

Abstract
Marfan syndrome (MFS) is a connective tissue disorder caused by mutations in fibrillin-1 (Fbn1). Although aortic rupture is the major cause of mortality in MFS, patients also experience pulmonary complications, which are poorly understood. Loss of basal nitric oxide (NO) production and vascular integrity has been implicated in MFS aortic root disease, yet their contribution to lung complications remains unknown. Because of its capacity to potentiate the vasodilatory NO/cyclic guanylate monophosphate signaling pathway, we assessed whether the phosphodiesterase-5 inhibitor, sildenafil (SIL), could attenuate aortic root remodeling and emphysema in a mouse model of MFS. Despite increasing NO-dependent vasodilation, SIL unexpectedly elevated mean arterial blood pressure, failed to inhibit MFS aortic root dilation, and exacerbated elastic fiber fragmentation. In the lung, early pulmonary artery dilation observed in untreated MFS mice was delayed by SIL treatment, and the severe emphysema-like alveolar destruction was prevented. In addition, improvements in select parameters of lung function were documented. Subsequent microarray analyses showed changes to gene signatures involved in the inflammatory response in the MFS lung treated with SIL, without significant down-regulation of connective tissue or transforming growth factor-β signaling genes. Because phosphodiesterase-5 inhibition leads to improved lung histopathology and function, the effects of SIL against emphysema warrant further investigation in the settings of MFS despite limited efficacy on aortic root remodeling.
Marfan syndrome (MFS) is an autosomal dominant connective tissue disorder (CTD) caused by mutations in the fibrillin-1 (Fbn1) gene.1 Fibrillin-1 is a part of the extracellular matrix (ECM) and acts as a major structural component of elastic fibers in large arteries. Loss of fibrillin-1 integrity leads to degeneration of elastic fibers, which predisposes vessels to microdissections, aneurysms, and eventual vessel rupture.2 In addition, MFS patients experience significant pulmonary complications associated with degradation of the lung parenchyma, including emphysematous-like tissue destruction, spontaneous pneumothorax, upper airway collapsibility, and pulmonary artery trunk dilation.3, 4, 5 Currently, there are no MFS-specific options to prevent or attenuate the severity of lung emphysema and associated remodeling in this population as pharmacotherapy generally focuses on improving aortic root stability. The two main medications for the prophylactic management of aortic complications are as follows: losartan, an angiotensin II type 1 receptor blocker capable of reducing afterload, blood pressure (BP), and pathogenic transforming growth factor (TGF)-β signaling; and atenolol, a β-adrenoreceptor blocker that lowers heart rate and pulse wave velocity. Despite encouraging preclinical data, the overall efficacy of these medications at slowing the rate of aortic root widening and reducing all-cause mortality is controversial.6, 7 Interestingly, others have shown robust effects against lung remodeling with losartan in two models of MFS,4, 8 although it remains to be determined whether this will successfully translate to the clinic.
Endothelial dysfunction is generally characterized by an attenuated release of vasodilatory mediators9 and has been linked to the pathogenesis of pulmonary disorders, such as pulmonary arterial hypertension and chronic obstructive pulmonary disease.10, 11 Nitric oxide (NO) is one of the key vasodilatory mediators often found to be down-regulated in endothelial dysfunction. It can be released constitutively by both the vascular endothelium and the lung epithelium,12 resulting in blunted smooth muscle contractility and attenuated vascular and bronchial tone (Sandoo et al13 and Ricciardolo14). However, a more enigmatic and ill-defined aspect of the biology of NO is its capacity to protect underlying tissues from inflammation and pathologic remodeling.15, 16 Aberrant endothelial function has been reported in MFS patients,17 which could take part in the severe remodeling typically observed in their aorta and pulmonary artery trunk, a process akin to accelerated arteriolar aging.18 Interestingly, our research group has shown that losartan and atenolol differ in their effect on endothelial function in MFS tissues and that losartan-induced increase in NO bioavailability is responsible for its anti-aortic root remodeling effects.19, 20 Because of the key similarities between vascular and pulmonary structures, it is plausible that increased NO signaling could attenuate MFS-associated lung pathology.
The predominant secondary messenger of NO that initiates vasodilation is phosphodiesterase-5 (PDE5), an enzyme abundantly expressed in the pulmonary vasculature that degrades cGMP.21 The PDE5 inhibitor, sildenafil (SIL), can potentiate NO-induced vasodilation,22 decrease pulmonary vascular resistance and bronchodilation, and improve the 6-minute walk distance in patients with pulmonary arterial hypertension (Barnett et al23). In addition, SIL was found to preserve or even improve ventilation-perfusion matching, a marker of impaired lung perfusion and oxygenation, in interstitial lung disease,24 as well as improve pulmonary hemodynamics and exercise tolerance in patients with chronic obstructive pulmonary disease–associated pulmonary arterial hypertension.25, 26, 27, 28 Hence, amplification of NO signaling via PDE5 inhibition can promote lung tissue homeostasis. Whether this is caused by simple dilation of the pulmonary arterial tree or a more complex vasodilation-independent antiremodeling effect is unknown. Thus, the aim of this study was to determine the therapeutic potential of PDE5 inhibition on the vascular and pulmonary complications of MFS syndrome in a preclinical model. We describe how treatment with SIL can prevent emphysematous-like destruction of the lung parenchyma and improve respiratory mechanics despite a limited anti-aortic root remodeling effect in the C1039G model of MFS. Our data suggest that potentiation of cGMP-dependent NO signaling with SIL could help attenuate pathologic lung remodeling in settings of MFS and perhaps other CTDs.
Materials and Methods
Animals
Male and female Marfan mice harboring the Fbn1 C1039G+/- mutation (MFS; position 1041 in mice) and littermate control [wild-type (WT); Fbn1 C1039G+/+] mice were bred and housed in the Genetically Engineered Models facility at the Centre for Heart Lung Innovation, University of British Columbia (Vancouver, BC, Canada). All animals were housed on a standard 12-hour light/dark cycle, were fed standard laboratory chow (LabDiet #5001; LabDiet, St. Louis, MO), and were maintained using breeding and procedures approved by the University of British Columbia Animal Care Committee.
Drug Treatment
Sildenafil citrate (Pfizer, New York, NY) treatment was provided via drinking water (50 mg/kg per day) starting at 6 weeks of age and continuing until 24 weeks. Dosage was titrated to body weight and volume of water consumed per cage (averaged per day). Water was replaced, and doses were adjusted every week for the first 4 weeks of treatment and every 2 weeks thereafter. Control animals consumed regular water. Experiments were performed in two different cohorts, 6 to 7 months apart, with all values combined unless specified otherwise.
Echocardiography
Echocardiographic measurements were performed on anesthetized mice (0.75% v/v isoflurane, 1.5 L O2) using a VisualSonics Vevo 2100 system (Fujifilm, Tokyo, Japan) with an MS-550D 40-MHz probe by a technician blinded to genotype and treatment group. Aortic root measurements were averaged from multiple measurements taken in both M- and B-modes at the level of the sinus of Valsalva, as previously described.4, 20 Data for treated and untreated mice were analyzed at both 12 and 24 weeks of age.
BP Measurements
Systemic BP was noninvasively measured before sacrifice (at 24 weeks) using the tail cuff system (CODA2; Kent Scientific, Torrington, CT). Briefly, mice were lightly anesthetized (0.75% v/v isoflurane, 1.5 L O2) and placed on a warming tray with the tail inserted into an inflatable cuff, where systolic BP and diastolic BP were measured. Mean arterial pressure (MAP) was calculated as follows: (1/3 × systolic BP) + (2/3 × diastolic BP).
Tissue Preparation and Processing
Mice were euthanized at 24 weeks of age under terminal anesthesia (5% v/v isoflurane, 1.5 L O2), followed by cervical dislocation. The heart, lungs, and ascending aorta were excised; separated from the descending aorta distal to the left subclavian artery; and fixed in 10% neutral buffer formalin. Lungs were dissected away from the hearts, as per standard methods, across the vasculature and airways. Hearts were cut away from the aorta via a section through the basal ventricles parallel to the atrioventricular plane, and both segments were embedded cut side down in paraffin. Sections were analyzed every 50 μm through the aortic root. As previously described,20 elastic fiber integrity was assessed at the sinus of Valsalva on slides stained with Movat's pentachrome by a cardiac pathologist blinded to genotype and treatment status (M.S.). Elastic fiber fragmentation was scored on a scale of 0 to 3, wherein 0 indicates no fragmentation; 1, mild fragmentation (0% to 20%); 2, moderate fragmentation (20% to 40%); and 3, extensive fragmentation (>40%). Aortic root medial thickness was quantified on histologic sections using Aperio ImageScope software version 11.2.0.780 (Leica Biosystems, Nussloch, Germany). After separating from the heart, the right lung was placed in RNAlater (Thermo Fisher Scientific, Waltham, MA) and stored at −20°C until required. Histology of the left lung was performed, as previously described, via inflation, paraffin embedding, and staining of sections with Masson's trichrome; then, airspace enlargement was analyzed by mean linear intercept.20 Serial lung sections stained with hematoxylin and eosin were used to visualize cellular aggregates.
Measurements of Lung Function
Lung function was assessed in a separate cohort of mice (aged 24 weeks) using the FlexiVent (SCIREQ, Montréal, QC, Canada). Mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine, tracheotomized with an 18-gauge blunted needle, and mechanically ventilated at a respiratory rate of 150 breaths/minute and a tidal volume of 10 mL/kg, with a pressure limit of 30 cm H2O. Muscle paralysis was achieved using pancuronium (2 mg/kg; Sandoz, Boucherville, QC, Canada) to prevent respiratory efforts during the measurement. The following sequence of measures was repeated three times and averaged for analysis using FlexiWare Software version 7.6 (SCIREQ): deep inflation, Snapshot-150, Quick Prime-3, and pressure/volume-loop to obtain measures for compliance, resistance, tissue damping, and lung inspiratory capacity, respectively.
Lung RNA Extraction and Gene Microarray
As previously described,29 RNA from lung samples stored in RNAlater at −20°C was extracted using the RNeasy Plus Universal Mini kit (number 73404; Qiagen, Hilden, Germany), following manufacturer's instructions. RNA levels and purity were assessed using the NanoVue spectrophotometer (GE Health Care, Chicago, IL), and RNA degradation was assessed using gel electrophoresis. Whole transcript expression of RNA samples (n = 4 to 6 for each group) was determined using the GeneChip Clariom S Assay HT mouse (Affymetrix) by Genome Québec (Montréal, QC, Canada). Microarray data analyses were performed using R statistical software version 1.1.463 using the R packages affy, samr, and limma (R Core Team, Vienna, Austria). Data are available at http://www.ncbi.nlm.nih.gov (accession number GSE128481).
ImmuCC Cell Predictions
The computational algorithm (ImmuCC)30 was used to predict the tissue composition of 25 immune cells in untreated and treated WT and MFS lungs using the microarray expression data generated above. The 25 immune cell types were grouped into the 15 major cell types for visualization.30 The algorithm was performed using R statistical software version 3.5.3. A workflow schematic of this process has been added in Supplemental Figure S1.
Isometric Force Myography
The descending thoracic aorta was dissected from the thoracic cage and cleaned of fat and connective tissue in ice-cold Krebs solution (118 mmol/L NaCl, 22.5 mmol/L NaHCO3, 4 mmol/L KCl, 1.2 mmol/L NaH2PO4, 2 mmol/L CaCl2, 2 mmol/L MgCl2, 11 mmol/L dextrose, and 0.01 mmol/L ibuprofen). Segments of the thoracic aorta (2 mm thick) were mounted isometrically in a small vessel myograph (AS Danish Myotechnology, Aarhus, Denmark) to measure smooth muscle cell (SMC) contractility. Thoracic aorta segments were left to equilibrate for 30 minutes at 37°C in Krebs solution, aerated continuously with 95% O2/5% CO2. Vessels were stretched to the optimal tension (6.0 mN) for 30 minutes, as previously described,19 and thereafter challenged twice with 30 mmol/L KCl to determine the viability and reactivity of aortic SMCs, followed by a concentration-dependent dose-response with phenylephrine (3 nmol/L to 100 μmol/L). Percentage contraction was calculated as the percentage increase or decrease in force with respect to untreated WT mice, where the maximum recorded response was arbitrarily set to 100% contraction. To study the effect of NO on SMC contractility, vessels were incubated with Nω-nitro-l-arginine methyl ester (L-NAME; 200 mmol/L), a NO inhibitor, for 30 minutes before addition of phenylephrine (3 nmol/L to 100 μmol/L).
Statistical Analysis
Statistical analyses and table figures were prepared using GraphPad Prism Software version 6.01 (GraphPad Software, San Diego, CA). Two-way analysis of variance was used to compare the means between each genotype and treatment grouping. Sidak's post-hoc test was used to correct for multiple comparisons. Data are presented as the means ± SEM, unless otherwise specified, with P < 0.05 considered significant. For analysis of the gene expression data, only genes with a false discovery rate of <0.05 and fold changes of >1.2 or <0.8 were considered statistically significant.
Results
SIL Treatment Attenuates Early Pulmonary Artery Dilation despite Unabated Aortic Root Widening and Elevated BP in MFS Tissues
In accordance with previously published data,20 it was observed by echocardiography that 12-week–old MFS mice exhibit significantly larger pulmonary artery and aortic root diameters compared with WT controls in the absence of treatment (P < 0.01) (Figure 1, A and C). At 24 weeks, untreated WT and MFS pulmonary artery diameters did not significantly differ (Figure 1B), whereas MFS mice still showed larger aortic root diameters compared with WT controls (Figure 1D). The significant difference in pulmonary artery diameter at 12 weeks was mitigated by SIL treatment, but there was no significant effect on pulmonary artery diameter at 24 weeks or on aortic root diameter at either 12 or 24 weeks (Figure 1, A–D). Due to a lack of anti-aortic root remodeling effects, BP and heart function were also investigated. Chronic SIL treatment in MFS mice led to an unexpected but statistically significant increase in systemic systolic BP and diastolic BP (P < 0.05) (Figure 1E) as well as mean arterial blood pressure (MAP) (Supplemental Figure S2A), an effect not observed in WT mice. Further echocardiographic evaluation demonstrated increased stroke volume and left ventricular diastolic diameter in untreated MFS mice when compared with WT controls, with no effect on other measured parameters (P < 0.05) (Supplemental Table S1). SIL treatment had no effect on left ventricular heart function (Supplemental Table S1), suggesting both a pulmonary artery–specific effect and increased afterload, which could rationalize a lack of anti-aortic root widening effect of SIL on MFS tissues.
Figure 1.

Pulmonary artery (PA) and aortic root measurements at 12 (A and C) and 24 (B and D) weeks, and systolic and diastolic blood pressure (BP) at 24 weeks (E) in wild-type (WT) and Marfan syndrome (MFS) mice treated with either SIL or vehicle. Data are expressed as means ± SEM (A–E). n = 12 to 22 mice per group (A–E). ∗∗P < 0.01, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001 versus WT mice in the same treatment group; †P < 0.05 versus untreated MFS mice (two-way analysis of variance with Sidak post-hoc test).
SIL Exacerbates Aortic Wall Remodeling in PDE5-Expressing MFS Tissues despite Potentiation of Systemic NO-Dependent Vasodilation
After euthanasia, histology of aortic root tissues revealed that MFS mice treated with SIL exhibit greater elastic fiber fragmentation and medial thickening when compared with untreated MFS mice (Figure 2, A–C). Aortic root sections showed robust PDE5 staining throughout medial SMC layers (Figure 2D) by immunochemistry. Ex vivo myography using aortic tissues confirmed a potentiation of vasodilatory NO signaling by SIL, as quantified by decreased KCl-induced contractility (Supplemental Figure S2B) and attenuated phenylephrine-induced contractility in an L-NAME–sensitive manner (Figure 2, E–G). These ex vivo data confirm the unabated increase in elastic fiber degradation and medial thickening during the aortic root widening process in MFS mice treated with SIL despite potentiated vasodilatory NO signaling, which suggests that the anti-aortic root remodeling properties of NO are vasodilation and PDE5 independent.
Figure 2.

A–D: Elastic fiber fragmentation (A), medial thickening (B), and representative Movat's pentachrome staining (C) in wild-type (WT) and Marfan syndrome (MFS) mice with or without SIL treatment; and representative phosphodiesterase-5 (PDE5) staining in untreated WT and MFS mice (D). Inset: No primary antibody control, for PDE5. E and F: Concentration-response curves of phenylephrine (PE) contraction before (E) and after (F) Nω-nitro-l-arginine methyl ester (L-NAME; 100 μmol/L) in WT and MFS thoracic aorta segments with or without sildenafil treatment. G: Maximum rate of contraction (Emax) values of before and after L-NAME exposed vessels are summarized. Values are expressed as a percentage of WT mice. A, B, and G: Gray shading indicates mice treated with sildenafil. Data are expressed as means ± SEM (A, B, and G). n = 5 to 12 mice per group (A and B); n = 5 to 8 mice per group (G). ∗∗P < 0.01 versus WT in the same treatment group; †P < 0.05 versus MFS treated (two-way analysis of variance with Sidak post-hoc test); ‡‡‡‡P < 0.0001. Scale bars: 60 μm (C); 100 μm (D, main images and inset).
SIL Treatment Prevents Emphysematous Lung Pathology and Improves Key Lung Function Parameters in MFS Mice
C1039G MFS mice present with an emphysematous phenotype (Figure 3A). A 35% greater mean linear intercept was observed in untreated MFS mice compared with WT controls, a marker of increased airspace widening (P < 0.001) (Figure 3, A and B). WT mice did not show a significant effect of SIL on mean linear intercept (Figure 3B), whereas MFS mice showed a significantly lower mean linear intercept in response to SIL compared with untreated MFS mice (Figure 3, B and C). Because of the apparent anti-emphysematous effect of SIL, lung function experiments were performed in an additional cohort of mice (Figure 4). Consistent with profound emphysema, pressure-volume loop dynamics, including compliance, resistance, tissue damping, lung inspiratory capacity (Figure 4, A–E), hysteresis, and elastance (data not shown), were negatively affected in pathogenic MFS lungs. In MFS lung, SIL significantly reduced pulmonary compliance (a measure of elastic tissue distensibility) by 17% (P < 0.05) and prevented MFS-associated changes in resistance (Figure 4, B and C). Although tissue damping, a measure of resistance and alveolar energy dissipation, was unaffected by SIL treatment (Figure 4D), total lung capacity was mildly improved (13%), albeit not statistically significant (Figure 4E).
Figure 3.

Representative lung histology from vehicle and SIL-treated wild-type (WT) and Marfan syndrome (MFS; A and C) mice and combined mean linear intercept measurements (B). B: Gray shading indicates mice treated with sildenafil. Data are expressed as means ± SEM (B). n= 6 to 12 mice per group (B). ∗∗∗P < 0.001 versus WT in the same treatment groups; ††P < 0.01 versus untreated MFS (two-way analysis of variance with Sidak post-hoc test). Scale bars = 300 μm (A and C).
Figure 4.

Lung function in both wild-type (WT) and Marfan syndrome (MFS) mice treated with either sildenafil or vehicle. Average pressure-volume loops (A), compliance (B), resistance (C), tissue damping (D), and inspiratory capacity (E) were assessed using FlexiVent. A: Error bars were removed for simplicity. B–E: Gray shading indicates mice treated with sildenafil. Data are expressed as means ± SEM (A–E). n = 5 to 13 mice per group (A–E). ∗P < 0.05, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001 versus WT in the same treatment group; †P < 0.05 versus untreated MFS (two-way analysis of variance with Sidak post-hoc test). s, seconds.
Effect of SIL Treatment on MFS-Associated Lung Gene Signature Changes
Affymetrix mRNA expression analyses (n = 5 to 6) were performed to characterize how SIL protected the lung parenchyma against MFS emphysema. Relative to untreated WT mice, untreated MFS mice showed 636 significantly elevated and 626 significantly down-regulated genes, mostly related to the ECM (Figure 5 and Supplemental Table S2). There was increased expression of fibronectin (Fn1; 1.55-fold expression in MFS), collagen type IV (Col4a1; 1.45-fold), collagen type VI (Col6a2; 1.45-fold), and TGF-β–related genes, such as latent TGF-β–binding protein 3 (Ltbp3; 1.37-fold) (Figure 5A). Interestingly, endothelial cell–specific molecule 1 (Esm1), which has proangiogenic activities and is released by endothelial cells in response to cytokine activation, was significantly down-regulated in MFS lung tissues (0.5-fold), and phosphodiesterase-10a (Pde10a) was reduced to 0.68-fold in MFS lungs (Figure 5A). Select genes involved in immune activation were also down-regulated, such as IL-1β (Il1b; 0.65-fold) and tumor necrosis factor ligand superfamily member 4 (Tnfsf4; 0.71-fold) (Figure 5A). Changes to genes involved in epithelial development and blood vessel morphogenesis were also noted (Supplemental Figure S3). Comparison of lung gene expression between SIL-treated and untreated MFS mice revealed changes in the mRNA expression of genes involved in diverse pathways (671 significantly up-regulated and 581 down-regulated genes) (Figure 5B and Supplemental Table S3). Most interestingly, the observed improvement in lung pathology of SIL-treated MFS mice was associated with changes in multiple immune response–related pathways, including cytokine expression and microbial response, without evident down-regulation of pathways related to ECM or TGF-β signaling (Figure 5B). Significantly up-regulated genes included IL-1α (Il1a; 1.77-fold), Il1b (1.55-fold), IL-6 (Il6; 1.64-fold), Cxcl13 (1.50-fold), immune-responsive gene 1 (Irg1; 2.35-fold), macrophage scavenger receptor 1 (Msr1; 2.15-fold), and CD274 (Cd274; 1.68-fold) and CD276 (Cd276; 1.75-fold) (Figure 5B).
Figure 5.

Lung mRNA expression profile comparison between untreated and treated Marfan syndrome (MFS) and wild-type (WT) mice using the Affymetrix GeneChip assay. A: Heat map of select affected genes in untreated WT and MFS lung, with spectrum of green to red based on row z-score going from low to high expression. B: Heat map of select affected genes in treated and untreated MFS lung, with spectrum of green to red based on row z-score going from low to high expression. n = 5 to 6 mice per group (A and B).
To account for the striking changes in cytokine expression between treated and untreated MFS samples, microarray data were further analyzed using ImmuCC,31 a computational algorithm used to predict the relative tissue composition of 25 different immune cell types. Despite MFS SIL lung containing more bulk tissue than untreated MFS lung (Figure 3, B and C), the predicted immune cell profiles were similar between the two cohorts. The five most abundant cell types of the innate and adaptive immune system and their predicted quantities in all tissue groups are shown in Supplemental Figure S4, A and B. Visualization of comparative lung sections stained with hematoxylin and eosin also shows no obvious signs of inflammatory infiltrates within the airway epithelium or alveolar structures (Supplemental Figure S4C).
Discussion
Although aortic dilation and remodeling are the primary clinically relevant symptoms of MFS, because of a significantly increased risk of life-threatening aortic rupture, changes to the respiratory system can be severe. To our knowledge, the current study is the first to assess histopathologic and mechanical outcomes of MFS animals with paired gene expression changes in lung tissues. We report that the heterozygous C1039G mutation of Fbn1 leads to increased emphysema-like remodeling of the alveolar structure and impaired lung function. This was associated with up-regulation of genes associated with ECM and TGF-β signaling and down-regulation of genes known to be involved in inflammatory responses and vascular function. Despite an absence of improved aortic root stability, SIL attenuated lung markers of emphysema and improved lung function without down-regulation of ECM gene expression initially found to be up-regulated in MFS lungs. Our data highlight the potential therapeutic effect of PDE5 inhibition in emphysema induced by Fbn1 deficiency.
PDE5 Inhibition–Dependent Vasodilation and MFS-Associated Aortic Root Pathology
Previous studies have shown that up-regulation of endothelial NO production can improve aortic root outcomes in MFS mice independently of BP lowering.20 Although the signaling behind NO's anti-inflammatory and antiremodeling properties is complex and poorly understood, it is generally agreed that the bulk of its vasodilatory properties are dependent on the accumulation of cGMP in medial SMCs. Despite observations that the pulmonary vasculature expresses much higher levels of PDE5 than its systemic counterpart, we show that SIL can promote vasodilation of WT and MFS aorta, as previously shown in rats by others,32 indicating functional PDE5 expression that can be inhibited to amplify the vasodilatory secondary messenger of NO. However, treatment with SIL failed to improve aortic root pathology in MFS mice, which suggests that the vasodilatory signaling of NO is independent of its anti-aortic root remodeling properties in MFS. SIL treatment led to a surprising increase in mean arterial blood pressure in MFS but not WT mice, despite increased NO vasodilation, further lending credence to reports describing abnormalities in the vascular NO activation pathway in MFS.17 Although we have previously shown that specific targeting of NO signaling was more effective at reducing MFS aortic root widening20 than reduction of BP alone, it is to be determined if the BP-elevating effect of SIL in MFS mice we observed may have increased the hemodynamic stress at the level of the aortic root, potentially leading to widening and remodeling. Whether the inhibition of pulmonary artery dilation with SIL is part of the failed protection of aortic tissues and compensatory increase of BP is also unknown and requires further investigation. Hence, the study of SIL in the presence of BP-lowering medications warrants further investigation.
A potential culprit behind the unexpected vascular phenotypes observed with SIL is inflammation-associated production of NO from inducible NO synthase (iNOS). Indeed, MFS aortas appear unexpectedly enriched in iNOS,33 but whether SIL can amplify iNOS-derived NO release and lead to agonist-independent vasodilation in the absence of protective, antivascular remodeling is unknown. From a purely quantitative standpoint, iNOS-derived levels of NO tend to greatly supersede those derived from other NOS isoforms, in addition to triggering different biological responses to endothelial NOS–derived NO (Forstermann and Sessa34), particularly in settings of vascular inflammation. Another concept that cannot be ignored is the importance of cGMP-independent NO signaling events, such as cysteine nitrosylation.35 Considered a reversible covalent modification of proteins, the role of NO-dependent nitrosylation in MFS aortic remodeling is unknown and would not have been amplified by SIL. Finally, vascular SMCs also express many PDEs other than PDE5, which might be responsible for the anti-aortic root remodeling effects of endothelial NO we and others have shown,20, 36 and these would likely not be amplified by SIL.
SIL in a CTD Model of Emphysema
Because of the irreversible nature of alveolar destruction in humans and its high incidence in chronic obstructive pulmonary disease and CTDs, the identification of medications that can prevent or attenuate the rate of parenchymal destruction is of significant interest. SIL is shown to preserve vascular integrity and attenuate lung remodeling in models of cigarette smoke–induced chronic obstructive pulmonary disease,37 which highlighted not only the protective role of the NO-PDE5 axis but also the dynamic interplay between vasculature and alveolar structures. Although the early inhibition of pulmonary artery dilation with SIL could be foreseen on the basis of its documented therapeutic properties, the underlying mechanism behind SIL's antiemphysema effect in settings of Fbn1 deficiency is of relevance in terms of identifying therapies for CTD patient management. The lungs of MFS mice had significantly dysregulated expression of key connective tissue and ECM genes, which supports the lung manifestation of this connective tissue pathology and the associated changes in lung integrity. Similar gene modulation was observed by others in the vasculature of Fbn1-mutant mice38 and in the skin of MFS patients.39, 40 In addition, some genes involved in TGF-β signaling were affected, a pathway that has been proposed to be a major contributor to the development of lung pathology in MFS mice.41 Although treatment with SIL led to significant improvements in lung histopathology, it remains to be determined which PDE5-expressing lung cell type the attenuation of MFS mean linear intercept changes with SIL is caused by and whether SIL increased tissue protection or heightened regeneration, a topic of renewed interest in regenerative medicine. A study looking at the effects of PDE inhibition on differentiation of lung fibroblasts to myofibroblasts showed that the NO-cGMP pathway plays an important role in TGF-β–induced differentiation during lung fibrosis.42 However, the mild improvement in lung function was not associated with the restoration of normal ECM- or TGF-β–related gene expression. The fact that SIL treatment was able to improve lung pathology without affecting these two major pathways, which are proposed to be the major contributors to the development of MFS pathology, indicates that their role in lung disease is unclear. Whether similar results would be observed in MFS mice treated with other PDE5 inhibitors or whether SIL is specific in its effects are currently unknown. NO-potentiating drugs, like losartan, have been shown to attenuate lung pathology as well as exhibit anti-aortic root remodeling properties in mild (C1039G)8 and severe (mgR)4 mouse models of MFS. In other mouse models of emphysema, statins, which are notorious for pleiotropic effects, including endothelial function activation, can promote lung repair,43 whereas both l-arginine (an NOS substrate) and L-NAME (a general NOS inhibitor) can differentially ameliorate lung pathology, further stressing the complexity of NO signaling in lung remodeling.44 This is particularly accurate when taken into the context of potential chronic iNOS overexpression in MFS lung vasculature and alveolar structure.45
SIL Treatment and Inflammatory Gene Response
Improvement of MFS lung pathology was found to be associated with increased expression of several genes involved in the production and secretion of inflammatory cytokines as well as other key players in the inflammatory response. An association study looking at the expression of inflammatory genes in the skin and the plasma levels of inflammatory cytokines of MFS patients found that they were associated with the progression of aortic root pathology.40 However, few studies have previously explored lung pathology or inflammation in MFS, and none has investigated MFS lung expression profiles. In non-MFS tissues, such as the pulmonary artery,46 the brain,47, 48, 49 and the lung,50, 51 SIL treatment has been shown to decrease inflammatory cytokine levels and dampen the innate and adaptive immune response through inhibition of the transcription of NF-κB, an important inflammatory signaling molecule.52 In an acrolein-induced model of lung inflammation, SIL was shown to significantly decrease cytokine release and inflammatory cell infiltration.51 Therefore, the unexpected up-regulation of inflammatory genes after SIL treatment requires further investigation. A potential reason for the up-regulation of IL-1β expression is caused by SIL's inhibitory effect on IL-1β–induced NO synthesis, simply leading to a lack of negative feedback and a compensatory increase in Il1b expression, which then leads to the activation of further inflammatory processes.53 On the basis of our data, the increased expression of inflammatory cytokines in SIL-treated MFS lung are, thus, likely to result from increased signaling of occupying cells or their expression by other local tissue groups. Regardless, SIL treatment in MFS lung appeared to mitigate the deficiencies in inflammatory cytokine expression observed in untreated MFS tissues, and these deficiencies are perhaps more indicative of a WT inflammatory response.
In conclusion, the significant improvement of lung pathology in MFS mice after SIL treatment suggests that pharmacologic management of pulmonary complications in patients may be possible. How SIL treatment is able to modulate the inflammatory response in MFS lungs is not clear and requires further investigation. In all, it is clear that SIL's beneficial effects in the lung are not associated with significant changes in connective tissue and TGF-β gene expression. Studies aimed at testing how SIL can protect the lung of MFS or CTD patients are warranted.
Acknowledgments
We thank Tatjana Ponomarev and Lubos Bohunek (St. Paul's Hospital, University of British Columbia) for help with animal experiments.
P.B., Z.W., and N.M. conceived and designed the experiments; P.B., Z.W., N.M., A.Y.T., J.L., M.S., and M.C.M. performed the experiments, interpreted the results, prepared the figures, and wrote the manuscript; N.M., Z.W., A.Y.T., J.L., S.B., and T.-L.H. analyzed the data; all authors approved the final version of the manuscript; P.B., Z.W., and M.E. funded the study.
Footnotes
Supported by Heart and Stroke Foundation grant G15-9211, Canadian Institutes of Health Research grant PJT159511, a Rare Disease Foundation grant 2462, a Canada Foundation for Innovation/British Columbia Knowledge Development Fund grant 35921 (P.B.), and Mitacs/Providence Airways Centre grant IT13604 (Z.W., P.B.).
Z.W. and N.M. contributed equally to this work.
Disclosures: None declared.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2019.05.003.
Supplemental Data
Supplemental Figure 1.

Workflow schematic of computational immune cell predictions from lung microarray data using a computational algorithm (ImmuCC) and CIBERSORT. DC, dendritic cell; NK, natural killer; SVR, support vector regression; Th17, type 17 helper T; Th2, type 2 helper T; TReg, regulatory T.
Supplemental Figure 2.

A and B: Mean arterial blood pressure (MAP; A) and response to KCl (30 mmol/L; B) in thoracic aortic segments in both wild-type (WT) and Marfan syndrome (MFS) mice with or without SIL treatment. A and B: Gray shading indicates mice treated with sildenafil. B: Values are expressed as a percentage of WT mice. Data are expressed as means ± SEM (A and B). n = 5 to 6 per group (B). †P < 0.05 versus untreated MFS mice (two-way analysis of variance with Sidak's post-hoc test).
Supplemental Figure 3.
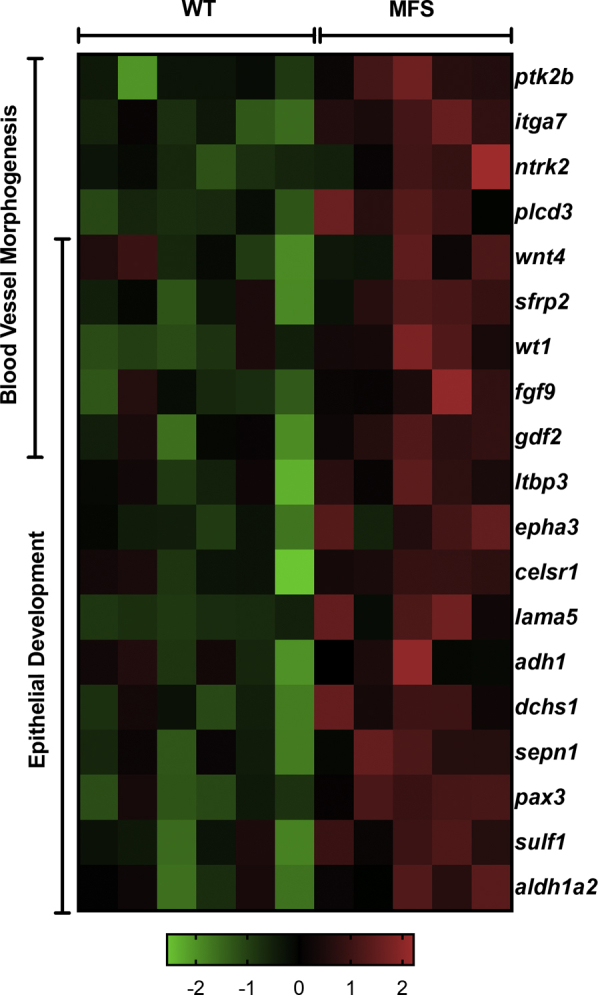
Lung mRNA expression profile comparison between untreated Marfan syndrome (MFS) and wild-type (WT) mice using the Affymetrix GeneChip assay. Heat map of select affected genes in the blood vessel morphogenesis and epithelial development pathways, spectrum of green to red based on row z-score, going from low to high expression. n = 5 to 6 mice per group.
Supplemental Figure 4.

The computational algorithm (ImmuCC) was used to predict the tissue composition of 25 immune cell types in untreated and treated wild-type (WT) and Marfan syndrome (MFS) lungs using microarray expression data. A and B: Graphs display the five most abundant cell types of the innate and adaptive immune system, respectively, visualized using CIBERSORT. Gray shading indicates mice treated with sildenafil. C: Representative images of hematoxylin and eosin–stained MFS and MFS SIL-treated lungs. Scale bars: 300 μm (C, main images); 200 μm (C, insets). NK, natural killer; PAP, predicted absolute proportion.
References
- 1.Dietz H.C., Cutting G.R., Pyeritz R.E., Maslen C.L., Sakai L.Y., Corson G.M., Puffenberger E.G., Hamosh A., Nanthakumar E.J., Curristin S.M. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 2.Pyeritz R.E. The Marfan syndrome. Annu Rev Med. 2000;51:481–510. doi: 10.1146/annurev.med.51.1.481. [DOI] [PubMed] [Google Scholar]
- 3.Ishii H., Shima R., Kinoshita Y., Kushima H. Marfan syndrome presenting with diffuse emphysematous change of the lung. BMJ Case Rep. 2018;2018 doi: 10.1136/bcr-2017-224056. bcr-2017-224056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee J.-J., Galatioto J., Rao S., Ramirez F., Costa K.D. Losartan attenuates degradation of aorta and lung tissue micromechanics in a mouse model of severe Marfan syndrome. Ann Biomed Eng. 2016;44:2994–3006. doi: 10.1007/s10439-016-1616-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uriarte J.J., Meirelles T., Gorbenko Del Blanco D., Nonaka P.N., Campillo N., Sarri E., Navajas D., Egea G., Farre R. Early impairment of lung mechanics in a murine model of Marfan syndrome. PLoS One. 2016;11:e0152124. doi: 10.1371/journal.pone.0152124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lacro R.V., Dietz H.C., Sleeper L.A., Yetman A.T., Bradley T.J., Colan S.D., Pearson G.D., Selamet Tierney E.S., Levine J.C., Atz A.M., Benson D.W., Braverman A.C., Chen S., De Backer J., Gelb B.D., Grossfeld P.D., Klein G.L., Lai W.W., Liou A., Loeys B.L., Markham L.W., Olson A.K., Paridon S.M., Pemberton V.L., Pierpont M.E., Pyeritz R.E., Radojewski E., Roman M.J., Sharkey A.M., Stylianou M.P., Wechsler S.B., Young L.T., Mahony L. Atenolol versus losartan in children and young adults with Marfan's syndrome. N Engl J Med. 2014;371:2061–2071. doi: 10.1056/NEJMoa1404731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forteza A., Evangelista A., Sanchez V., Teixido-Tura G., Sanz P., Gutierrez L., Gracia T., Centeno J., Rodriguez-Palomares J., Rufilanchas J.J., Cortina J., Ferreira-Gonzalez I., Garcia-Dorado D. Efficacy of losartan vs. atenolol for the prevention of aortic dilation in Marfan syndrome: a randomized clinical trial. Eur Heart J. 2016;37:978–985. doi: 10.1093/eurheartj/ehv575. [DOI] [PubMed] [Google Scholar]
- 8.Habashi J.P., Judge D.P., Holm T.M., Cohn R.D., Loeys B.L., Cooper T.K., Myers L., Klein E.C., Liu G., Calvi C., Podowski M., Neptune E.R., Halushka M.K., Bedja D., Gabrielson K., Rifkin D.B., Carta L., Ramirez F., Huso D.L., Dietz H.C. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giaid A., Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–221. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 10.Peinado V.I., Gomez F.P., Barbera J.A., Roman A., Angels Montero M., Ramirez J., Roca J., Rodriguez-Roisin R. Pulmonary vascular abnormalities in chronic obstructive pulmonary disease undergoing lung transplant. J Heart Lung Transplant. 2013;32:1262–1269. doi: 10.1016/j.healun.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Dinh-Xuan A.T., Higenbottam T.W., Clelland C.A., Pepke-Zaba J., Cremona G., Butt A.Y., Large S.R., Wells F.C., Wallwork J. Impairment of endothelium-dependent pulmonary-artery relaxation in chronic obstructive lung disease. N Engl J Med. 1991;324:1539–1547. doi: 10.1056/NEJM199105303242203. [DOI] [PubMed] [Google Scholar]
- 12.Pechkovsky D.V., Zissel G., Goldmann T., Einhaus M., Taube C., Magnussen H., Schlaak M., Muller-Quernheim J. Pattern of NOS2 and NOS3 mRNA expression in human A549 cells and primary cultured AEC II. Am J Physiol Lung Cell Mol Physiol. 2002;282:L684–L692. doi: 10.1152/ajplung.00320.2000. [DOI] [PubMed] [Google Scholar]
- 13.Sandoo A., van Zanten J.J.C.S.V., Metsios G.S., Carroll D., Kitas G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J. 2010;4:302–312. doi: 10.2174/1874192401004010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ricciardolo F.L.M. Multiple roles of nitric oxide in the airways. Thorax. 2003;58:175–182. doi: 10.1136/thorax.58.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kubes P., Suzuki M., Granger D.N. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma A., Sellers S., Stefanovic N., Leung C., Tan S.M., Huet O., Granville D.J., Cooper M.E., de Haan J.B., Bernatchez P. Direct endothelial nitric oxide synthase activation provides atheroprotection in diabetes-accelerated atherosclerosis. Diabetes. 2015;64:3937–3950. doi: 10.2337/db15-0472. [DOI] [PubMed] [Google Scholar]
- 17.Wilson D.G., Bellamy M.F., Ramsey M.W., Goodfellow J., Brownlee M., Davies S., Wilson J.F., Lewis M.J., Stuart A.G. Endothelial function in Marfan syndrome: selective impairment of flow-mediated vasodilation. Circulation. 1999;99:909–915. doi: 10.1161/01.cir.99.7.909. [DOI] [PubMed] [Google Scholar]
- 18.Salvi P., Grillo A., Marelli S., Gao L., Salvi L., Viecca M., Di Blasio A.M., Carretta R., Pini A., Parati G. Aortic dilatation in Marfan syndrome: role of arterial stiffness and fibrillin-1 variants. J Hypertens. 2018;36:77–84. doi: 10.1097/HJH.0000000000001512. [DOI] [PubMed] [Google Scholar]
- 19.Chung A.W.Y., Au Yeung K., Cortes S.F., Sandor G.G.S., Judge D.P., Dietz H.C., van Breemen C. Endothelial dysfunction and compromised eNOS/Akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br J Pharmacol. 2007;150:1075–1083. doi: 10.1038/sj.bjp.0707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sellers S.L., Milad N., Chan R., Mielnik M., Jermilova U., Huang P.L., de Crom R., Hirota J.A., Hogg J.C., Sandor G.G., Van Breemen C., Esfandiarei M., Seidman M.A., Bernatchez P. Inhibition of Marfan syndrome aortic root dilation by losartan: role of angiotensin II receptor type 1-independent activation of endothelial function. Am J Pathol. 2018;188:574–585. doi: 10.1016/j.ajpath.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Butrous G. The role of phosphodiesterase inhibitors in the management of pulmonary vascular diseases. Glob Cardiol Sci Pract. 2014;2014:257–290. doi: 10.5339/gcsp.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kass D.A., Takimoto E., Nagayama T., Champion H.C. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc Res. 2007;75:303–314. doi: 10.1016/j.cardiores.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 23.Barnett C.F., Machado R.F. Sildenafil in the treatment of pulmonary hypertension. Vasc Health Risk Manag. 2006;2:411–422. doi: 10.2147/vhrm.2006.2.4.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lederer D.J., Bartels M.N., Schluger N.W., Brogan F., Jellen P., Thomashow B.M., Kawut S.M. Sildenafil for chronic obstructive pulmonary disease: a randomized crossover trial. COPD. 2012;9:268–275. doi: 10.3109/15412555.2011.651180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blanco I., Gimeno E., Munoz P.A., Pizarro S., Gistau C., Rodriguez-Roisin R., Roca J., Barbera J.A. Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am J Respir Crit Care Med. 2010;181:270–278. doi: 10.1164/rccm.200907-0988OC. [DOI] [PubMed] [Google Scholar]
- 26.Galie N., Ghofrani H.A., Torbicki A., Barst R.J., Rubin L.J., Badesch D., Fleming T., Parpia T., Burgess G., Branzi A., Grimminger F., Kurzyna M., Simonneau G. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 27.Pepke-Zaba J., Gilbert C., Collings L., Brown M.C.J. Sildenafil improves health-related quality of life in patients with pulmonary arterial hypertension. Chest. 2008;133:183–189. doi: 10.1378/chest.07-0592. [DOI] [PubMed] [Google Scholar]
- 28.Singh T.P., Rohit M., Grover A., Malhotra S., Vijayvergiya R. A randomized, placebo-controlled, double-blind, crossover study to evaluate the efficacy of oral sildenafil therapy in severe pulmonary artery hypertension. Am Heart J. 2006;151:851.e1–851.e5. doi: 10.1016/j.ahj.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 29.Lechasseur A., Jubinville E., Routhier J., Berube J.-C., Hamel-Auger M., Talbot M., Lamothe J., Aubin S., Pare M.-E., Beaulieu M.-J., Bosse Y., Duchaine C., Morissette M.C. Exposure to electronic cigarette vapors affects pulmonary and systemic expression of circadian molecular clock genes. Physiol Rep. 2017;5 doi: 10.14814/phy2.13440. e13440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Z., Huang A., Sun J., Jiang T., Qin F.X.-F., Wu A. Inference of immune cell composition on the expression profiles of mouse tissue. Sci Rep. 2017;7:40508. doi: 10.1038/srep40508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang M., Koitabashi N., Nagayama T., Rambaran R., Feng N., Takimoto E., Koenke T., O'Rourke B., Champion H.C., Crow M.T., Kass D.A. Expression, activity, and pro-hypertrophic effects of PDE5A in cardiac myocytes. Cell Signal. 2008;20:2231–2236. doi: 10.1016/j.cellsig.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang H.H.C., van Breemen C., Chung A.W.Y. Vasomotor dysfunction in the thoracic aorta of Marfan syndrome is associated with accumulation of oxidative stress. Vascul Pharmacol. 2010;52:37–45. doi: 10.1016/j.vph.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 34.Forstermann U., Sessa W.C. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. 837a–837d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stamler J.S. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 36.Gibson C., Nielsen C., Alex R., Cooper K., Farney M., Gaufin D., Cui J.Z., van Breemen C., Broderick T.L., Vallejo-Elias J., Esfandiarei M. Mild aerobic exercise blocks elastin fiber fragmentation and aortic dilatation in a mouse model of Marfan syndrome associated aortic aneurysm. J Appl Physiol. 2017;123:147–160. doi: 10.1152/japplphysiol.00132.2017. [DOI] [PubMed] [Google Scholar]
- 37.Dominguez-Fandos D., Valdes C., Ferrer E., Puig-Pey R., Blanco I., Tura-Ceide O., Paul T., Peinado V.I., Barbera J.A. Sildenafil in a cigarette smoke-induced model of COPD in the guinea-pig. Eur Respir J. 2015;46:346–354. doi: 10.1183/09031936.00139914. [DOI] [PubMed] [Google Scholar]
- 38.Kim K.L., Choi C., Suh W. Analysis of disease progression-associated gene expression profile in fibrillin-1 mutant mice: new insight into molecular pathogenesis of Marfan syndrome. Biomol Ther (Seoul) 2014;22:143–148. doi: 10.4062/biomolther.2014.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao Z., Jaeger J.C., Ruzzo W.L., Morale C.Z., Emond M., Francke U., Milewicz D.M., Schwartz S.M., Mulvihill E.R. A Marfan syndrome gene expression phenotype in cultured skin fibroblasts. BMC Genomics. 2007;8:319. doi: 10.1186/1471-2164-8-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radonic T., de Witte P., Groenink M., de Waard V., Lutter R., van Eijk M., Jansen M., Timmermans J., Kempers M., Scholte A.J., Hilhorst-Hofstee Y., van den Berg M.P., van Tintelen J.P., Pals G., Baars M.J.H., Mulder B.J.M., Zwinderman A.H. Inflammation aggravates disease severity in Marfan syndrome patients. PLoS One. 2012;7:e32963. doi: 10.1371/journal.pone.0032963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neptune E.R., Frischmeyer P.A., Arking D.E., Myers L., Bunton T.E., Gayraud B., Ramirez F., Sakai L.Y., Dietz H.C. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 42.Dunkern T.R., Feurstein D., Rossi G.A., Sabatini F., Hatzelmann A. Inhibition of TGF-beta induced lung fibroblast to myofibroblast conversion by phosphodiesterase inhibiting drugs and activators of soluble guanylyl cyclase. Eur J Pharmacol. 2007;572:12–22. doi: 10.1016/j.ejphar.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 43.Pinho-Ribeiro V., Melo A.C., Kennedy-Feitosa E., Graca-Reis A., Barroso M.V., Cattani-Cavalieri I., Carvalho G.M.C., Zin W.A., Porto L.C., Gitirana L.B., Lanzetti M., Valença S.S. Atorvastatin and simvastatin promoted mouse lung repair after cigarette smoke-induced emphysema. Inflammation. 2017;40:965–979. doi: 10.1007/s10753-017-0541-5. [DOI] [PubMed] [Google Scholar]
- 44.Valenca S.S., Rueff-Barroso C.R., Pimenta W.A., Melo A.C., Nesi R.T., Silva M.A.S., Porto L.C. L-NAME and L-arginine differentially ameliorate cigarette smoke-induced emphysema in mice. Pulm Pharmacol Ther. 2011;24:587–594. doi: 10.1016/j.pupt.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 45.Oller J., Mendez-Barbero N., Ruiz E.J., Villahoz S., Renard M., Canelas L.I., Briones A.M., Alberca R., Lozano-Vidal N., Hurle M.A., Milewicz D., Evangelista A., Salaices M., Nistal J.F., Jimenez-Borreguero L.J., De Backer J., Campanero M.R., Redondo J.M. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat Med. 2017;23:200–212. doi: 10.1038/nm.4266. [DOI] [PubMed] [Google Scholar]
- 46.Bogdan S., Seferian A., Totoescu A., Dumitrache-Rujinski S., Ceausu M., Coman C., Ardelean C.-M., Dorobantu M., Bogdan M. Sildenafil reduces inflammation and prevents pulmonary arterial remodeling of the monocrotaline -induced disease in the Wistar rats. Maedica (Buchar) 2012;7:109–116. [PMC free article] [PubMed] [Google Scholar]
- 47.Nunes A.K., Raposo C., Luna R.L., Cruz-Hofling M.A., Peixoto C.A. Sildenafil (Viagra(R)) down regulates cytokines and prevents demyelination in a cuprizone-induced MS mouse model. Cytokinel. 2012;60:540–551. doi: 10.1016/j.cyto.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 48.de Santana Nunes A.K., Raposo C., Bjorklund U., da Cruz-Hofling M.A., Peixoto C.A., Hansson E. Sildenafil (Viagra((R))) prevents and restores LPS-induced inflammation in astrocytes. Neurosci Lett. 2016;630:59–65. doi: 10.1016/j.neulet.2016.07.029. [DOI] [PubMed] [Google Scholar]
- 49.Raposo C., Nunes A.K., Luna R.L., Araujo S.M., da Cruz-Hofling M.A., Peixoto C.A. Sildenafil (Viagra) protective effects on neuroinflammation: the role of iNOS/NO system in an inflammatory demyelination model. Mediators Inflamm. 2013;2013:321460. doi: 10.1155/2013/321460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Toward T.J., Smith N., Broadley K.J. Effect of phosphodiesterase-5 inhibitor, sildenafil (Viagra), in animal models of airways disease. Am J Respir Crit Care Med. 2004;169:227–234. doi: 10.1164/rccm.200211-1372OC. [DOI] [PubMed] [Google Scholar]
- 51.Wang T., Liu Y., Chen L., Wang X., Hu X.-R., Feng Y.-L., Liu D.-S., Xu D., Duan Y.-P., Lin J., Ou X.-M., Wen F.-Q. Effect of sildenafil on acrolein-induced airway inflammation and mucus production in rats. Eur Respir J. 2009;33:1122–1132. doi: 10.1183/09031936.00055908. [DOI] [PubMed] [Google Scholar]
- 52.Kniotek M., Boguska A. Sildenafil can affect innate and adaptive immune system in both experimental animals and patients. J Immunol Res Egypt. 2017;2017:4541958. doi: 10.1155/2017/4541958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim K.-O., Park S.-Y., Han C.-W., Chung H.K., Yoo D.-H., Han J.-S. Effect of sildenafil citrate on interleukin-1beta-induced nitric oxide synthesis and iNOS expression in SW982 cells. Exp Mol Med. 2008;40:286–293. doi: 10.3858/emm.2008.40.3.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.