

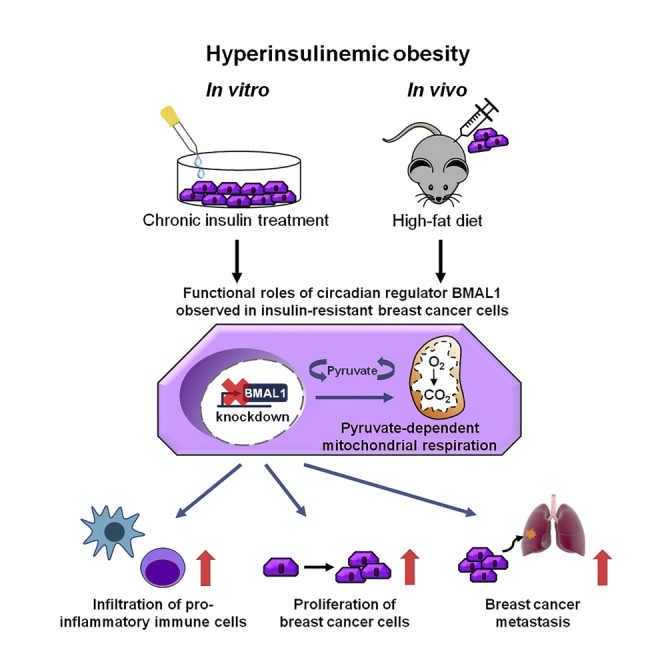
Summary
The epidemiological association between disrupted circadian rhythms and metabolic diseases is implicated in increased risk of human breast cancer and poor therapeutic outcomes. To define a metabolic phenotype and the underlying molecular mechanism, we applied chronic insulin treatment (CIT) to an in vitro model of triple-negative breast cancer to directly address how BMAL1, a key circadian transcription factor, regulates cancer cell respiration and governs tumor progression. At the cellular level, BMAL1 suppresses the flexibility of mitochondrial substrate usage and the pyruvate-dependent mitochondrial respiration induced by CIT. We established an animal model of diet-induced obesity/hyperinsulinemia and observed that BMAL1 functions as a tumor suppressor in obese, but not lean, mice. Downregulation of BMAL1 is associated with higher risk of metastasis in human breast tumors. In summary, loss of BMAL1 in tumors confers advantages to cancer cells in both intrinsic mitochondrial metabolism and extrinsic inflammatory tumor microenvironment during pre-diabetic obesity/hyperinsulinemia.
Subject Areas: Biological Sciences, Cell Biology, Chronobiology, Cancer
Graphical Abstract
Highlights
-
•
Circadian regulator BMAL1 rewires metabolism in a chronic insulin-treated TNBC model
-
•
Pyruvate links BMAL1 to mitochondrial bioenergetics
-
•
BMAL1 suppresses tumor proliferation and metastasis in hyperinsulinemic obese mice
-
•
BMAL1 influences tumor microenvironment in high-fat-diet-fed mice
Biological Sciences; Cell Biology; Chronobiology; Cancer
Introduction
Rhythmic oscillations in cellular activity are coordinated by genes that comprise the core circadian machinery (Bell-Pedersen et al., 2005). The notion that circadian rhythm disruptions lead to an increased likelihood of mammary tumor development is supported by studies that female workers taking frequent night shifts have disrupted internal clocks and an increased risk of developing breast cancer (BC) (Davis et al., 2001, Schernhammer et al., 2001, Wegrzyn et al., 2017). It has been reported that circadian genes have anti-tumor roles in multiple cancer types (de Assis et al., 2018, Papagiannakopoulos et al., 2016, Tang et al., 2017) and may act as tumor suppressors by regulating a wide variety of pathophysiological events, including cellular metabolism and pathogenesis of cancer (Cederroth et al., 2019, Lesicka et al., 2018, Lin and Farkas, 2018). Indeed, disrupted circadian rhythm likely affects multiple hallmarks of cancer, including proliferative signaling maintenance, cell death resistance, replicative immortality, invasion and metastasis activation, and energy metabolism reconfiguration (Hanahan and Weinberg, 2011).
Epidemiological data also suggest that disrupted circadian rhythm is strongly associated with metabolic syndrome (Verlande and Masri, 2019). Multiple factors common to metabolic syndrome and its related disorders have been shown to promote BC tumor growth and progression (Hursting et al., 2012). For example, hyperinsulinemia, or elevated circulating insulin levels, is a hallmark of obesity and pre-diabetes and is associated with higher BC incidence and poor prognosis (Del Giudice et al., 1998, Goodwin et al., 2002, Gunter et al., 2009, Lawlor et al., 2004, Lipscombe et al., 2006, Tsujimoto et al., 2017). Moreover, multiple reports indicate that metabolic diseases differentially impact individual subtypes of BC (Capasso et al., 2014, Garcia-Estevez and Moreno-Bueno, 2019, Yang et al., 2011). Overall, despite current studies demonstrating a close relationship among circadian disruption, metabolic syndrome, and cancer progression, the mechanisms linking these together are not yet systematically established.
Circadian clocks are orchestrated by a gene network through transcriptional regulation (Partch et al., 2014). Circadian-controlled genes are transcribed after initiation by the transcription factor heterodimer complex comprising Brain and Muscle Arnt-Like (BMAL) and Circadian Locomotor Output Cycles Kaput (CLOCK) (Sahar and Sassone-Corsi, 2009). Together with BMAL1 (gene name: ARNTL) and CLOCK, the core circadian genes comprise the machinery that implements the oscillations in cellular activity (Bell-Pedersen et al., 2005). Therefore, BMAL1 is an essential clock gene affecting all rhythmic behaviors as the loss of BMAL1 ensures that the circadian heterodimer is disrupted (Bunger et al., 2000). A number of reports have documented the crosstalk between BMAL1 function and insulin signaling, mitochondrial function, and lipid metabolism in non-cancer tissues (Breit et al., 2018, Dang et al., 2016, Jacobi et al., 2015, Liu et al., 2016, Luciano et al., 2018, Luo et al., 2019, Marcheva et al., 2010, McGinnis et al., 2017, Zhang et al., 2014). In addition, BMAL1 transcriptionally activates genes important for its own feedback loop (Sato et al., 2006) as well as many cellular processes, including mitochondrial dynamics, cell cycle regulation, and stress responses (Hatanaka et al., 2010, Jacobi et al., 2015, Rey et al., 2011). Global and liver-specific knockout of BMAL1 resulted in hyperlipidemia and increased lipoprotein production (Pan et al., 2016). However, the functional role of BMAL1 in BC cell metabolism and tumor progression remains largely unknown.
Understanding how circadian disruption and metabolic dysregulation influence BC can help elucidate the underlying biological mechanisms, especially in the basal-like (BL)/triple-negative BC (TNBC) subtype for which there is currently no effective therapeutic target. In this study, we developed two independent cell models, in vitro and in vivo, to investigate how the deficiency of intrinsic BMAL1 affects cellular metabolic homeostasis and tumor progression of TNBC. We demonstrate a previously unrecognized effect by tumor-intrinsic BMAL1 on shaping the mitochondrial fuel flexibility and anti-tumor landscape. Our observations provide insights into the mechanism underlying BMAL1-mediated TNBC suppression and evidence that BMAL1 downregulation in combination with hyperinsulinemic obesity exacerbates TNBC progression and aggressiveness. We discuss the implications of using BMAL1 as a marker, along with diet management, for cancer prevention and therapeutics.
Results
Developing an In Vitro Cell Model to Investigate BMAL1 Function in TNBC
Circadian rhythms not only vary among different organisms but also can be unique in different tissue organs within the same organism (Yoo et al., 2004). Investigation across cancers originated from different tissues suggested that altered expression of clock genes often shows cancer type-specific pattern and is associated with oncogenic pathways, clinical outcomes, and molecular subtypes (Ye et al., 2018). To examine the expression profile of canonical core circadian genes across different cancer types, we investigated The Cancer Genome Atlas (TCGA) Pan-Cancer datasets. Consistent with the corroborated link between circadian disruption and BC (Blakeman et al., 2016), the expression of ARNTL/BMAL1 was significantly down-regulated in primary tumor samples compared with normal tissues in multiple cancer types, including BC (Figure S1A). As BC is highly heterogeneous and can be classified into different molecular subtypes (Perou et al., 2000), subsequent analysis using unsupervised hierarchical clustering revealed that the BL subtype has a distinct circadian gene expression profile among the PAM50 molecular subtypes (Figure 1A). Most tumors of BL subtype do not express receptors for estrogen, progesterone, and HER2, the major characteristics of TNBC. This subtype is more aggressive and has a poorer prognosis than other BC subtypes (Subik et al., 2010, Toft and Cryns, 2011). The average expression of the core circadian genes (Figure 1B) and ARNTL/BMAL1 (Figure 1C) in the BL and HER2-enriched subtypes was significantly lower compared with the other subtypes. We thereby sought to further study BMAL1 deficiency with the focus on BL/TNBC because circadian rhythm disruption may affect BC in a subtype-dependent manner.
Figure 1.

Expression of Core Circadian Genes Is Altered in BC
(A) The 1,097 BC patient samples from the TCGA database were analyzed via unsupervised hierarchical clustering to show circadian expression profiles among the PAM50 subtypes: Basal-like, HER2-enriched, Luminal A, Luminal B, and Normal-like.
(B and C) The patient samples were analyzed for average expression of circadian genes (B) and ARNTL expression (C), grouped by BC subtypes.
(D) Immunoblot analysis (left panel) of markers of active insulin signaling—phosphorylated AKT and phosphorylated IRβ—were examined to reflect relative levels of insulin signaling. GAPDH serves as a loading control. Quantification of phosphorylated AKT/total AKT and phosphorylated IRβ/total IRβ is shown relative to GAPDH levels, and signal in untreated cells is set to 1 (right panel). CIT: chronic insulin treatment.
(E) Cells were entrained by serum shock. Total RNA was collected every 4 h and analyzed by qRT-PCR. Data are shown as mean ± SD; n = 3; one-way ANOVA with Tukey's post hoc test. The GAPDH mRNA level of untreated cells at 0 h is set to 1.
(B, C, and E) N/S p > 0.05; *p > 0.05; **p < 0.05; ***p < 0.001. See also Figures S1 and S2.
To identify a suitable in vitro cell model, we evaluated a panel of 51 BC cell lines. The gene expression profiles are in general agreement with our observations from the TCGA clinical samples, confirming that circadian gene expression levels differ among the BC subtypes. Among the BL/TNBC cell lines, MDA-MB-231 was selected for its low- to mid-range abundance of circadian gene expression (Figure S1B) and ARNTL message (Figure S1C). Next, to develop a metabolic phenotype, MDA-MB-231 cells were continuously passaged in media supplemented with insulin for more than 10 passages, referred to as chronic insulin treatment (CIT). To mimic the insulin levels in a post-meal, fed state during pre-diabetes (high insulin with normal glucose), CIT cells treated with 10 or 100 nM insulin were assayed to verify that these cells were no longer sensitive to additional insulin stimulation. As shown in Figure 1D, no strong increase in insulin signaling activation was observed in serum/insulin-deprived (24 h) CIT cells stimulated with a high concentration of insulin at 100 nM, indicating the development of insulin resistance. To examine the effect of CIT on circadian outputs, a serum shock procedure was applied to stimulate and synchronize oscillations of circadian genes (Balsalobre et al., 1998). Cells grown without insulin exhibited a standard ARNTL mRNA oscillation, whereas we observed decreased amplitude for short-term insulin treatment and an early peak for CIT (Figure 1E). Of note, we also tested the same assay with additional cell lines having different abundance of endogenous BMAL1 (Figure S2A). Alteration of ARNTL mRNA oscillation was again observed in another TNBC cell line BT549 (Figure S2B), as well as progesterone receptor-positive MCF7 cells (Figure S2C), suggesting that the effect of short-term insulin and CIT on ARNTL mRNA oscillation is common. However, the alteration pattern may vary with different cell types.
The Interplay between BMAL1 and Mitochondrial Adaptations to CIT
The molecular interplay between circadian rhythms and cellular metabolism has been delineated as circadian genes control the nicotinamide adenine dinucleotide (NAD+) salvage pathway (Nakahata et al., 2009). Thus, we conducted oscillating circadian-controlled NAD+ assays (Ramsey et al., 2009) with untreated and CIT cells. CIT cells showed a faster peak time and a higher steady-state NAD+/NADH ratio than those in insulin-responsive MDA-MB-231 cells (Figure S3A), demonstrating the links among insulin signaling, circadian output, and cellular metabolism. Also, the oxidation of NADH to NAD+ links the tricarboxylic acid (TCA) cycle to ATP generation in mitochondria. Based on the above results, we tested the hypothesis that mitochondrial activity may partake in BMAL1 regulation. In ρ0 cells (depleted of mitochondrial DNA), we observed a significant decrease in the steady-state BMAL1 protein levels (Figure S3B) and mRNA levels of ARNTL/BMAL1, along with its direct target NAMPT, irrespective of CIT (Figure S3C). Unlike in MDA-MB-231 cells (Figure 1E), there was no detectable difference in ARNTL message oscillation between untreated and CIT ρ0 cells (Figure S3D). We concluded that the mitochondrial activity is indispensable for the expression and CIT-mediated regulation of BMAL1 and its downstream NAD+-regulating target gene.
Next, we tested the state of mitochondrial respiration in the context of decreased BMAL1 expression. ARNTL-targeting small interfering RNA (siRNA) (siBMAL1) was designed and used to decrease BMAL1 abundance by nearly 90% (Figure 2A). In both untreated and CIT MDA-MB-231 cells, ARNTLknockdown produced higher levels of intracellular ATP (Figure 2B). Moreover, following CIT, BMAL1-knockdown cells produced a higher mitochondria-specific reactive oxygen species signal (reflecting electrons leaking from the electrochemical gradient) compared with control cells (Figure 2C). Together, these data suggested that BMAL1 regulates bioenergetic homeostasis and possibly other respiration outputs.
Figure 2.

BMAL1 Mediates Pyruvate Utilization during Chronic Insulin Treatment
(A) Representative immunoblot showing the CIT effect on BMAL1 in MDA-MB-231 cells. BMAL1-targeting or control siRNA was transiently transfected into untreated or CIT cells. Owing to different levels of protein abundance, BMAL1 blot with longer exposure time, compared with GAPDH blot, is shown.
(B and C) (B) Intracellular levels of ATP and (C) mitochondrial superoxide were assayed in MDA-MB-231 cells grown without insulin (Untreated) or over 10 passages of insulin (100 nM, CIT) and then transiently transfected with control or BMAL1-targeting siRNA. Measurements are normalized to cell number. n = 3; independent samples t test.
(D) Intracellular pyruvate was measured in MDA-MB-231 cells not treated or chronically treated with insulin (100 nM, CIT) and transfected with control or BMAL1-targeting siRNA. Pyruvate levels are normalized to cell number. n = 3.
(E) One representative oxygen consumption rate (OCR) profile of Mito Stress Test with MDA-MB-231 cells containing control or BMAL1-targeting siRNA in media ± pyruvate.
(F) Pyruvate is required for maximal respiration, calculated from (E). n ≥ 3.
(G) The relative levels of mRNA encoding pyruvate-regulating enzymes from untreated or CIT MDA-MB-231 cells transfected with control or BMAL1/ARNTL-targeting siRNA, quantified using qRT-PCR. n = 3.
(H) The basal respiration in untreated or CIT MDA-MB-231 cells transfected with control, BMAL1/ARNTL, or PKM-targeting siRNA were analyzed using Mito Stress Test. Measurements are normalized to cell number. n = 3; one-way ANOVA with Tukey's post hoc.
(I) The capacity to oxidize pyruvate (left panel), glutamine (middle panel), and fatty acids (right panel) in control or siBMAL1-transfected, untreated, or CIT MDA-MB-231 cells was analyzed using the Mito Fuel Flex Test. n = 3; one-way ANOVA with Tukey's post hoc.
(B–I) Data are shown as mean ± SD; *p < 0.05; **p < 0.01; ***p < 0.001. (B–D and F–I) one-way ANOVA with Tukey's post hoc. See also Figures S3–S6.
To pinpoint the involvement of BMAL1 in mitochondrial bioenergetics, oxygen consumption rate (OCR) was measured in untreated or CIT MDA-MB-231 cells. In control-knockdown cells, CIT increased OCR in a dose-dependent manner (Figure S3E), and an opposite OCR pattern was observed in BMAL1-knockdown cells (Figure S3F). BMAL1 knockdown increased basal respiration over control knockdown in untreated cells (Figure S3G, left panel). Also, BMAL1 was required for CIT to increase maximum respiration, a measurement of respiratory capacity, in a dose-dependent manner (Figure S3G, right panel). Given the fact that BMAL1 suppresses basal OCR in untreated cells and promotes maximal OCR in CIT cells, BMAL1 conceivably plays a key role in the mitochondrial metabolic adaptation to CIT.
We then investigated how BMAL1-mediated mitochondrial adaptations affect other metabolic parameters by measuring the levels of neutral lipid droplets, a nutrient source for cancer (Petan et al., 2018). Before assaying, MDA-MB-231 cells were starved of glucose to promote fatty acid mobilization (Rambold et al., 2015). We observed that BMAL1 knockdown increased the amounts of lipid droplets in both untreated and CIT cells (Figure S4A). Also, another functional consequence of CIT and BMAL1knockdown was an increase in the sensitivity to metformin (Figure S4B), a biguanide that causes oxidative stress by inhibiting mitochondrial complex I (Mayer et al., 2015). Last, by measuring extracellular acidification rate, we concluded that neither BMAL1 nor CIT impacted glycolytic activity (Figures S4C–S4E) in MDA-MB-231 cells.
Pyruvate Links BMAL1 to Mitochondrial Bioenergetics
Pyruvate is a key link between glycolysis and TCA cycle (Figure S5A). Based on the above-mentioned results, we hypothesized that BMAL1 serves a distinct role in regulating pyruvate utilization in untreated and CIT cells. To address this possibility in different BC cells, intracellular pyruvate levels were measured in MDA-MB-231, along with BT549 and MCF7 cells. As we observed in MDA-MB-231 cells (shown in Figure 2A), CIT did not affect steady-state level of BMAL1 in either BT549 or MCF7 cells (Figure S5B). However, CIT increased pyruvate levels in control (without BMAL1 knockdown) MDA-MB-231 (Figure 2D) and BT-549 (Figure S5C) cells compared with their untreated cells. Also, pyruvate level was not affected by BMAL1 knockdown in untreated MDA-MB-231 cells (Figure 2D). In contrast, BMAL1 knockdown in CIT cells decreased pyruvate levels in all three (MDA-MB231, BT549, and MCF7) cell lines (Figures 2D, S5C, and S5E), suggesting that BMAL1 suppresses pyruvate utilization during CIT adaptation. We then measured OCR in the presence and absence of external pyruvate in MDA-MB-231 cells (Figure 2E). As expected, the increased maximal respiration by BMAL1 knockdown depended on external pyruvate availability (Figure 2F).
Because BMAL1 is a transcription factor, we posited that BMAL1 regulates genes that metabolize pyruvate (Deng et al., 2018). We found that BMAL1 knockdown reduced the expression of pyruvate kinase (PKM) and pyruvate carboxylase (PC) (converting phosphoenolpyruvate to pyruvate and converting pyruvate to oxaloacetate, as shown in Figure S5A) in untreated MDA-MB-231 cells (Figure 2G). In comparison, BMAL1 knockdown also markedly decreased PKM, PC, and PDK4 mRNA levels in CIT BT549 (Figure S5D), but only showed relatively minor effect in MCF7 cells (Figure S5F), suggesting a context-dependent role of BMAL1 in regulating pyruvate metabolism enzyme expression in different BC cells. To test if the effects of BMAL1 on OCR are mediated through its regulation of PKM abundance, we applied PKM-targeting siRNA to lower PKM expression in MDA-MB-231 cells and assayed for mitochondrial respiration activity. Consistently, PKM knockdown increased OCR in untreated cells, but decreased OCR in CIT MDA-MB-231 cells, mimicking the effects seen in BMAL1knockdown (Figure 2H). The decreased PC activity could also render pyruvate less available to mitochondria (Perry et al., 2015). Together, these findings underscored a specific role of BMAL1 in regulating pyruvate metabolism.
To corroborate that BMAL1 affects substrate utilization, specific inhibitors were used to block three major mitochondrial fuels: glutamine, long-chain fatty acids, and pyruvate. Mito Fuel Flex assays demonstrated that BMAL1 knockdown altered substrate capacity, or the ability of cells to increase oxidation of particular fuel to compensate for inhibition of alternative fuels. Specifically, BMAL1knockdown increased glutamine and fatty acid oxidation capacity in both untreated and CIT cells (Figure 2I, middle and right panels), indicating that BMAL1 suppresses glutamine and fatty acid utilization in MDA-MB-231 cells. However, BMAL1 knockdown increased the capacity of pyruvate oxidation under CIT, but decreased it in the insulin-sensitive cells (Figure 2I, left panel). Finally, compromised BMAL1 decreased pyruvate (Figure S6A), but not glutamine (Figure S6B) and fatty acid (Figure S6C), dependency in mitochondria of CIT cells. These results demonstrated that BMAL1 regulates substrate usage and that CIT cells require BMAL1 to suppress substrate flexibility. This is consistent with our observations that BMAL1 knockdown mediates reduction of message abundance of key pyruvate metabolism-regulating enzymes (Figure 2G). Altogether, we surmised that CIT and BMAL1 counterbalanced the regulation of mitochondrial activity. This observed dual regulation of pyruvate by both BMAL1 and insulin would explain how insulin context alters the effect by BMAL1 on tumor cell metabolism.
BMAL1 Suppresses HFD-Induced Tumor Growth In Vivo
To further establish whether BMAL1 regulates tumor progression in vivo, we used wild-type female C57BL/6 mice coupled with mouse mammary tumor E0771 cells orthotopically implanted in syngeneic lean and obese mice (Figure S7A). The advantage of using an allografted syngeneic mouse tumor model over an MDA-MB-231-xenografted NSG mouse model is that C57BL/6 mice are better responsive to high-fat diet (HFD) feeding (NSG mice are, but only with litter reduction, Behan et al., 2013) and that we are able to study the interaction between cancer cells and their surrounding tumor microenvironment in an immunocompetent animal model. The E0771 cells were derived from a spontaneously developed TNBC in C57BL/6 mice (Ewens et al., 2005, Yang et al., 2017). Parental E0771 cells are poorly metastatic when compared with 4T1 cells (Johnstone et al., 2015) and have homozygous mutations in Trp53 and Kras genes (Yang et al., 2011) with a detectable BMAL1 expression (Figure 3A, inset). When mice reached young adulthood at the age of 8 weeks, they were separated into two groups: HFD or low-fat diet (LFD) (Dutta and Sengupta, 2016). These two diets had equal calories and sucrose for consistent chow palatability and to ensure consistent caloric intake. HFD comprised 60% fat, whereas LFD had 10%. Fasting insulin and glucose levels were measured before starting the diets and 4 weeks after consuming either HFD or LFD. The results showed that LFD did not cause a notable change in fat mass and fasting insulin levels compared with measurements before starting the diet; however, both increased in HFD-fed mice without affecting fasting glucose levels (Figure S7B). To test whether the lack of hyperglycemia in HFD-fed mice is due to hyperinsulinemia, which mimics pre-diabetic state, an oral glucose tolerance test was performed. Following glucose feeding after a starvation period, blood glucose levels spiked faster and remained high longer in HFD-fed mice compared with the mice with LFD (Figure S7C). The HFD-fed mice also displayed higher body weight gained than LFD-fed mice, even though food intake did not differ (Figures S7D and S7E).
Figure 3.

BMAL1 Suppresses HFD-Promoted Tumor Growth
(A) Palpable parental (left panel) or shBMAL1 (right panel) tumors were measured in LFD- and HFD-fed mice. n = 4–6; independent samples' t test at each time point. Inset features immunoblot analysis of parental E0771 and E0771 with stable knockdown of BMAL1. Italicized numbers indicate relative BMAL1 expression level.
(B) Tumor volumes from parental and BMAL1-knockdown E0771 cells in LFD- and HFD-fed mice were analyzed at day 28 after implantation (left panel). Representative tumor images are shown (right panel). n = 4–6.
(C) Formalin-fixed tissue sections from tumors derived from parental and BMAL1-knockdown E0771 cells in LFD- and HFD-fed mice were stained with anti-Ki67 antibody. % Ki67+ cells were calculated as Ki67-positive cells divided by total cells (left panel). Representative images of stained tumor tissues are shown (right panel). n = 3. Scale bar, 200 μm; red scale bar, 100 μm (inset).
(D) Number of lung nodules of parental or BMAL1-knockdown E0771 cells per mouse were counted in LFD- and HFD-fed mice (left panel). Representative image of lung lobes with metastatic nodules are shown (right panel); black scale bar, 3 mm; white scale bar in inset, 2 mm. n ≥ 4.
(E) Relative maximum OCR was calculated for primary E0771 tumors of engrafted cells at passage 2 after extraction from tumors in LFD- and HFD-fed mice. n ≥ 4.
(B–E) Data are shown as mean ± SD; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; one-way ANOVA with Tukey's post hoc. See also Figure S7.
After confirming HFD-induced obesity/hyperinsulinemia, the fourth mammary fat pad on the right side of lean or obese mice was implanted with parental E0771 cells or E0771 cells with stable BMAL1 knockdown. The mice were maintained on their respective diet for an additional 4 weeks. In general, HFD accelerated tumor progression over LFD (Figure 3A). Upon tumor harvest, BMAL1-knockdown E0771 tumors were not significantly different in size from parental tumors in lean mice, whereas they were larger than the corresponding tumors of parental cells in hyperinsulinemic mice (Figure 3B). The E0771/shBMAL1-derived tumors from HFD-fed mice showed the largest sizes among these four groups (Figure 3B, right panel). There was no notable difference in Ki67, a marker for cell proliferation, signals between tumors from the parental E0771 cells of LFD- and HFD-fed mice; however, BMAL1 knockdown decreased Ki67 signals in tumors harvested from lean, but not hyperinsulinemic, obese mice (Figure 3C). E0771/shBMAL1-derived tumors from LFD-fed mice had the least proliferative potential. We next examined whole lungs from obese and lean tumor-harboring mice as lungs are a common site of metastasis from E0771-derived mammary tumors (Yousefi et al., 2018). Likewise, the obese tumor-bearing mice harbored more lung tumor nodules than the lean group (Figure 3D). In addition, BMAL1knockdown decreased the lung tumor nodules in LFD-fed tumor-bearing mice, yet significantly increased it in HFD-fed mice. Altogether, BMAL1 serves a non-canonical role of BMAL1 to suppress tumor progression and/or metastasis under hyperinsulinemic obese context.
Last, primary tumor cells from the orthotopically engrafted E0771 tumors were isolated, cultured, and assessed for mitochondrial respiratory function. It is important to note that, recapitulating findings in MDA-MB-231 cells in vitro, BMAL1 knockdown increased OCR in E0771 cells recovered from tumors of lean mice, whereas BMAL1 knockdown decreased OCR in E0771 cells from hyperinsulinemic obese mice (Figure 3E). This observation validates the importance of the combined effect of BMAL1-mediated metabolic adaptation and hyperinsulinemia as a determinant of TNBC progression and aggressiveness. In addition, using two different knockdown approaches in cells from two different species yielded comparable conclusion about the role of BMAL1 on mitochondrial bioenergetics, and these separate approaches likely ruled out potential off-target effect(s) of BMAL1 knockdown.
BMAL1 Influences Tumor Microenvironment in Hyperinsulinemic Obese Mice
To further explore how BMAL1 and HFD-induced obesity interactively regulate tumor progression, we examined how the tumor was influenced by the microenvironment. Infiltration of F4/80 macrophage, a major component of the tumor microenvironment, was assessed by immunohistochemistry (Goswami et al., 2017, Hu et al., 2016, Netea-Maier et al., 2018). Inside the parental E0771-derived tumors, there were no notable differences in F4/80 signals regardless of diet; however, the E0771/shBMAL1-derived tumors in the hyperinsulinemic obese mice, compared with lean mice, exhibited significantly increased F4/80 signals (Figure 4A). Other prominent immune cells featured inside tumors are the CD8+ T cells, which play a protective role and are associated with better cancer prognoses and outcomes (Fu and Jiang, 2018). Tumors from hyperinsulinemic obese mice exhibited significantly decreased CD8+ T cell signals in the parental tumors than those in the lean mice (Figure 4B). Notably, there were no differences in CD8+ T cell recruitment in shBMAL1 tumors regardless of diet (Figure 4B).
Figure 4.

Tumor-Expressed BMAL1 Influences Immune Cell Infiltration
(A) F4/80 signal was quantified as the average area of F4/80 staining in non-overlapping sections of entire tumor and compared with other samples at the same magnification. n = 4 (left panel). Representative images of tissue sections are shown (right panel). Scale bar, 500 μm. Red scale bar, 125 μm (inset).
(B) CD8 signal was quantified as the average area of CD8 staining in non-overlapping sections of entire tumor section and compared with other samples at the same magnification. n = 4. (A and B) Data shown as mean ± SEM; *p < 0.05; **p < 0.01; one-way ANOVA with Tukey's post hoc. Representative images of stained tissue are shown (right panel). Scale bar, 250 μm. Red scale bar, 100 μm (inset).
(C) Gene Set Enrichment Analysis shows that expression of hallmark OxPhos gene set in TCGA BC primary tumors (n = 1,097) is inversely correlated with BMAL1 expression.
(D and E) Kaplan-Meier curves show distal metastasis-free survival rate of patients selected according to BMAL1 expression from two independent datasets GEO: GSE20685 (D) and GSE11121 (E).
(F) Hyperinsulinemia and BMAL1 regulate both internal metabolism and external microenvironment of TNBC. The intrinsic metabolism of TNBC reveals that BMAL1 and hyperinsulinemia have parallel yet distinct regulations of pyruvate and mitochondrial metabolism. The external TNBC microenvironment shows that BMAL1 and hyperinsulinemia in concert control immune cell recruitment and infiltration. The dotted line indicates a possible mechanism for immune cell recruitment via mitochondria-induced reactive oxygen species.
We further performed Gene Set Enrichment Analysis of human primary breast tumor data from TCGA and found that the oxidative phosphorylation (OxPhos) pathway is significantly upregulated in patient tumors with downregulated ARNTL (Figure 4C). Such observation is consistent with our findings of increased OCR during BMAL1 knockdown (Figures S3E and S3F). Together with Figure S1C, our previously reported increase in OxPhos gene expression in TNBC and HER2-enriched BCs (Cheng et al., 2016) could be attributed to decreased ARNTL expression. In addition, although the metabolic states were not available, our analyses of two independent public datasets for human BC (Kao et al., 2011, Schmidt et al., 2008) revealed that reduced ARNTL expression is associated with worse distal metastasis-free survival (Figures 4D and 4E).
Discussion
Although HFD-induced obesity drives tumor growth through multiple mechanisms (Nunez et al., 2008), our findings support a new paradigm that reduced expression of ARNTL, a key component of the circadian clock, accelerates TNBC tumor growth and lung metastasis in an HFD-induced hyperinsulinemic obese context. Virtually, circadian clock disruption is detrimental to metabolic homeostasis in all cells. To underscore the importance of BMAL1 in the adaptation to hyperinsulinemia in BC cells, here we report that in two different TNBC cells in two distinct models, in vitro and in vivo, BMAL1 loss (BMAL1 crisis) decreases or increases mitochondrial respiration depending on hyperinsulinemia. In parallel, intrinsic BMAL1 oscillation is suppressed by short-term insulin exposure in a mitochondria-dependent manner in MDA-MB-231 cells. We propose a model in which the reduced BMAL1 expression cooperates with HFD-induced hyperinsulinemic obesity to accelerate TNBC tumor progression and lung metastasis through increased mitochondrial fuel flexibility and reshaped recruitment of macrophages and CD8+ T cells (Figure 4F).
Cancer cells exhibit metabolic plasticity, rewiring their metabolism to satisfy the demands of rapid cell proliferation and survival (Ghaffari et al., 2015). Mitochondria comprise a metabolic hub that is responsible for energy production, and also produce building blocks for cell growth by catabolizing glucose, amino acids, and fatty acids. Although we observed that circadian gene expression and mitochondrial activity appear to be interactive and counter-balanced, the detailed underlying mechanism is still not clear. It is plausible that metabolites from the mitochondria directly affect the transcriptional regulation of circadian genes. In addition, post-translational modifications also play a key role in regulating the expression of circadian genes, such as histone acetylation being required at promoters of BMAL1-CLOCK target genes (DiTacchio et al., 2011). Here, we introduce a link between circadian signaling and mitochondrial function by showing that BMAL1 transcriptionally regulates PKM expression. It hints that endogenous pyruvate production functions as a linchpin for BMAL1-mediated metabolic adaptations. As such, the combined effect of BMAL1 and hyperinsulinemic obesity on reshaping the tumor microenvironment is conceivably mediated by regulating intracellular pyruvate utilization. Our model on the combined effect of intracellular pyruvate and on promoting tumor progression is supported by reports that pyruvate metabolism is important in maintaining colon cancer stem cell proliferation (Schell et al., 2017) and that PKM2-null mice display increased HFD-induced metabolic stress (Dayton et al., 2016).
Inflammation plays a key role in hyperinsulinemic obesity and metabolic disease (Chawla et al., 2011, Lumeng and Saltiel, 2011). In our model, it appears that cancer cell-intrinsic BMAL1 acts as a critical suppressor of F4/80 macrophage tumor infiltration under hyperinsulinemic obesity. Cancer cell BMAL1-mediated suppression of F4/80 macrophage infiltration thus has important implications for how anti-tumor immunity is impaired during hyperinsulinemic obesity. A causative role of immune cells in promoting tumor progression has been demonstrated (Wellenstein and de Visser, 2018) as depleting inflammatory immune cells leads to rapid improvement in insulin sensitivity and glucose tolerance, which is associated with decreased local and systemic inflammation in obese mice (Nishimura et al., 2009). The role of BMAL1 in immune cell recruitment is not yet elucidated. The observed decrease in CD8+ T cell recruitment in tumors harvested from HFD-fed mice is in agreement with findings in another report (Wang et al., 2019). Considering such a decrease was not observed in BMAL1-knockdown tumors, one plausible function of BMAL1 is to prevent T cell exhaustion during obesity. Together, these data show that the immune cells surrounding the tumor are likely affected by the tumor cells' metabolic flexibility. Cancer-associated fibroblasts have been shown to secrete pyruvate to support metabolism in the tumor (Sakamoto et al., 2019) and the BMAL1-suppressed pyruvate utilization we observed in CIT cells may contribute to the tumor microenvironment landscape. Furthermore, macrophages consume exogenous pyruvate, resulting in increased PD-L1 expression and suppressed anti-tumor immune cells (Watanabe et al., 2017), providing a plausible mechanism of tumor-secreted pyruvate and uptake by immune cells.
In summary, our experimental results showed that BMAL1 suppresses TNBC progression in multiple ways: by maintaining the clock machinery and circadian rhythms, by suppressing hyperinsulinemia-dysregulated mitochondrial activity and pyruvate utilization, and by suppressing pro-inflammatory components of the immune microenvironment in the obese context. The latter roles could be attributed to non-canonical circadian functions of BMAL1. In this context, our results provide a compelling case for BMAL1 expression to be examined in patients with TNBC who also present with hyperinsulinemia in the clinic. TNBC tumors with BMAL1 crisis are predicted to have worse outcomes, especially during obesity/hyperinsulinemia. BMAL1 appears to act as a tumor suppressor in TNBC during obesity/hyperinsulinemia, underscoring the importance of understanding BMAL1-mediated mitochondrial metabolic reprogramming during CIT adaptation, which could be exploited pharmacologically or with diet management for better patient outcomes in the future.
Limitations of the Study
There is ample evidence that the cellular circadian clock regulates more than just cell-intrinsic metabolic processes. Based on consistent observations using two distinct models of TNBC, both in vitro and in vivo, we propose a paradigm that BMAL1 serves as a metabolic switch on hyperinsulinemia-regulated mitochondrial activity to govern TNBC tumor progression. Indeed, there is a need to better link circadian biology to cancer risk. Although our findings provide insights regarding the non-canonical BMAL1 function, additional studies are needed to validate how BMAL1 loss (BMAL1 crisis) influences most cases of BC in different metabolic states and whether these insights can be leveraged into an effective therapy. Also, although we demonstrated that BMAL1 crisis confers advantages to cancer cells in both intrinsic metabolism and extrinsic inflammatory microenvironment during hyperinsulinemia, the underlying signaling mechanism among BMAL1 crisis, hyperinsulinemia, and tumorigenesis in vivo remains to be established.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank the members of Dr. Ann's laboratory and Drs. Rama Natarajan and Lei Jiang for helpful discussions on the manuscript. We thank Dr. Joe Gray (Oregon Health & Science University) for providing breast cancer cell line gene expression data. This work was supported in part by funds from the National Cancer Institute of United States grant R01CA220693 (D.K.A. and V.L.S.), Department of Defense of United States grant BC141351 (M.A.L.), City of Hope Center for Cancer and Aging predoctoral fellowship (C.A.R.), and P30CA033572 (supporting research work carried out at City of Hope Core Facilities).
Author Contributions
Conceptualization, C.A.R. and D.K.A.; Methodology, C.A.R., C.O., Y.C., V.L.S., and D.K.A.; Investigation, C.A.R., C.O., Y.Q., Y.C., and C.-T.C.; Data analysis, C.A.R., C.O., Y.Q., and Y.C.; Resources, M.A.L. and D.K.A.; Manuscript Writing, C.A.R, C.O., Y.Q., M.A.L., V.L.S., and D.K.A.; Funding Acquisition, C.A.R., M.A.L., V.L.S., and D.K.A.
Declaration of Interests
The authors declare no competing interests.
Published: February 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100839.
Supplemental Information
References
- Balsalobre A., Damiola F., Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–937. doi: 10.1016/s0092-8674(00)81199-x. [DOI] [PubMed] [Google Scholar]
- Behan J.W., Ehsanipour E.A., Sheng X., Pramanik R., Wang X., Hsieh Y.T., Kim Y.M., Mittelman S.D. Activation of adipose tissue macrophages in obese mice does not require lymphocytes. Obesity. 2013;21:1380–1388. doi: 10.1002/oby.20159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell-Pedersen D., Cassone V.M., Earnest D.J., Golden S.S., Hardin P.E., Thomas T.L., Zoran M.J. Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat. Rev. Genet. 2005;6:544–556. doi: 10.1038/nrg1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeman V., Williams J.L., Meng Q.J., Streuli C.H. Circadian clocks and breast cancer. Breast Cancer Res. 2016;18:89. doi: 10.1186/s13058-016-0743-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breit A., Miek L., Schredelseker J., Geibel M., Merrow M., Gudermann T. Insulin-like growth factor-1 acts as a zeitgeber on hypothalamic circadian clock gene expression via glycogen synthase kinase-3beta signaling. J. Biol. Chem. 2018;293:17278–17290. doi: 10.1074/jbc.RA118.004429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger M.K., Wilsbacher L.D., Moran S.M., Clendenin C., Radcliffe L.A., Hogenesch J.B., Simon M.C., Takahashi J.S., Bradfield C.A. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso I., Esposito E., de Laurentiis M., Maurea N., Cavalcanti E., Botti G., Petrillo A., Montella M., D'Aiuto M., Coppola C. Metabolic syndrome-breast cancer link varies by intrinsic molecular subtype. Diabetol. Metab. Syndr. 2014;6:105. doi: 10.1186/1758-5996-6-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederroth C.R., Albrecht U., Bass J., Brown S.A., Dyhrfjeld-Johnsen J., Gachon F., Green C.B., Hastings M.H., Helfrich-Forster C., Hogenesch J.B. Medicine in the fourth dimension. Cell Metab. 2019;30:238–250. doi: 10.1016/j.cmet.2019.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A., Nguyen K.D., Goh Y.P. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 2011;11:738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C.T., Kuo C.Y., Ouyang C., Li C.F., Chung Y., Chan D.C., Kung H.J., Ann D.K. Metabolic stress-induced phosphorylation of KAP1 Ser473 blocks mitochondrial fusion in breast cancer cells. Cancer Res. 2016;76:5006–5018. doi: 10.1158/0008-5472.CAN-15-2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang F., Sun X., Ma X., Wu R., Zhang D., Chen Y., Xu Q., Wu Y., Liu Y. Insulin post-transcriptionally modulates Bmal1 protein to affect the hepatic circadian clock. Nat. Commun. 2016;7:12696. doi: 10.1038/ncomms12696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S., Mirick D.K., Stevens R.G. Night shift work, light at night, and risk of breast cancer. J. Natl. Cancer Inst. 2001;93:1557–1562. doi: 10.1093/jnci/93.20.1557. [DOI] [PubMed] [Google Scholar]
- Dayton T.L., Gocheva V., Miller K.M., Israelsen W.J., Bhutkar A., Clish C.B., Davidson S.M., Luengo A., Bronson R.T., Jacks T. Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes Dev. 2016;30:1020–1033. doi: 10.1101/gad.278549.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Assis L.V.M., Kinker G.S., Moraes M.N., Markus R.P., Fernandes P.A., Castrucci A.M.L. Expression of the circadian clock gene BMAL1 positively correlates with antitumor immunity and patient survival in metastatic melanoma. Front. Oncol. 2018;8:185. doi: 10.3389/fonc.2018.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Giudice M.E., Fantus I.G., Ezzat S., McKeown-Eyssen G., Page D., Goodwin P.J. Insulin and related factors in premenopausal breast cancer risk. Breast Cancer Res. Treat. 1998;47:111–120. doi: 10.1023/a:1005831013718. [DOI] [PubMed] [Google Scholar]
- Deng W., Zhu S., Zeng L., Liu J., Kang R., Yang M., Cao L., Wang H., Billiar T.R., Jiang J. The circadian clock controls immune checkpoint pathway in sepsis. Cell Rep. 2018;24:366–378. doi: 10.1016/j.celrep.2018.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiTacchio L., Le H.D., Vollmers C., Hatori M., Witcher M., Secombe J., Panda S. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science. 2011;333:1881–1885. doi: 10.1126/science.1206022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S., Sengupta P. Men and mice: relating their ages. Life Sci. 2016;152:244–248. doi: 10.1016/j.lfs.2015.10.025. [DOI] [PubMed] [Google Scholar]
- Ewens A., Mihich E., Ehrke M.J. Distant metastasis from subcutaneously grown E0771 medullary breast adenocarcinoma. Anticancer Res. 2005;25:3905–3915. [PubMed] [Google Scholar]
- Fu C., Jiang A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front. Immunol. 2018;9:3059. doi: 10.3389/fimmu.2018.03059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Estevez L., Moreno-Bueno G. Updating the role of obesity and cholesterol in breast cancer. Breast Cancer Res. 2019;21:35. doi: 10.1186/s13058-019-1124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaffari P., Mardinoglu A., Nielsen J. Cancer metabolism: a modeling perspective. Front. Physiol. 2015;6:382. doi: 10.3389/fphys.2015.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin P.J., Ennis M., Pritchard K.I., Trudeau M.E., Koo J., Madarnas Y., Hartwick W., Hoffman B., Hood N. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J. Clin. Oncol. 2002;20:42–51. doi: 10.1200/JCO.2002.20.1.42. [DOI] [PubMed] [Google Scholar]
- Goswami K.K., Ghosh T., Ghosh S., Sarkar M., Bose A., Baral R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell. Immunol. 2017;316:1–10. doi: 10.1016/j.cellimm.2017.04.005. [DOI] [PubMed] [Google Scholar]
- Gunter M.J., Hoover D.R., Yu H., Wassertheil-Smoller S., Rohan T.E., Manson J.E., Li J., Ho G.Y., Xue X., Anderson G.L. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 2009;101:48–60. doi: 10.1093/jnci/djn415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hatanaka F., Matsubara C., Myung J., Yoritaka T., Kamimura N., Tsutsumi S., Kanai A., Suzuki Y., Sassone-Corsi P., Aburatani H. Genome-wide profiling of the core clock protein BMAL1 targets reveals a strict relationship with metabolism. Mol. Cell. Biol. 2010;30:5636–5648. doi: 10.1128/MCB.00781-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W., Li X., Zhang C., Yang Y., Jiang J., Wu C. Tumor-associated macrophages in cancers. Clin. Transl. Oncol. 2016;18:251–258. doi: 10.1007/s12094-015-1373-0. [DOI] [PubMed] [Google Scholar]
- Hursting S.D., Digiovanni J., Dannenberg A.J., Azrad M., Leroith D., Demark-Wahnefried W., Kakarala M., Brodie A., Berger N.A. Obesity, energy balance, and cancer: new opportunities for prevention. Cancer Prev. Res. 2012;5:1260–1272. doi: 10.1158/1940-6207.CAPR-12-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobi D., Liu S., Burkewitz K., Kory N., Knudsen N.H., Alexander R.K., Unluturk U., Li X., Kong X., Hyde A.L. Hepatic Bmal1 regulates rhythmic mitochondrial dynamics and promotes metabolic fitness. Cell Metab. 2015;22:709–720. doi: 10.1016/j.cmet.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone C.N., Smith Y.E., Cao Y., Burrows A.D., Cross R.S., Ling X., Redvers R.P., Doherty J.P., Eckhardt B.L., Natoli A.L. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis. Model. Mech. 2015;8:237–251. doi: 10.1242/dmm.017830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao K.J., Chang K.M., Hsu H.C., Huang A.T. Correlation of microarray-based breast cancer molecular subtypes and clinical outcomes: implications for treatment optimization. BMC Cancer. 2011;11:143. doi: 10.1186/1471-2407-11-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor D.A., Smith G.D., Ebrahim S. Hyperinsulinaemia and increased risk of breast cancer: findings from the British Women's Heart and Health Study. Cancer Causes Control. 2004;15:267–275. doi: 10.1023/B:CACO.0000024225.14618.a8. [DOI] [PubMed] [Google Scholar]
- Lesicka M., Jablonska E., Wieczorek E., Seroczynska B., Siekierzycka A., Skokowski J., Kalinowski L., Wasowicz W., Reszka E. Altered circadian genes expression in breast cancer tissue according to the clinical characteristics. PLoS One. 2018;13:e0199622. doi: 10.1371/journal.pone.0199622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.H., Farkas M.E. Altered circadian rhythms and breast cancer: from the human to the molecular level. Front. Endocrinol. 2018;9:219. doi: 10.3389/fendo.2018.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe L.L., Goodwin P.J., Zinman B., McLaughlin J.R., Hux J.E. Increased prevalence of prior breast cancer in women with newly diagnosed diabetes. Breast Cancer Res. Treat. 2006;98:303–309. doi: 10.1007/s10549-006-9166-3. [DOI] [PubMed] [Google Scholar]
- Liu J., Zhou B., Yan M., Huang R., Wang Y., He Z., Yang Y., Dai C., Wang Y., Zhang F. CLOCK and BMAL1 regulate muscle insulin sensitivity via SIRT1 in male mice. Endocrinology. 2016;157:2259–2269. doi: 10.1210/en.2015-2027. [DOI] [PubMed] [Google Scholar]
- Luciano A.K., Zhou W., Santana J.M., Kyriakides C., Velazquez H., Sessa W.C. CLOCK phosphorylation by AKT regulates its nuclear accumulation and circadian gene expression in peripheral tissues. J. Biol. Chem. 2018;293:9126–9136. doi: 10.1074/jbc.RA117.000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng C.N., Saltiel A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Q., Xiao Y., Alex A., Cummins T.R., Bhatwadekar A.D. The diurnal rhythm of insulin receptor substrate-1 (IRS-1) and Kir4.1 in diabetes: implications for a clock gene Bmal1. Invest. Ophthalmol. Vis. Sci. 2019;60:1928–1936. doi: 10.1167/iovs.18-26045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheva B., Ramsey K.M., Buhr E.D., Kobayashi Y., Su H., Ko C.H., Ivanova G., Omura C., Mo S., Vitaterna M.H. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–631. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M.J., Klotz L.H., Venkateswaran V. Metformin and prostate cancer stem cells: a novel therapeutic target. Prostate Cancer Prostatic Dis. 2015;18:303–309. doi: 10.1038/pcan.2015.35. [DOI] [PubMed] [Google Scholar]
- McGinnis G.R., Tang Y., Brewer R.A., Brahma M.K., Stanley H.L., Shanmugam G., Rajasekaran N.S., Rowe G.C., Frank S.J., Wende A.R. Genetic disruption of the cardiomyocyte circadian clock differentially influences insulin-mediated processes in the heart. J. Mol. Cell. Cardiol. 2017;110:80–95. doi: 10.1016/j.yjmcc.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y., Sahar S., Astarita G., Kaluzova M., Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–657. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea-Maier R.T., Smit J.W.A., Netea M.G. Metabolic changes in tumor cells and tumor-associated macrophages: a mutual relationship. Cancer Lett. 2018;413:102–109. doi: 10.1016/j.canlet.2017.10.037. [DOI] [PubMed] [Google Scholar]
- Nishimura S., Manabe I., Nagasaki M., Eto K., Yamashita H., Ohsugi M., Otsu M., Hara K., Ueki K., Sugiura S. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- Nunez N.P., Perkins S.N., Smith N.C., Berrigan D., Berendes D.M., Varticovski L., Barrett J.C., Hursting S.D. Obesity accelerates mouse mammary tumor growth in the absence of ovarian hormones. Nutr. Cancer. 2008;60:534–541. doi: 10.1080/01635580801966195. [DOI] [PubMed] [Google Scholar]
- Pan X., Bradfield C.A., Hussain M.M. Global and hepatocyte-specific ablation of Bmal1 induces hyperlipidaemia and enhances atherosclerosis. Nat. Commun. 2016;7:13011. doi: 10.1038/ncomms13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papagiannakopoulos T., Bauer M.R., Davidson S.M., Heimann M., Subbaraj L., Bhutkar A., Bartlebaugh J., Vander Heiden M.G., Jacks T. Circadian rhythm disruption promotes lung tumorigenesis. Cell Metab. 2016;24:324–331. doi: 10.1016/j.cmet.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partch C.L., Green C.B., Takahashi J.S. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014;24:90–99. doi: 10.1016/j.tcb.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou C.M., Sorlie T., Eisen M.B., van de Rijn M., Jeffrey S.S., Rees C.A., Pollack J.R., Ross D.T., Johnsen H., Akslen L.A. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Perry R.J., Camporez J.G., Kursawe R., Titchenell P.M., Zhang D., Perry C.J., Jurczak M.J., Abudukadier A., Han M.S., Zhang X.M. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petan T., Jarc E., Jusovic M. Lipid droplets in cancer: guardians of fat in a stressful world. Molecules. 2018;23:1941. doi: 10.3390/molecules23081941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold A.S., Cohen S., Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell. 2015;32:678–692. doi: 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey K.M., Yoshino J., Brace C.S., Abrassart D., Kobayashi Y., Marcheva B., Hong H.K., Chong J.L., Buhr E.D., Lee C. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey G., Cesbron F., Rougemont J., Reinke H., Brunner M., Naef F. Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol. 2011;9:e1000595. doi: 10.1371/journal.pbio.1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahar S., Sassone-Corsi P. Metabolism and cancer: the circadian clock connection. Nat. Rev. Cancer. 2009;9:886–896. doi: 10.1038/nrc2747. [DOI] [PubMed] [Google Scholar]
- Sakamoto A., Kunou S., Shimada K., Tsunoda M., Aoki T., Iriyama C., Tomita A., Nakamura S., Hayakawa F., Kiyoi H. Pyruvate secreted from patient-derived cancer-associated fibroblasts supports survival of primary lymphoma cells. Cancer Sci. 2019;110:269–278. doi: 10.1111/cas.13873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T.K., Yamada R.G., Ukai H., Baggs J.E., Miraglia L.J., Kobayashi T.J., Welsh D.K., Kay S.A., Ueda H.R., Hogenesch J.B. Feedback repression is required for mammalian circadian clock function. Nat. Genet. 2006;38:312–319. doi: 10.1038/ng1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell J.C., Wisidagama D.R., Bensard C., Zhao H., Wei P., Tanner J., Flores A., Mohlman J., Sorensen L.K., Earl C.S. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol. 2017;19:1027–1036. doi: 10.1038/ncb3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schernhammer E.S., Laden F., Speizer F.E., Willett W.C., Hunter D.J., Kawachi I., Colditz G.A. Rotating night shifts and risk of breast cancer in women participating in the nurses' health study. J. Natl. Cancer Inst. 2001;93:1563–1568. doi: 10.1093/jnci/93.20.1563. [DOI] [PubMed] [Google Scholar]
- Schmidt M., Bohm D., von Torne C., Steiner E., Puhl A., Pilch H., Lehr H.A., Hengstler J.G., Kolbl H., Gehrmann M. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68:5405–5413. doi: 10.1158/0008-5472.CAN-07-5206. [DOI] [PubMed] [Google Scholar]
- Subik K., Lee J.F., Baxter L., Strzepek T., Costello D., Crowley P., Xing L., Hung M.C., Bonfiglio T., Hicks D.G. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer. 2010;4:35–41. [PMC free article] [PubMed] [Google Scholar]
- Tang Q., Cheng B., Xie M., Chen Y., Zhao J., Zhou X., Chen L. Circadian clock gene Bmal1 inhibits tumorigenesis and increases paclitaxel sensitivity in tongue squamous cell carcinoma. Cancer Res. 2017;77:532–544. doi: 10.1158/0008-5472.CAN-16-1322. [DOI] [PubMed] [Google Scholar]
- Toft D.J., Cryns V.L. Minireview: basal-like breast cancer: from molecular profiles to targeted therapies. Mol. Endocrinol. 2011;25:199–211. doi: 10.1210/me.2010-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto T., Kajio H., Sugiyama T. Association between hyperinsulinemia and increased risk of cancer death in nonobese and obese people: a population-based observational study. Int. J. Cancer. 2017;141:102–111. doi: 10.1002/ijc.30729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlande A., Masri S. Circadian clocks and cancer: timekeeping governs cellular metabolism. Trends Endocrinol. Metab. 2019;30:445–458. doi: 10.1016/j.tem.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Aguilar E.G., Luna J.I., Dunai C., Khuat L.T., Le C.T., Mirsoian A., Minnar C.M., Stoffel K.M., Sturgill I.R. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2019;25:141–151. doi: 10.1038/s41591-018-0221-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R., Shirai T., Namkoong H., Zhang H., Berry G.J., Wallis B.B., Schaefgen B., Harrison D.G., Tremmel J.A., Giacomini J.C. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J. Clin. Invest. 2017;127:2725–2738. doi: 10.1172/JCI92167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn L.R., Tamimi R.M., Rosner B.A., Brown S.B., Stevens R.G., Eliassen A.H., Laden F., Willett W.C., Hankinson S.E., Schernhammer E.S. Rotating night-shift work and the risk of breast cancer in the nurses' health studies. Am. J. Epidemiol. 2017;186:532–540. doi: 10.1093/aje/kwx140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellenstein M.D., de Visser K.E. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity. 2018;48:399–416. doi: 10.1016/j.immuni.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Yang X.R., Chang-Claude J., Goode E.L., Couch F.J., Nevanlinna H., Milne R.L., Gaudet M., Schmidt M.K., Broeks A., Cox A. Associations of breast cancer risk factors with tumor subtypes: a pooled analysis from the Breast Cancer Association Consortium studies. J. Natl. Cancer Inst. 2011;103:250–263. doi: 10.1093/jnci/djq526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Yang H.H., Hu Y., Watson P.H., Liu H., Geiger T.R., Anver M.R., Haines D.C., Martin P., Green J.E. Immunocompetent mouse allograft models for development of therapies to target breast cancer metastasis. Oncotarget. 2017;8:30621–30643. doi: 10.18632/oncotarget.15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Xiang Y., Ozguc F.M., Kim Y., Liu C.J., Park P.K., Hu Q., Diao L., Lou Y., Lin C. The genomic landscape and pharmacogenomic interactions of clock genes in cancer chronotherapy. Cell Syst. 2018;6:314–328.e2. doi: 10.1016/j.cels.2018.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S.H., Yamazaki S., Lowrey P.L., Shimomura K., Ko C.H., Buhr E.D., Siepka S.M., Hong H.K., Oh W.J., Yoo O.J. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl. Acad. Sci. U S A. 2004;101:5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi M., Nosrati R., Salmaninejad A., Dehghani S., Shahryari A., Saberi A. Organ-specific metastasis of breast cancer: molecular and cellular mechanisms underlying lung metastasis. Cell. Oncol. 2018;41:123–140. doi: 10.1007/s13402-018-0376-6. [DOI] [PubMed] [Google Scholar]
- Zhang D.Q., Tong X., Arthurs B., Guha A., Rui L.Y., Kamath A., Inoki K., Yin L. Liver clock protein BMAL1 promotes de novo lipogenesis through insulin-mTORC2-AKT signaling. J. Biol. Chem. 2014;289:25925–25935. doi: 10.1074/jbc.M114.567628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.