV infection.
To understand the immune response against IDV in mice, we analyzed 36 genes involved in innate and adaptive responses using transcriptomics. Previously, the immune response was analyzed only in the calf model, using both transcriptomics and proteomics (29). IDV seems to induce a mild innate response in mice. Here, we observed differences between the mouse and calf models. First, we observed a lower induction of proinflammatory gene expression in mice than in calves. The greater IDV fitness in calves than that in mice could explain these differences, although we cannot exclude the possibility that the immune response in calves contributes to the clinical outcome.
Differences between the calf and mouse models could also be explained by the type of samples and the times of sample collection; bronchoalveolar lavage fluid was used for calves versus lung and spleen homogenates used for mice, and the immune response was measured at 2, 4, and 6 dpi in mice but at 2, 7, and 14 dpi in calves. The methodology and biostatistical analyses used for the two studies (microfluidic quantitative PCR [qPCR] on the platform BioMark for calves and classical real-time RT-PCR on a LightCycler for mice) were different. Indeed, the immune response was monitored for each time point with the same calves, and the statistical analysis was performed using a linear mixed model with random effect for group, considering interactions between time and status (infected or control). In the mouse model, one mouse was used at each time point, modifying the data analysis.
Another difference between the two models was the pattern of gene overexpression after IDV infection. A higher number of genes was overexpressed in calves than in mice. The highest fold changes between infected and control mice were observed for IFN-γ, IL-1β, and CCL5 at 2 dpi and TLR7 at 6 dpi in the lungs, and for RIG-I, CCL2, and TGF-β1 at 4 dpi and NF-κB at 4 to 6 dpi in the spleen. In calves, an overexpression of RIG-I, TGF-β1, IFN-γ, and CCL2 at the different time points was observed. Moreover, an increase in TLR, chemokine, cytokine, and pathway molecule transcript levels was also observed in calves. No overexpression of NF-κB, IL-1β, and CCL5 was, however, recorded in this model, but the different methodologies could be responsible for this observation (29).
TLR7 is an endosomal TLR and recognizes single-stranded RNA (ssRNA), whereas RIG-I recognizes RNA bearing a 5′-triphosphate end (53). The activation of TLR7 and RIG-I led to the recruitment of transcriptional factors such as NF-κB and then the production of cytokines. IDV infection induced an increase in NF-κB and in IL-1β, TGF-β1, CCL2, and CCL5 transcript levels in the lungs or spleen of mice. These molecules are produced by epithelial cells, neutrophils, and monocytes during the first step of the immune response. CCL2 and CCL5 are proinflammatory chemokines involved in the recruitment of T cells and monocytes. TGF-β1 is involved in the regulation of immune response, both inflammatory and regulatory. Recently, it has been demonstrated that TGF-β1 acts as a proviral molecule in the lungs of mice infected by IAV by inhibiting the type I interferon response (54). The early overexpression of IFN-γ and CCL5 suggests the involvement of NK cells during IDV infection. Indeed, CLL5 is known to recruit NK cells in the lungs of mice infected with IAV (55). It has been demonstrated that NK cells are important during the early stage of IAV infection by secreting different cytokines, including IFN-γ, and also acting as cytotoxic cells. The production of IFN-γ by NK cells also stimulates the activation of macrophages (56, 57).
Finally, at 6 dpi, mild overexpression of Gata-3 and T-bet transcription factors suggests the activation of mixed and local Th1/Th2 lymphocyte responses, as observed in the calf model (29). Contrary to the observation in the calf model, we did not detect IFN-γ or IL-13 overexpression in the latest stage of IDV infection. Their secretion in mice may occur at a later point (after 6 dpi).
In order to confirm the mild inflammation induced by IDV infection, we inoculated NF-κB-luciferase mice. This mouse model was previously used for IAV infection. Indeed, NF-κB-luciferase mice infected with 1.105 PFU of A/WSN/1933 (H1N1) developed clinical signs and presented a high level of inflammation, with detection of luciferase starting at 1 dpi, reaching a peak at 2 to 3 dpi in the respiratory tract, and showing systemic inflammation after 3 dpi (58).
Here, we did not observe clinical signs during IDV infection. Moreover, we only observed a mild and two-phase response after infection at 2 and 5 dpi. The inflammation observed at 2 and 5 dpi in NF-κB-luciferase mice seems to correlate with the time pattern of cytokine expression observed in in DBA/2 mice and viral replication at 4 dpi. Moreover, the intensity of inflammation in NF-κB-luciferase mice correlates with the mild overexpression of proinflammatory genes in DBA/2 mice. Unfortunately, a transcriptomic method cannot allow us to discriminate which cells are secreting the cytokines. Analyses with flow cytometry could help us gain a better understanding of the immune response after IDV infection in mice. In NF-κB-luciferase mice, we can hypothesize that the inflammation observed at 2 dpi could be induced by the infected epithelium of respiratory organs. These cells are the first target of influenza virus and induce a rapid production of antiviral, proinflammatory, and chemotactic molecules after TLR induction. Airway epithelial cells produce IFN-I and cytokines such as IL-1, IL-6, and CCL5 during the first 3 to 6 h postinfection to target the virus and recruit and polarize the immune cells (59). The second peak of inflammation at 5 dpi in NF-κB-luciferase mice could be induced by the other cells involved in innate immunity, such as monocytes, macrophages, and neutrophils, which also secrete proinflammatory cytokines during infection.
In infected DBA/2 mice, we did not detect overexpression of IFN-I, but we observed an increase in IRF3 and 2′,5′-OAS transcript levels, suggesting the involvement of this pathway during IDV infection. Type I IFNs are critical in the anti-IAV response in mice (40, 60). In order to determine the role of these proteins during IDV infection, Ifnar1−/− mice, which do not express the receptor for IFN-I, were infected with D/5920. These mice did not present clinical signs or higher level of viral replication than with DBA/2 mice.
Four hypotheses could explain our results. The first is that IFN-I are not involved in the immune response against IDV. Another possibility is that IFN-I would be involved in the immune response but the virus would inhibit them rapidly after secretion, hence the absence of overexpression or differences in Ifnar1−/− mice. IAV escape from the immune system occurs thanks to the NS1 protein. It has been demonstrated that NS1 inhibits IFN-I by the inhibition of STAT1 and STAT2 phosphorylation and upregulation of the JAK/STAT inhibitors SOCS1 and SOCS3 (61).
Our results are supported by the results obtained in the calf model. Indeed, no overexpression of IFN-I or IFN-I pathway molecules was observed in calves infected with D/5920. Salem et al. observed overexpression of SOCS1 and SOCS3 (29). In DBA/2 mice, we unfortunately failed to detect SOCS1 mRNA in both the lungs and spleen. Moreover, we did not also analyze SOCS3 transcripts; therefore, we are unable to further explore this hypothesis. Thus, analyses are required to understand the involvement of the IFN-I response during IDV infection. The third hypothesis is the involvement of type III interferons (IFN-III) instead of IFN-I during IDV infection. These cytokines are also induced during IAV infection and stimulate the same interferon-stimulated genes (ISGs) as IFN-I (62). Here, we did not observe overexpression of IFN-λ in the lungs and spleen of DBA/2 mice, suggesting that IFN-III are not major players in the immunity against IDV. It is also possible that SOCS1 and SOCS3 are able to inhibit these interferons, as IFN-III use the same signaling pathway as IFN-I (62). Finally, since the Ifnar1−/− mice used here were not generated on a DBA/2 strain (but on a C57Bl/6 strain), one cannot rule out that the genetic background plays a role on mouse susceptibility to IDV infection (hypothesis 4). However, 10 wild-type C57Bl/6 mice were infected with IDV as a control, and virus replication was observed in the respiratory tract with titers similar to those in DBA/2 mice (data not shown).
In summary, despite the absence of clinical signs, mice were susceptible to IDV infection. IDV replicated mainly in the upper respiratory tract, corroborating the notion that IDV presents a respiratory tropism. IDV might also induce viremia and show a slight enteric tropism. Little is known about the immune response against IDV. Susceptible hosts, including mice, seroconverted after infection, suggesting a strong antibody response (IgG), but the induced innate and cellular responses are still to be fully assessed. Taken together, mice are a convenient and relevant small-animal model to study IDV viral replication and will help better understand the phenotypes associated with different strains of the recently discovered pathogen. It could also be a useful model to study the immune response through transgenic mice and the availability of reagents. Disease, however, cannot be reproduced in DBA/2 mice, and further studies are warranted to develop a more relevant model to study IDV pathogenesis.
MATERIALS AND METHODS
Cells and viruses.
Swine testis cells (ATCC) were grown in Dulbecco’s modified Eagle medium (DMEM) complemented with 10% fetal bovine serum and penicillin-streptomycin (Dutscher) and incubated for 24 h at 37°C with 5% CO2. In this study, we used the viral strain D/bovine/France/5920/2014 (D/5920) (29). Viruses were grown on ST cells in Opti-MEM medium complemented with penicillin-streptomycin (Dutscher) at 37°C and 5% CO2 for 5 days. Viruses were stored at –80°C until further used. The virus titers were determined by the TCID50, as described below.
Mice.
Six- and 10-week-old female DBA/2JRj mice were purchased from Janvier Labs (Le Genest-Saint-Isle, France). Twelve-week-old Ifnar1−/− (C57BL/6 backbone) mice were bred at IBPS (Toulouse). Ten-week-old NF-κB-luciferase BALB/c mice were bred at INRA-VIM (Jouy-en-Josas). The DBA/2 and Ifnar1−/− mice were housed at the Veterinary School of Toulouse in animal biosafety level 2 (ABSL‐2) facilities and had food and water provided ad libitum. NF-κB-luciferase BALB/c mice were housed at INRA of Jouy-en-Josas (France) in ABSL-2 facilities as well. Experimentations were conducted in accordance with European and French legislations on Laboratory Animal Care and Use (French Decree 2001-464 and European Directive CEE86/609), and the animal protocol was approved by the ethics committee Sciences et Santé Animale, committee number 115 (protocol no. 2018030212288103) and by the Animal Care and Use Committee at Centre de Recherche de Jouy-en-Josas (COMETHEA) (protocol no. 2015100910396112v1).
Experimental design.
(i) Infection of DBA/2 mice. Six-week-old female DBA/2JRj mice were separated in two groups, with 11 noninfected (controls) mice and 36 infected mice. Mice were lightly anesthetized with a ketamine-xylazine combination and infected with 30 μl of virus intranasally or with PBS as a control. Clinical observations were recorded daily, and weight was measured every 2 days. Any animal showing a weight loss greater than 30% or signs of suffering was humanely euthanized. Mice were infected intranasally with 105 TCID50 of D/5920. Necropsies were performed at 2, 4, 6, and 8 dpi (5 infected mice per day), and brain, nasal turbinate, tracheal, lung, spleen, liver, kidney, and intestinal (duodenum, jejunum, and colon) tissues were sampled. Blood was also collected at 2, 6, and 14 dpi. All the mice were euthanized at 14 dpi.
(ii) Comparison between DBA/2 and Ifnar1−/− mice. Ten-week-old female DBA/2JRj mice and 12-week-old Ifnar1−/− mice were separated into two groups, with 11 noninfected (controls) and 11 infected DBA/2 mice and 7 noninfected (3 females and 4 males) and 10 infected (5 males and 5 females) Ifnar1−/− mice. Mouse infection and monitoring were performed as described above.
Necropsies were performed at 2, 4, and 6 dpi for DBA/2 mice with 3 noninfected and 3 infected mice per day. For Ifnar1−/− mice, necropsies were performed only at 4 dpi (1 female noninfected and 2 females infected, and 2 males noninfected and 2 males infected). The brain, nasal turbinates, trachea, lungs, spleen, liver, kidneys, and intestines were collected and treated as described in Materials and Methods. Blood was collected at 14 dpi before the remaining mice were euthanized.
(iii) Infection of NF-κB-luciferase mice. Ten-week-old NF-κB-luciferase mice were separated into two groups, with 5 noninfected (controls) and 8 infected mice. Infection was performed under anesthesia, and 105 TCID50 of D/5920 were inoculated intranasally in mice. Control mice were inoculated with PBS. Weight loss and clinical signs were recorded daily. Bioluminescence measurements were performed from 1 to 7 dpi. Mice were anesthetized with isoflurane (2% in oxygen as a carrier gas), and luminescence was measured 2 min after intranasal inoculation of 50 μl of PBS containing luciferin (0.75 mg kg−1; Sigma). Luciferase activity was measured using the Living Image software (version 4.0; PerkinElmer). Bioluminescence images were acquired for 1 min with f/stop = 1 and binning = 8. A digital false-color photon emission image of the mouse was generated, and photons were counted within the whole-body area. Photon emission was measured as radiance in p s−1 cm−2 sr−1 (58).
(iv) Adaptation of IDV in DBA/2 mice. Six-week-old female DBA/2JRj mice were infected with 105 TCID50 of D/5920 intranasally. Necropsies were performed at 4 dpi for three mice, and nasal turbinates, trachea, and lungs were collected and the tissues homogenized. The pure homogenate was used to intranasally infect three naive mice (30 μl/mouse) and titrated in parallel using the TCID50 method. After five serial passages, 16 DBA/2 mice were separated in two groups, with 8 noninfected (controls) mice and 8 mice infected intranasally with 30 μl of mouse-passaged IDV (D/5920-M5). Clinical observations were recorded daily, and necropsies were performed at 4 dpi (3 mice infected and 3 mice noninfected; the organs collected were the brain, nasal turbinates, trachea, lungs, spleen, liver, kidneys, and intestines). Blood was collected at 14 dpi before euthanasia of the remaining mice.
Sample treatments.
Whole organs were dissociated using the tissue homogenizer Precellys 24 (Ozyme), resuspended in 500 μl of PBS, centrifuged, and stored at –80°C until further analysis. Blood was also taken at the end of the experiments (14 dpi) for serology. Blood was taken at 2 dpi and 6 dpi to study viremia.
Determination of virus titers by TCID50.
All samples from infected mice were titrated using the 50% tissue culture infective dose (TCID50), as described elsewhere (63), using ST cells and with a 5-day culture. The titers were determined using the Reed and Muench method (64). A positivity threshold was set at 1.5 log10 TCID50/g of organ.
Real-time PCR to study the immune response.
The real-time RT-PCR for immune response quantification was performed using the iTaq Universal SYBR green one-step kit (Bio-Rad), and the primer sequences are available upon request. For the relative quantification, we used a housekeeping gene (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]) to normalize the amount of the target gene. We used the following calibrator formula: 2−ΔΔCT. Here, CT is the cycle threshold, and ΔΔCT represents the following: ΔCT (sample) ([CTcytokine gene − CThousekeeping gene] of infected mice) − ΔCT (calibrator) ([CTcytokine gene – CThousekeeping gene] of noninfected mice). The mean ΔCT values were used for analysis, and the results are represented as the mean ± standard deviation (SD).
Hemagglutination inhibition assay.
Sera were treated with receptor-destroying enzyme (RDE; Seika), following the manufacturer’s instructions. Then, they were hemadsorbed on packed horse red blood cells. The HI assay was performed as previously described (29), with 4 hemagglutination units (UHA) of D/bovine/France/5920/2014 or D/bovine/Nebraska/9-5/2012 and 1% horse red blood cells.
Histology and immunochemistry.
At 2 dpi and 6 dpi, organs were collected and fixed in 10% paraformaldehyde for 2 days. Nasal turbinates were decalcified in EDTA for 1 week. After fixation, tissues were routinely processed in paraffin blocks, sectioned at 3 μm, and stained with hematoxylin and eosin for microscopic examination. Lesions were assessed histologically and graded as follows: −, no lesion; +, light lesions; ++, moderate lesions; and +++, marked lesions.
Immunoreaction was performed on the paraffin-embedded sections with an in-house polyclonal rabbit anti-IDV antibody (0.05% pronase retrieval solution, 10 min at 37°C; antibody dilution 1/1,000, incubation overnight at 4°C) after a blocking step with normal goat serum (1/10 dilution; catalog no. X0907; Dako). The anti-IDV antibody was revealed with a biotinylated polyclonal goat anti-rabbit immunoglobulin conjugated with horseradish peroxidase (HRP; LSAB2 system-HRP, catalog no. K0675; Dako) and the diaminobenzidine chromogen of the HRP (catalog no. TA-125-HDX; Thermo Scientific).
Sequencing.
Viral RNA was extracted from organ homogenates or from cell culture supernatant for the inoculum, as described above, and cDNA was generated using the RevertAid reverse transcriptase (RT) kit (Thermo Fisher), according to the manufacturer’s instructions. PCR was performed for each gene segment using Phusion high-fidelity polymerase (Thermo Fisher) and previously published primers (12). The PCR products were purified using the QIAquick gel extraction kit (Qiagen), according to the manufacturer’s instructions. Sanger sequencing was performed using the GATC Biotech platform (Germany), and the sequences were aligned with ClustalW, available on BioEdit (65), and compared with reference D/5920 sequences.
Statistics.
The variations in the weights of the mice were analyzed using a two-way analysis of variance (ANOVA), followed by the Bonferroni multiple-comparison test. The viral (TCID50) titers were compared with a one-way ANOVA, followed by the Tukey multiple-comparison test. The fold changes in mRNA transcript levels for the immune response were compared using a one-way ANOVA, followed by Dunn’s multiple-comparison test. Statistical tests were performed using GraphPad Prism 5.0. A P value of ≤0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Jean-Marc Delmas and Marion Robine (ENVT) for their help in the animal facility and Séverine Boullier for providing us with horse blood.
This work was funded by the Agence Nationale pour la Recherche (ANR), project FLUD. J. Oliva is supported by a PhD scholarship of the French Ministry of Higher Education and Research. We thank the MIMA2 platform for access to IVIS200, which was acquired with funds from the Ile de France région (SESAME).
REFERENCES
- 1.Hause BM, Ducatez M, Collin EA, Ran Z, Liu R, Sheng Z, Armien A, Kaplan B, Chakravarty S, Hoppe AD, Webby RJ, Simonson RR, Li F. 2013. Isolation of a novel swine influenza virus from Oklahoma in 2011 which is distantly related to human influenza C viruses. PLoS Pathog 9:e1003176. doi: 10.1371/journal.ppat.1003176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hause BM, Collin EA, Liu R, Huang B, Sheng Z, Lu W, Wang D, Nelson EA, Li F. 2014. Characterization of a novel influenza virus in cattle and swine: proposal for a new genus in the Orthomyxoviridae family. mBio 5:e00031-14. doi: 10.1128/mBio.00031-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferguson L, Eckard L, Epperson WB, Long L-P, Smith D, Huston C, Genova S, Webby R, Wan X-F. 2015. Influenza D virus infection in Mississippi beef cattle. Virology 486:28–34. doi: 10.1016/j.virol.2015.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo J, Ferguson L, Smith DR, Woolums AR, Epperson WB, Wan X-F. 2017. Serological evidence for high prevalence of influenza D viruses in Cattle, Nebraska, United States, 2003–2004. Virology 501:88–91. doi: 10.1016/j.virol.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang M, Hill JE, Fernando C, Alexander TW, Timsit E, van der Meer F, Huang Y. 2019. Respiratory viruses identified in western Canadian beef cattle by metagenomic sequencing and their association with bovine respiratory disease. Transbound Emerg Dis 66:1379–1386 doi: 10.1111/tbed.13172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitra N, Cernicchiaro N, Torres S, Li F, Hause BM. 2016. Metagenomic characterization of the virome associated with bovine respiratory disease in feedlot cattle identified novel viruses and suggests an etiologic role for influenza D virus. J Gen Virol 97:1771–1784. doi: 10.1099/jgv.0.000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murakami S. 2016. Influenza D virus infection in herd of cattle, Japan. Emerg Infect Dis 22:1517–1519. doi: 10.3201/eid2208.160362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horimoto T, Hiono T, Mekata H, Odagiri T, Lei Z, Kobayashi T, Norimine J, Inoshima Y, Hikono H, Murakami K, Sato R, Murakami H, Sakaguchi M, Ishii K, Ando T, Otomaru K, Ozawa M, Sakoda Y, Murakami S. 2016. Nationwide distribution of bovine influenza D virus infection in Japan. PLoS One 11:e0163828. doi: 10.1371/journal.pone.0163828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhai S, Zhang H, Chen S, Zhou X, Lin T, Liu R, Lv D, Wen X, Wei W, Wang D, Li F. 2017. Influenza D virus in animal species in Guangdong Province, southern China. Emerg Infect Dis 23:1392–1396. doi: 10.3201/eid2308.170059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang W-M, Wang S-C, Peng C, Yu J-M, Zhuang Q-Y, Hou G-Y, Liu S, Li J-P, Chen J-M. 2014. Identification of a potential novel type of influenza virus in bovine in China. Virus Genes 49:493–496. doi: 10.1007/s11262-014-1107-3. [DOI] [PubMed] [Google Scholar]
- 11.Mekata H, Yamamoto M, Hamabe S, Tanaka H, Omatsu T, Mizutani T, Hause BM, Okabayashi T. 2017. Molecular epidemiological survey and phylogenetic analysis of bovine influenza D virus in Japan. Transbound Emerg Dis 65:e355–e360. doi: 10.1111/tbed.12765. [DOI] [PubMed] [Google Scholar]
- 12.Ducatez MF, Pelletier C, Meyer G. 2015. Influenza D virus in cattle, France, 2011–2014. Emerg Infect Dis 21:368–371. doi: 10.3201/eid2102.141449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oliva J, Eichenbaum A, Belin J, Gaudino M, Guillotin J, Alzieu J-P, Nicollet P, Brugidou R, Gueneau E, Michel E, Meyer G, Ducatez MF. 2019. Serological evidence of influenza D virus circulation among cattle and small ruminants in France. Viruses 11:516. doi: 10.3390/v11060516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foni E, Chiapponi C, Baioni L, Zanni I, Merenda M, Rosignoli C, Kyriakis CS, Luini MV, Mandola ML, Bolzoni L, Nigrelli AD, Faccini S. 2017. Influenza D in Italy: towards a better understanding of an emerging viral infection in swine. Sci Rep 7:11660. doi: 10.1038/s41598-017-12012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiapponi C, Faccini S, De Mattia A, Baioni L, Barbieri I, Rosignoli C, Nigrelli A, Foni E. 2016. Detection of influenza D virus among swine and cattle, Italy. Emerg Infect Dis 22:352–354. doi: 10.3201/eid2202.151439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flynn O, Gallagher C, Mooney J, Irvine C, Ducatez M, Hause B, McGrath G, Ryan E. 2018. Influenza D virus in cattle, Ireland. Emerg Infect Dis 24:389–391. doi: 10.3201/eid2402.170759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snoeck C, Oliva J, Pauly M, Losch S, Wildschutz F, Muller C, Hübschen J, Ducatez M. 2018. Influenza D virus circulation in cattle and swine, Luxembourg, 2012–2016. Emerg Infect Dis 24:1388–1389. doi: 10.3201/eid2407.171937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dane H, Duffy C, Guelbenzu M, Hause B, Fee S, Forster F, McMenamy MJ, Lemon K. 2019. Detection of influenza D virus in bovine respiratory disease samples, U.K. Transbound Emerg Dis 66:2184–2187. doi: 10.1111/tbed.13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salem E, Cook E, Lbacha H, Oliva J, Awoume F, Aplogan G, Couacy Hymann E, Muloi D, Deem S, Alali S, Zouagui Z, Fèvre E, Meyer G, Ducatez M. 2017. Serologic evidence for influenza C and D virus among ruminants and camelids, Africa, 1991–2015. Emerg Infect Dis 23:1556–1559. doi: 10.3201/eid2309.170342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murakami S, Odagiri T, Melaku SK, Bazartseren B, Ishida H, Takenaka-Uema A, Muraki Y, Sentsui H, Horimoto T. 2019. Influenza D virus infection in dromedary camels, Ethiopia. Emerg Infect Dis 25:1224–1226. doi: 10.3201/eid2506.181158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collin EA, Sheng Z, Lang Y, Ma W, Hause BM, Li F. 2015. Cocirculation of two distinct genetic and antigenic lineages of proposed influenza D virus in cattle. J Virol 89:1036–1042. doi: 10.1128/JVI.02718-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quast M, Sreenivasan C, Sexton G, Nedland H, Singrey A, Fawcett L, Miller G, Lauer D, Voss S, Pollock S, Cunha CW, Christopher-Hennings J, Nelson E, Li F. 2015. Serological evidence for the presence of influenza D virus in small ruminants. Vet Microbiol 180:281–285. doi: 10.1016/j.vetmic.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nedland H, Wollman J, Sreenivasan C, Quast M, Singrey A, Fawcett L, Christopher-Hennings J, Nelson E, Kaushik RS, Wang D, Li F. 2017. Serological evidence for the co-circulation of two lineages of influenza D viruses in equine populations of the Midwest United States. Zoonoses Public Health 65:e148–e154. doi: 10.1111/zph.12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckard L. 2016. Assessment of the zoonotic potential of a novel bovine influenza virus. PhD dissertation. University of Tennessee Health Science Center, Memphis, TN. [Google Scholar]
- 25.White SK, Ma W, McDaniel CJ, Gray GC, Lednicky JA. 2016. Serologic evidence of exposure to influenza D virus among persons with occupational contact with cattle. J Clin Virol 81:31–33. doi: 10.1016/j.jcv.2016.05.017. [DOI] [PubMed] [Google Scholar]
- 26.Borkenhagen LK, Mallinson KA, Tsao RW, Ha S-J, Lim W-H, Toh T-H, Anderson BD, Fieldhouse JK, Philo SE, Chong K-S, Lindsley WG, Ramirez A, Lowe JF, Coleman KK, Gray GC. 2018. Surveillance for respiratory and diarrheal pathogens at the human-pig interface in Sarawak, Malaysia. PLoS One 13:e0201295. doi: 10.1371/journal.pone.0201295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belser JA, Katz JM, Tumpey TM. 2011. The ferret as a model organism to study influenza A virus infection. Dis Model Mech 4:575–579. doi: 10.1242/dmm.007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferguson L, Olivier AK, Genova S, Epperson WB, Smith DR, Schneider L, Barton K, McCuan K, Webby RJ, Wan X-F. 2016. Pathogenesis of influenza D virus in cattle. J Virol 90:5636–5642. doi: 10.1128/JVI.03122-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salem E, Hägglund S, Cassard H, Corre T, Näslund K, Foret C, Gauthier D, Pinard A, Delverdier M, Zohari S, Valarcher J-F, Ducatez M, Meyer G. 2019. Pathogenesis, host innate immune response and aerosol transmission of influenza D virus in cattle. J Virol 93:e01853-18. doi: 10.1128/JVI.01853-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sreenivasan C, Thomas M, Sheng Z, Hause BM, Collin EA, Knudsen DEB, Pillatzki A, Nelson E, Wang D, Kaushik RS, Li F. 2015. Replication and transmission of the novel bovine influenza D virus in a guinea pig model. J Virol 89:11990–12001. doi: 10.1128/JVI.01630-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferguson L, Luo K, Olivier AK, Cunningham FL, Blackmon S, Hanson-Dorr K, Sun H, Baroch J, Lutman MW, Quade B, Epperson W, Webby R, DeLiberto TJ, Wan X-F. 2018. Influenza D virus infection in feral swine populations, United States. Emerg Infect Dis 24:1020–1028. doi: 10.3201/eid2406.172102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouvier NM, Lowen AC. 2010. Animal models for influenza virus pathogenesis and transmission. Viruses 2:1530–1563. doi: 10.3390/v20801530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pica N, Iyer A, Ramos I, Bouvier NM, Fernandez-Sesma A, García-Sastre A, Lowen AC, Palese P, Steel J. 2011. The DBA.2 mouse is susceptible to disease following infection with a broad, but limited, range of influenza A and B viruses. J Virol 85:12825–12829. doi: 10.1128/JVI.05930-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ibricevic A, Pekosz A, Walter MJ, Newby C, Battaile JT, Brown EG, Holtzman MJ, Brody SL. 2006. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol 80:7469–7480. doi: 10.1128/JVI.02677-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miao H, Hollenbaugh JA, Zand MS, Holden-Wiltse J, Mosmann TR, Perelson AS, Wu H, Topham DJ. 2010. Quantifying the early immune response and adaptive immune response kinetics in mice infected with influenza A virus. J Virol 84:6687–6698. doi: 10.1128/JVI.00266-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Groves HT, McDonald JU, Langat P, Kinnear E, Kellam P, McCauley J, Ellis J, Thompson C, Elderfield R, Parker L, Barclay W, Tregoning JS. 2018. Mouse models of influenza infection with circulating strains to test seasonal vaccine efficacy. Front Immunol 9:126. doi: 10.3389/fimmu.2018.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding Y, Cao Z, Cao L, Ding G, Wang Z, Xiao W. 2017. Antiviral activity of chlorogenic acid against influenza A (H1N1/H3N2) virus and its inhibition of neuraminidase. Sci Rep 7:45723. doi: 10.1038/srep45723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boon ACM, deBeauchamp J, Hollmann A, Luke J, Kotb M, Rowe S, Finkelstein D, Neale G, Lu L, Williams RW, Webby RJ. 2009. Host genetic variation affects resistance to infection with a highly pathogenic H5N1 influenza A virus in mice. J Virol 83:10417–10426. doi: 10.1128/JVI.00514-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evseenko VA, Bukin EK, Zaykovskaya AV, Sharshov KA, Ternovoi VA, Ignatyev GM, Shestopalov AM. 2007. Experimental infection of H5N1 HPAI in BALB/c mice. Virol J 4:77. doi: 10.1186/1743-422X-4-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arimori Y, Nakamura R, Yamada H, Shibata K, Maeda N, Kase T, Yoshikai Y. 2013. Type I interferon limits influenza virus-induced acute lung injury by regulation of excessive inflammation in mice. Antiviral Res 99:230–237. doi: 10.1016/j.antiviral.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 41.Kamal RP, Katz JM, York IA. 2014. Molecular determinants of influenza virus pathogenesis in mice, p 243–274. In Compans RW, Oldstone MBA (ed), Influenza pathogenesis and control, vol I. Springer, Cham, Switzerland. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Almeida RS, Domingues HG, Coswig LT, D'Arce RCF, de Carvalho RF, Arns CW. 2004. Detection of bovine respiratory syncytial virus in experimentally infected BALB/c mice. Vet Res 35:189–197. doi: 10.1051/vetres:2004003. [DOI] [PubMed] [Google Scholar]
- 43.Rameix-Welti M-A, Le Goffic R, Hervé P-L, Sourimant J, Rémot A, Riffault S, Yu Q, Galloux M, Gault E, Eléouët J-F. 2014. Visualizing the replication of respiratory syncytial virus in cells and in living mice. Nat Commun 5:5104. doi: 10.1038/ncomms6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barnard DL. 2009. Animal models for the study of influenza pathogenesis and therapy. Antiviral Res 82:A110–A122. doi: 10.1016/j.antiviral.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu L, Bao L, Li F, Lv Q, Ma Y, Zhou J, Xu Y, Deng W, Zhan L, Zhu H, Ma C, Shu Y, Qin C. 2011. Adaption of seasonal H1N1 influenza virus in mice. PLoS One 6:e28901. doi: 10.1371/journal.pone.0028901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim E-H, Park S-J, Kwon H-I, Kim SM, Kim Y, Song M-S, Choi E-J, Pascua PNQ, Choi Y-K. 2015. Mouse adaptation of influenza B virus increases replication in the upper respiratory tract and results in droplet transmissibility in ferrets. Sci Rep 5:15940. doi: 10.1038/srep15940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koçer ZA, Krauss S, Stallknecht DE, Rehg JE, Webster RG. 2012. The potential of avian H1N1 influenza A viruses to replicate and cause disease in mammalian models. PLoS One 7:e41609. doi: 10.1371/journal.pone.0041609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bao L, Xu L, Zhu H, Deng W, Chen T, Lv Q, Li F, Yuan J, Xu Y, Huang L, Li Y, Liu J, Yao Y, Yu P, Chen H, Qin C. 2014. Transmission of H7N9 influenza virus in mice by different infective routes. Virol J 11:185. doi: 10.1186/1743-422X-11-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song H, Qi J, Khedri Z, Diaz S, Yu H, Chen X, Varki A, Shi Y, Gao GF. 2016. An open receptor-binding cavity of hemagglutinin-esterase-fusion glycoprotein from newly-identified influenza D virus: basis for its broad cell tropism. PLoS Pathog 12:e1005505. doi: 10.1371/journal.ppat.1005505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klein A, Krishna M, Varki NM, Varki A. 1994. 9-O-Acetylated sialic acids have widespread but selective expression: analysis using a chimeric dual-function probe derived from influenza C hemagglutinin-esterase. Proc Natl Acad Sci U S A 91:7782–7786. doi: 10.1073/pnas.91.16.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mori I, Komatsu T, Takeuchi K, Nakakuki K, Sudo M, Kimura Y. 1995. Viremia induced by influenza virus. Microb Pathog 19:237–244. doi: 10.1016/S0882-4010(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 52.Zhang X, Outlaw C, Olivier AK, Woolums A, Epperson W, Wan X-F. 2019. Pathogenesis of co-infections of influenza D virus and Mannheimia haemolytica in cattle. Vet Microbiol 231:246–253. doi: 10.1016/j.vetmic.2019.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, Palese P, Shaw ML, Treanor J, Webster RG, García-Sastre A. 2018. Influenza. Nat Rev Dis Primers 4:3. doi: 10.1038/s41572-018-0002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Denney L, Branchett W, Gregory LG, Oliver RA, Lloyd CM. 2018. Epithelial-derived TGF-β1 acts as a pro-viral factor in the lung during influenza A infection. Mucosal Immunol 11:523–535. doi: 10.1038/mi.2017.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carlin LE, Hemann EA, Zacharias ZR, Heusel JW, Legge KL. 2018. Natural killer cell recruitment to the lung during influenza A virus infection is dependent on CXCR3, CCR5, and virus exposure dose. Front Immunol 9:781. doi: 10.3389/fimmu.2018.00781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schultz-Cherry S. 2015. Role of NK cells in influenza infection, p 109–120. In Oldstone MBA, Compans RW (ed), Influenza pathogenesis and control, vol II. Springer International Publishing, Cham, Switzerland. [Google Scholar]
- 57.Guo H, Kumar P, Malarkannan S. 2011. Evasion of natural killer cells by influenza virus. J Leukoc Biol 89:189–194. doi: 10.1189/jlb.0610319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vidy A, Maisonnasse P, Costa BD, Delmas B, Chevalier C, Goffic RL. 2016. The influenza virus protein PB1-F2 increases viral pathogenesis through neutrophil recruitment and NK cells inhibition. PLoS One 11:e0165361. doi: 10.1371/journal.pone.0165361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oslund KL, Baumgarth N. 2011. Influenza-induced innate immunity: regulators of viral replication, respiratory tract pathology & adaptive immunity. Future Virol 6:951–962. doi: 10.2217/fvl.11.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.García-Sastre A. 2011. Induction and evasion of type I interferon responses by influenza viruses. Virus Res 162:12–18. doi: 10.1016/j.virusres.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jia D, Rahbar R, Chan RWY, Lee SMY, Chan MCW, Wang BX, Baker DP, Sun B, Peiris JSM, Nicholls JM, Fish EN. 2010. Influenza virus non-structural protein 1 (NS1) disrupts interferon signaling. PLoS One 5:e13927. doi: 10.1371/journal.pone.0013927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jewell NA, Cline T, Mertz SE, Smirnov SV, Flaño E, Schindler C, Grieves JL, Durbin RK, Kotenko SV, Durbin JE. 2010. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol 84:11515–11522. doi: 10.1128/JVI.01703-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.World Health Organization. 2002. WHO manual on animal influenza diagnosis and surveillance. World Health Organization, Geneva, Switzerland: https://www.who.int/csr/resources/publications/influenza/en/whocdscsrncs20025rev.pdf. [Google Scholar]
- 64.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497. doi: 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
- 65.Hall T. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser (Oxf) :95–98. [Google Scholar]