

Abstract
The outstanding therapeutic progress achieved with modern pediatric regimens in childhood acute lymphoblastic leukemia (ALL) led efforts to explore whether a similar treatment approach could be equally effective and safe in older patients, starting initially with older adolescents and young adults (AYA), variably defined in different studies by an age between 15–18 and 25–39 years. Several comparative and noncomparative trials of this type have been carried out during the last two decades, enrolling thousands of patients. Almost without exception, the new strategy improved patients’ outcomes compared with traditional adult treatments in B-lineage and T-lineage Philadelphia (Ph) chromosome-negative B-ALL, while the use of tyrosine kinase inhibitors (TKI) led to comparative progress in Ph+ ALL, a former high-risk subset more typically observed in older age groups. At present, highly effective pediatric-based regimens warrant 5-year survival rates of 60–70% in AYA patients. In view of these data, the same approach was progressively extended to older patients, improving the results up to 55 years of age. Issues of treatment compliance and drug-related toxicity have thus far prevented a comparable therapeutic advancement in patients aged >55 years. This critical review updates and summarizes with pertinent examples this global, positive therapeutic change, and examines how to promote further progress with new targeted therapies that include novel immuno-therapeutics and other agents developed against the many molecular dysfunctions detectable in various ALL subsets. Substantial progress is expected to occur soon, bringing AYA survival figures very close to that of children, and also to improve the outcome of ALL at all ages.
Keywords: adolescent and young adults, ALL, therapy
Introduction
There are few survival graphs of comparable visual impact as those presented by Pui,1 and by Hunger and Mullighan,2 in their reviews illustrating the outstanding therapeutic progress achieved in childhood acute lymphoblastic leukemia (ALL) over the past 50 years. These impressive data documented how 5-year survival rates of 2628 and 39,697 patients, registered in six consecutive trials of St. Jude’s Hospital (SJH) and the Children’s Cancer Group/Children’s Oncology Group (CCG/COG) between 1962 and 2009 were improved from less than 10% to about 90%.1,2 Since we have no evidence that patient-related or disease-related risk factors varied significantly over this timespan, we can argue that this huge prognostic improvement was due primarily to improved treatment methods, namely upfront chemotherapy as first treatment, and, to some extent, allogeneic hematopoietic cell transplantation (HCT), as well as other salvage therapies applied to selected risk subsets and to the minority of children who fail frontline therapy, respectively. As the evidence became increasingly sound and widespread, modern pediatric regimens were successfully considered for ALL patients older than 15–18 years, that is older adolescents and young adults up to the age of 40 years (AYA). These patients were previously treated in adult centers with traditional, less intensive (and less effective) adult programs, yielding survival rates around 40%. The use of pediatric-based programs brought survival of AYAs close to 70%, resulting in a growing number of clinical studies, and eventually led to critical reviews and position papers by experts in favor of this new approach.3–5 In addition, these improved programs were used in older age groups, although with less encouraging results and greater toxicity reported beyond age 45–55 years.6,7 Here, we review the evidence and the most recent data supporting this epochal change, focusing on specific treatment elements for distinct ALL and risk subsets, the main toxicity issues related to the use of pediatric-based programs and drugs in AYAs and adults with ALL, and finally discussing the ongoing treatment modifications that will likely result in further therapeutic benefit.
A concise review of evidence
An excellent review by Siegel and colleagues summarized the results of 18 comparative and 9 noncomparative trials totaling 3154 and 1700 patients with Philadelphia-negative (Ph–) ALL, respectively.3 Of the 27 studies, 21 included AYA patients with a maximum age of 40 years, while older adults were included in the remaining 6 reports. Data analysis clearly favored pediatric-type rather than adult regimens in all but two comparisons, one from Finland and another from the M.D. Anderson Cancer Center (MDACC).8 The monocentric MDACC study reported on 106 AYA patients treated with an augmented Berlin-Frankfurt-Münster (BFM) pediatric regimen, whose outcome was similar to that of 102 patients receiving MDACC’s standard Hyper-CVAD regimen [cyclophosphamide (C)-vincristine (V)-doxorubicin-dexamethasone (Dex)] alternating with [methotrexate (MTX)-high dose (HD) cytarabine], which was also patterned after an older pediatric SJH regimen by adding the VAD combination to the original backbone in 1992.9 In this study, neither complete remission (CR) nor overall survival (OS) rates were affected by regimen choice, while the most significant prognostic factor was postinduction minimal (or measurable) residual disease (MRD) response (see below). The place of the Hyper-CVAD regimen in AYAs was extensively examined in a paper by Siegel and colleagues,10 in comparison with other pediatric-type COG and Dana Farber Cancer Institute (DFCI) regimens. This survey added rather strong evidence in favor of the pediatric schedules, also because the monocentric MDACC results with Hyper-CVAD were poorly reproducible elsewhere and in multi-institutional studies. Beside clinical trials, another valuable source of information is the analysis of unselected patient populations. In the United States (US), a large population-based survey (n = 1473 cases) demonstrated a significant survival advantage for AYAs receiving pediatric-based regimens at pediatric sites or National Cancer Institute/COG-designated centers compared with similar cases treated at community adult centers with either pediatric- or adult-type programs (hazard ratio = 0.53 and 0.51 for OS and leukemia-specific survival, respectively).11 Similar results, albeit on a smaller numerical scale, were provided by a Canadian survey.12 In agreement with these observations, the importance of participation into clinical trials was further stressed by Bleyer and colleagues on reviewing the effects of trial participation for the US AYA patient population.13 This analysis showed a clear and significant correlation between the sharp decrease in trial accrual rate registered between age 16 and 24 years (the so-called AYA ‘cliff’) and the corresponding 5-year leukemia-specific survival, with a loss of about 20% compared with trial patients in this age group. Similar conclusions were independently reached by an observational study from United Kingdom (UK),14 in which Hough and colleagues documented how 2-year survival estimate of younger AYAs (age 15–24 years, n = 511) was 17.9% better for UKALL2003 trial patients compared with nontrial patients (p < 0.0001).
Evidence-based considerations
Based on the evidence examined so far, the results of early pivotal trials,15–17 and many subsequent confirmatory studies (reviewed in the literaure3–5), AYAs aged 18–40 years with Ph– ALL are optimally treated with modern pediatric-based programs rather than traditional adult protocols. However, this choice requires careful application of the treatment protocol, which is usually more intensive and potentially more toxic in some parts than the less effective, but apparently less demanding, adult protocols. The main differences are listed in Table 1. As demonstrated over time, treatment adherence and expertise of the management team plays a pivotal role, and often represents an underestimated prognostic factor.18,19 This issue can, in part, explain the intercenter variability of therapeutic results, and may be of critical importance when applying highly intensive modern pediatric regimens, with particular regard to the prevention and management of toxic complications and the delivery of chemotherapy without undue dose reductions and delay. This concept was recently highlighted in a large US analysis,11 although it may not concern single centers of excellence for acute leukemia therapy such as the MDACC, contributing to explain the superior monocentric results obtained with standard adult therapy.20 Whatever the treatment setting, the issue of AYA therapy is further compounded by host- and disease-related characteristics, globally worse in AYAs compared with children. This implies the adoption of a different risk stratification system, and, according to protocol design, a more frequent therapy intensification with allogeneic HCT in CR patients expressing risk factors associated with high risk (HR) of relapse following intensive pediatric-type chemotherapy.
Table 1.
Main differences between pediatric-based and adult-type programs for Ph– ALL in AYA patients.
| Treatment phase | Characteristics of pediatric-based therapy (versus adult standard therapy) | Annotations |
|---|---|---|
| Chemotherapy (induction, consolidation, maintenance) | Corticosteroids: higher cumulative dose | Dexamethasone preferred (higher activity); Higher penetration into CNS; Toxicity: osteonecrosis (age-related), other [metabolism, hypertension, peptic ulcer, infections (fungal)] |
| Vincristine: higher injection no. and cumulative dose | Risk of neuropathy (higher doses) | |
| Asparaginase/Peg-ASP: higher cumulative dose | Peg-ASP recommended/preferred (minimum 4
injections); Careful association with other potentially hepatotoxic drugs; Toxicity (risk factors: age >45, liver steatosis, BMI >30): hepatic, metabolic, pancreatic coagulation/thrombosis, allergy |
|
| Antimetabolites: more intensive use and higher cumulative dose of MTX, 6-thiopurines, cytarabine | Higher MTX dose recommended/preferred (>1.5 g/m2, up to 3–5 g/m2) | |
| Anthracyclines: less intensive use | Lower risk of myelotoxicity and cardiomyopathy | |
| CNS prophylaxis | IT chemotherapy: intensified, higher injection no. | Single agent IT MTX, cytarabine or triple IT combination (MTX, cytarabine, corticosteroids) |
| Cranial prophylaxis: omitted or in high-risk subsets only | Higher activity of systemic CNS-active therapy and IT
prophylaxis; Better treatment compliance, lower risk of short- and long-term brain damage; Radiation-related risk of secondary brain neoplasms |
|
| Treatment intensity/adherence | Aim: higher overall intensity without undue dose reductions and treatment delay | Dedicated, well-trained staff (medical and
nonmedical); Compliance to intensive chemotherapy |
| Allogeneic HCT | First CR: according to MRD/risk-based strategy | More frequently used in AYA/adults (>15–18 years) compared with children |
| Salvage: standard procedure in second/later CR | – |
ALL, acute lymphoblastic leukemia; AYA, adolescents and young adults; BMI, body mass index; CNS, central nervous system; CR, complete remission; HCT, hematopoietic cell transplantation; IT, intrathecal; MRD, minimal residual disease; MTX, methotrexate; Peg-ASP, pegylated asparaginase; Ph–, Philadelphia chromosome-negative B-ALL.
Risk stratification for risk-oriented therapy
The HR AYA group consists of the few patients who are refractory to induction chemotherapy and of those who exhibit risk factors predictive of treatment failure, which is relapse subsequent to the achievement of CR. The patients with no obvious risk factors are defined standard risk (SR) and are usually excluded from treatment intensification with allogeneic HCT in first CR. This latter procedure is reserved to cases belonging to HR group, according to the risk definition adopted by each treatment protocol and study group (see below, Table 2),21 to overcome the high likelihood of relapse associated with chemotherapy alone. However, depending on exact protocol design, selected AYA patients with intermediate/HR features can be treated with intensive chemotherapy plus maintenance regimens without allogeneic HCT in first CR. As reported in Table 2, the criteria identifying HR and SR ALL vary slightly among clinical protocols, often combining patient’s age (acting as continuous rather than dichotomous prognostic factor), presenting white blood cell (WBC) count, disease genetics/cytogenetics,22,23 and, above all, status of MRD after induction and early consolidation steps (Figure 1).
Table 2.
Risk stratification criteria adopted for allogeneic HCT in European trials for CR1 patients with Ph– ALL in an age range between 15/25 and 55/65 years (all studies including AYAs).
| National study group (European survey) | Risk stratification criteria for
allogeneic HCT in CR1 (Ph– ALL) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Age | WBC | Phenotype | Cytogenetics | Genetics | MRD | BM blasts | Late CR | Other | |
| RALL (Russia) | >30 | t(4;11), t(1;19) | KMT2Ar | POS | |||||
| GMALL (Germany) | >30 (B) | Pro-B, early/mature-T | KMT2Ar | POS | yes | ||||
| HOVON (Netherlands) | >30 (B) >100 (T) | Adverse | POS | yes | |||||
| PALG (Poland) | >30 (B) >100 (T) | KMT2Ar | POS | CNS+ | |||||
| FALL (Finland) | >100 | abn11q23, hypodiploid | POS | D15 >25% | yes | ||||
| GIMEMA (Italy) | >100 | early/mature-T | adverse | KMT2Ar | POS | ||||
| UKALL (UK) | High count | adverse | adverse | POS | |||||
| SVALL (Sweden) | hypodiploid | KMT2Ar | POS | EOI >5% | |||||
| CELL (Czechia) | POS | ||||||||
| PETHEMA (Spain) | POS | ||||||||
| GRAALL (France) | POS | ||||||||
Modified after Giebel and colleagues.21
ALL, acute lymphoblastic leukemia; AYA, adolescents and young adults; BM, bone marrow; CNS, central nervous system; D, day; EOI, end of induction; HCT, hematopoietic cell transplantation; KMT2Ar, KMT2A-rearranged; Ph–, Philadelphia chromosome-negative B-ALL; POS, positive; WBC, white blood cell count (×109/l).
Figure 1.
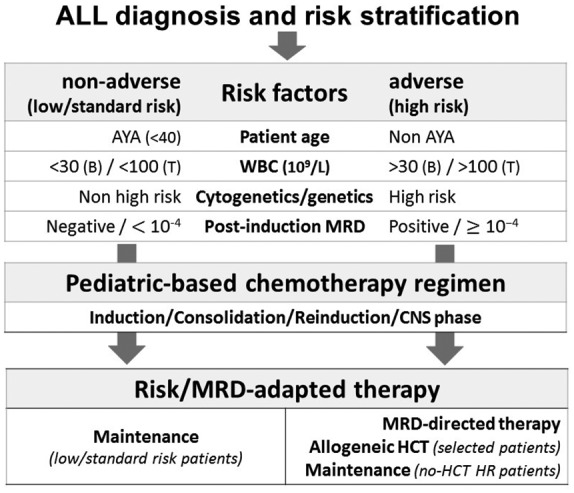
Prognosis to treatment relationships in AYA Ph– ALL. Different patient characteristics and clinicobiologic ALL subsets concur to determine the individual risk profile. Postinduction MRD analysis reflects patterns of chemosensitivity and refines the prognostic index, which is used to orientate postremission therapy reserving an allogeneic HCT to HR patients (see also Table 2 and Figure 2). MRD itself can be targeted with novel immunotherapeutics and other experimental agents (i.e. blinatumomab in CD19+ ALL, CAR-T cells, etc.). Regardless of risk class definition and transplantation policy (a decision related to specific protocol design), overall patient outcome is improved using pediatric-based rather than traditional adult chemotherapy protocols.
ALL, acute lymphoblastic leukemia; AYA, adolescents and young adults; HCT, hematopoietic cell transplantation; HR, high risk; MRD, minimal (or measurable) residual disease; Ph–, Philadelphia chromosome-negative B-ALL.
ALL cytogenetics and genetics
Patterns of ALL cytogenetics and genetics in AYAs have been reported in large cohorts of 7113 patients with B-precursor ALL,24 5202 unselected ALL patients,23 and 542 adult patients with Ph– ALL.25 Both studies assessing the frequency of Ph+ ALL demonstrated an almost linear correlation with age, with an incidence <10% at age 15–19 years that increased to 15–20% and 25–35% at 18–25 years and 25–40 years, respectively.23,24 With regard to another very HR subset, t(4;11)+/KMT2A-rearranged ALL, incidence was 11.8% at age 15–24 years and 19.8% at 25–44 years.25 Other HR abnormalities reported in AYAs were iAMP21 (relatively rare), 14q32/IGH rearrangements (3–5%), low hypodiploidy/near triplody (altogether 10% or less), and complex or monosomal karyotypes (5% and 8–10%). Conversely, the frequency of the favorable prognosis t(12;21)/ETV6-RUNX1+ ALL and high hyperdiploid ALL in AYAs decreased from about 35–40% each in children to <10% in teens (<5% at 20+ years) and to 20–25% (10–15% >25 years), respectively. The remainder of cases within the B-precursor subset constituted an intermediate risk category, which included t(1;19)/TCF3-PBX1+ ALL and all other cases (‘B-other’) that express secondary lesions known as copy alterations (CNA), such as deletions of IKZF1 and other lymphoid development genes, and a relatively frequent overexpression of CLRF2. In children and young adults (age <25 years), different CNA profiles were confirmed to exert a significant effect on the risk of relapse.26 The newly recognized HR entity known as Ph–like (or BCR-ABL1-like) ALL falls into the ‘B-other’ group, and can be identified through molecular screening. The estimated incidence of Ph–like ALL in AYAs aged 16–39 years with B-precursor ALL is 19–27%, a figure higher than that reported in younger and older patient groups, respectively.27
Postinduction MRD analysis
The MRD assay is of extreme prognostic relevance, being the sole or most significant risk factor for relapse confirmed in many studies by multivariable prognostic analysis.28–31 This is not entirely unexpected because MRD represents the disease itself and reflects disease sensitivity to early chemotherapy. Altogether, despite a partial lack of predictive power (some MRD-negative patients relapse), MRD represents the major prognostic information and decisional support for the allocation to allogeneic HCT or other targeted therapy, a view uniformly endorsed by both European and US experts.32,33 In a recent European survey, MRD positivity was the only risk factor supporting the decision to allograft shared by 11 national study Groups on adult ALL (including AYAs: lower age limit 15–18 years) (Table 2).21 In prospective therapy-oriented MRD studies that employed sensitive molecular markers for MRD detection, 37–48% of CR patients tested MRD positive at weeks 4–6 after an induction course, (any positivity or ⩾10−4), a proportion decreasing to 16–30% at weeks 10–12 after early consolidation.34–36 Corresponding MRD positivity rates in AYAs only are not known precisely. An operational limit of an MRD-based risk stratification is the lack of MRD data due to either a defective ALL cell collection for molecular probe generation, the lack of a specific and sensitive (⩾10−4) molecular probe, or an insufficient marrow sampling at MRD time-points critical for treatment decisions. While this issue may be of lesser concern using multiparameter flow cytometry assays, in molecular MRD-based trials, it caused the exclusion from study and optimal risk stratification of a high proportion of patients, between 23% and 31%.36,37
Combined genetic and MRD risk stratification
The prognostic significance of MRD was recently shown to vary in relation to the genetic/oncogenetic risk subset, in both adults35,38 and children.39,40 These studies documented that risk of relapse related to persistent or recurrent MRD varied significantly as a function of associated genetic/oncogenetic abnormalities. The first evidence in adult/AYA patients came from a Group for Research on Adult ALL (GRAALL) study,35 documenting the MRD-independent prognostic effect of a four-gene adverse classifier (B-precursor ALL: KMT2A rearrangements or IKZF1 deletion; T-ALL: unmutated NOTCH1/FWBX7 or abnormal RAS/PTEN expression). The recent large UKALL14 trial, enrolling patients with Ph– ALL aged 25–65 years, validated a robust prognostic index (PIUKALL) integrating WBC count, genetic risk class, and postinduction MRD. This allowed to predefine different PIUKALL groups with highly variable response to the planned risk-oriented treatment, ranging from an excellent relapse-free survival (RFS) of 90% on chemotherapy only to a relapse risk after myeloablative HCT as high as 42%.38 Moreover, as demonstrated in childhood ALL, current genetic/cytogenetic risk classifications can be further improved through the analysis of associated CNA profiles in patients who do not express a clear genetic risk marker, particularly in the ‘B-other’ intermediate risk group.26 A new combined risk model based on postinduction MRD and disease genetics incorporating a detailed CNA expression analysis, validated in a large UKALL pediatric cohort,26 is being prospectively assessed in the ALLTogether Consortium project, a very large International collaborative effort among several ALL study Groups, in which children and AYAs with Ph– ALL aged up to 25 years (UKALL) or 45 years Nordic Society of Pediatric Hematology and Oncology (NOPHO) are risk-stratified in this fashion for risk-adapted therapy.41 These emerging concepts need to be considered and will likely affect the risk-oriented design of new AYA and adult trials (Figure 2).
Figure 2.

Recent example of combined risk stratification by genetics and MRD in Ph– ALL (patterned after UKALL study25 and as adopted in ALLTogether study.41 The present risk classification is used within the international ALLTogether project by the NOPHO group for AYAs aged up to 45 years. In this study T-ALL is considered a uniform genetic risk group. In B-lineage ALL (B-ALL), genetics/cytogenetics defines good, intermediate risk and HR groups (with the notable absence of Ph–like ALL; see text for details), and the intermediate risk class is subdivided according to CNA (involved genes are indicated). By study design, final risk classification allows patients to be allocated to intensive chemotherapy and maintenance (standard/low and low/intermediate risk group, experimental interventions with new agents (HR/intermediate risk group) and allogeneic HCT or chimeric antigen receptor T cell (HR group).
ALL, acute lymphoblastic leukemia; AYA, adolescents and young adults; CNA, copy number alterations; HCT, hematopoietic cell transplantation; HR, high risk; MRD, minimal (or measurable) residual disease; Ph–, Philadelphia chromosome-negative B-ALL.
aHR genetics: t(4;11)/KMT2A rearrangements; near haploidy/low hypodiploidy, iAMP21, rearrangements affecting ABL1, ABL2, PDGFRB, and CSFR1 (except BCR-ABL1).
bGood CNA profile: no deletion IKZF1, CDKN2A/B, PAR1, BTG1, EBF1, PAX5, ETV6, RB1; isolated deletion ETV6, PAX5, BTG1; ETV6 deletion with single deletion BTG1, PAX5, CDNK2A/B.
Practicalities of treatment regimens for Ph– ALL in AYA patients
Although current pediatric-based regimens are preferable to standard adult programs, useful guidance for AYA treatment must consider some relevant examples from prospective clinical trials. Because patient age maintains a primary prognostic role, the current selection of recent study results in Ph– ALL (Tables 3 and 4) separates the typical AYA patient population (age ⩽40 years) from older patients treated in the same way or with similar age-adapted regimens, up to an age of 55–65 years.6–8,36,42–52 This analysis aims to address the following questions: is there a better regimen or drug combination? Which are the essentials of a pediatric-based regimen? Is there an upper age limit for a safe and effective use of this treatment scheme? How can we prevent and manage main drug-related toxicities? What is the perception and applicability of a risk-/MRD-based post-induction strategy in AYAs? Finally, which new therapeutic elements and strategies will allow further progress?
Table 3.
Results from recent, representative trials for Ph– ALL in AYA and adult patients (pediatric-based chemotherapy, risk/MRD-oriented consolidation and allogeneic HCT, >100 patients, outcome estimates a 3+ years). Trial order according to increasing patient age (median and range; upper age limit in each trial is indicated).
| Trial | No. | Age (years) | CR (%) | CRD/DFS (%) | OS (%) | EFS (%) | FUP | Annotations |
|---|---|---|---|---|---|---|---|---|
| Maximum patient age <25 years | ||||||||
| JALSG 202-U42 | 139 | 19 (16–24) | 97 | 71 | 74 | – | 4-year | Allo-HCT in t(4;11)+ |
| UKALL 200343 | 229 | 16–24 | 97 | – | 76.4 | 72.3 | 5-year | CR rate calculated upon induction failures (2.6%) |
| Maximum patient age <40 years | ||||||||
| GMALL 05/93, 07/0344 |
642 887 |
15–35 | 88 91 |
49 61 |
46 65 |
– | 5-year | 07/03: intensified Peg-ASP, Dex and HD
consolidation; allo-HCT in HR or MRD+; p < 0.05 for CRD and OS |
| MDACC augmented BFM8 |
106 | 22 (13–39) | 93 | 60 | 53 | – | 5-year | Allo-HCT in t(4;11)+ or MRD+; CRD/OS comparable with Hyper-CVAD |
| U.S. Intergroup C1040345 |
295 | 24 (17–39) | 89 | 66 | 73 | 59 | 3-year | – |
| Maximum patient age 45–65 years (all studies including AYAs) | – | |||||||
| NOPHO ALL200846 | 221 | 26 (18–45) | – | – | 78 | 74 | 5-year | Allo-HCT if day 29 MRD > 5% or day 79 MRD ⩾ 0.1% |
| DFCI 01-175647
DFCI 06-25448 |
92 110 |
28 (18–50) 32 (18–50) |
86 89 |
71 73 |
70 75 |
– | 4-year 3-year |
Allo-HCT in t(4;11)+, +8, Ph+; Intensified Peg-ASP consolidation (toxicity: reduced from 2500 to 2000 IU/m2 and from 16 to 10 doses) |
| GMALL 07/0349 | 1226 | 35 (15–55) | 91 | – | 60–67 | – | 3-year | Allo-HCT in HR or MRD+, intensified Peg-ASP (1000 versus 2000 IU/m2 in cohort 1 versus cohort 2), x7 in SR; Dex and HD consolidation |
| RALL 200950 | 250 | 30 (15–60) | 87 | 69.3 | 65.6 | – | 4-year | Allo-HCT in HR |
| GRAALL 20036
GRAALL 20057 |
225 787 |
31 (15–60) 36 (18–60) |
93.5 92 |
– – |
60 58.5 |
55 52 |
3.5-year 5-year |
2003: Allo-HCT in t(4;11)+, HR, MRD > 10−2, age
⩽55 years 2005: Allo-HCT in HR; phase III trial (hyper- versus standard Cy induction [comparable results except for patient >55 years (hyper-Cy favourable)] |
| PETHEMA HR-1151 | 126 | 38 (max. 60) | 86 | 40 (l-Asp) 58 (PEG-Asp) |
57 (l-Asp) 60 (PEG-Asp) |
– | 3-year | HR only, for allo-HCT if MRD+; comparable MRD response l-ASP versus Peg-ASP |
| JALSG ALL 202-O52 | 344 | 24–65 | 86 | 42 | – | 52 | 5-year | Phase III trial (MTX 0.5 versus 3 g/m2: DFS 32% versus 58%; p = 0.0218) |
| NILG 10/0736 | 163 | 41 (17–67) | 87 | 52 | 55 | – | 5-year | Allo-HCT in MRD+ or very HR; MRD highly predictive of outcome |
ALL, acute lymphoblastic leukemia; allo-HCT, allogeneic hematopoietic cell transplantation; AYA, adolescents and young adults; CR, complete remission; CRD, CR duration; Cy, cyclophosphamide; Dex, dexamethazone; DFS, disease-free survival; DFCI, Dana Farber Cancer Institute; EFS, event-free survival; FUP, follow up; GRAALL, Group for Research on Adult ALL; GroupGMALL, German Multicenter Group for Adult ALL; HD, high dose; HD dose consolidation (with MTX and cytarabine in GMALL trial); HR, high risk; JALSG, Japan Adult Leukemia Study Group; l-/Peg-ASP, l-asparaginase/pegylated asparaginase; MDACC, M.D. Anderson Cancer Center; MRD, minimal residual disease; MTX, methotrexate; NILG, Northern Italy Leukemia Group; NOPHO, Nordic Society of Pediatric Haematology and Oncology; OS, overall survival; PETHEMA, Programa Español de Tratamientos en Hematologia; Ph–, Philadelphia chromosome-negative B-ALL; RALL, Russian ALL Group; UKALL, United Kingdom ALL Study.
Table 4.
Results of prognostic analysis from recent, representative trials for Ph– ALL in AYA and adult patients (pediatric-based chemotherapy, MRD/risk-oriented consolidation and allogeneic HCT, >100 patients). Trial order according to increasing patient age. Selection of studies presented in Table 3, reporting detailed prognostic factor analysis.
| Trial | No. of patients | Age (years), median (range) | CR (%) | Outcomes according to risk factors (patient age, MRD, ALL subset/genetics, other) |
|---|---|---|---|---|
| UKALL 200343 | 229 | 16–24 | 97 | 5-year EFS: correlation with MRD risk class (p = 0.0001) |
| MDACC augmented BFM8 | 106 | 22 (13–39) | 93 | 5-year OS: day 29 MRD- 75% versus MRD+ 40%
(p = 0.004) 5-year CRD: day 29 MRD- 64% versus MRD+ 33% (p = 0.017) 5-year OS: day 84 MRD- 75% versus MRD+ 22% (p = 0.0004) 5-year CRD: day 84 MRD- 63% versus MRD+ 26% (p = 0.0018) |
| U.S. Intergroup C1040345 | 295 | 24 (17–39) | 89 | 3-year EFS: Ph–like 42% versus non-Ph–like 69%
(p = 0.008) 3-year OS: Ph–like 63% versus Non-Ph–like 81% (p = 0.0371) 3-year DFS: end of induction MRD- 85% versus MRD+ 54% (p = 0.001) |
| NOPHO ALL200846 | 221 | 26 (18–45) | – | 5-year EFS: SR 87%, IR 78%, HR 66%, HR to allo-HCT (including MRD+) 61% |
| DFCI 01-17547 | 92 | 28 (18–50) | 85 | 4-year DFS: T 87%, B Ph– 66%
(p = 0.14) 4-year EFS: T 77%, B Ph– 57% (p = 0.11) 4-year OS: T 76%, B Ph– 68% (p = 0.12) |
| DFCI 06-25448 | 110 | 32 (18–50) | 89 | 3-year OS: age 18–19 years 100% versus
20–29 years 85% versus 30–39 years 75%
versus 40–50 years 60% 3-year OS: T 78% versus B 81% 3-year OS: BMI underweight/normal 85% versus overweight 71% versus obese/morbidly obese 63% |
| GMALL 07/0349 | 1226 | 35 (15–55) | 91 | 3-year CRD: SR cohort 1 61% versus cohort 2 74%
(p = 0.02); AYA cohort 1 60%
versus cohort 2 78% 3-year OS: cohort 1 60% versus cohort 2 67%; SR cohort 1 68% versus cohort 2 80% (p = 0.02); AYA cohort 1 77% versus cohort 2 86% |
| RAALL 200950 | 250 | 30 (15–60) | 87 | 4-year DFS: age <30 years 71.5% versus
⩾30 years 61.2% (p = 0.1) 4-year OS: age <30 years 73.6% versus ⩾30 years 52.7% (p = 0.0009) |
| GRAALL 20036 | 225 | 31 (15–60) | 93.5 | 3.5-year CRD: age 15–45 years 61% versus
>45 years 53% (p = 0.21) 3.5-year OS: age 15–45 years 64% vs > 45 years 47% (p = 0.004) |
| GRAALL 20057 | 787 | 36 (18–60) | 92 | 5-year EFS: age ⩾55 years 25.8% versus <55 years 55.7% (p < 0.001); age 35–54 years 52.2% versus 18–34 years 58.7% (p = 0.019) |
| JALSG ALL 202-O52 | 344 | 24–65 | 86 | 5-year DFS: SR <40 years 71% versus SR >40 years 52% versus HR 27% (p = 0.001) |
| NILG 10/0736 | 163 | 41 (17–67) | 87 | 5-year DFS: week 4 MRD- 67% versus MRD+ 41%
(p = 0.041) 5-year DFS: week 10 MRD- 64% versus MRD+ 23% (p = 0.0001) 5-year DFS: B 48% versus T 61% 5-year OS: B 48% versus T 74% |
ALL, acute lymphoblastic leukemia; allo-HCT; allogeneic hematopoietic cell transplantation; AYA, adolescent and young adult patients; B, B-ALL; BMI, body mass index; CR, complete remission; CRD, CR duration; DFCI, Dana Farber Cancer Institute; DFS, disease-free survival; EFS, event-free survival; GMALL, German Multicenter Group for Adult ALL; GRAALL, Group for Research on Adult ALL; HCT, hematopoietic cell transplantation; HR, high-risk; IR, intermediate risk; JALSG, Japan Adult Leukemia Study Group; MDACC, M.D. Anderson Cancer Center; MRD, minimal residual disease; NILG, Northern Italy Leukemia Group; NOPHO, Nordic Society of Pediatric Haematology and Oncology; OS, overall survival; Ph, Philadelphia chromosome; RAALL, Russian Adult ALL Group; SR, standard risk; T, T-ALL; UKALL, United Kingdom ALL Study Group.
CR induction results
The examples of pediatric-based or fully pediatric therapy reported in Table 3 documented CR rates close to 100% in patients younger than 25 years, decreasing to about 90% in patients aged up to 40 years and to 85–90% in those aged up to 55–65 years. Some of these induction schedules have been already modified to include immunotherapy with anti-CD20 antibody rituximab for CD20+ ALL. This was the case with recent MDACC, German Multicenter ALL study Group (GMALL) and GRAALL trials,49,53–55 and must be taken into account when discussing improved treatment results. Details of induction failures were not always available, though, in general, incidence of both induction resistance and death were distributed equally and correlated with an increasing age. While it does not seem possible to claim the superiority of any induction schedule over another, some studies reported very low resistance rates after two or more induction courses, as in the NOPHO46 and GRAALL trials, this latter employing a HD cytarabine plus idarubicin combination in course-1-resistant patients.6,7
Survival results
Long-term outcome measures indicated (not in all studies) 5-year OS rates slightly above 60–70% in AYAs aged up to 35–40 years. The MDACC study using the BFM regimen gave a slightly inferior result (OS 53%), which was therefore superimposable to the Hyper-CVAD group.8 Disease-free survival (DFS) and event-free survival (EFS) data were close to OS ranges. The US Intergroup study adopted a reference COG regimen previously tested in patients aged 1–30 years, confirming its feasibility in patients aged 17–39 years, with good EFS and OS results at 3 years, and a significant improvement in prognosis over an historical data set.45
Outcomes by patient age and other prognostic factors
Results from each study were comparatively better in younger patients and in patients with more favorable risk profile, such as MRD-negative post-induction response (Table 4). In studies including older patients, up to 60 or 65 years (median patient age between 30 and 41 years), the general results were improved compared with historical data, but were not as good as in AYA studies. OS, DFS and EFS rates were between 55% and 60% (GRAALL; Programa Español de Tratamientos en Hematología, PETHEMA), with a significant reduction in therapeutic benefit above 45–55 years of age [GRAALL, Northern Italy Leukemia Group (NILG) and Russian ALL study Group (RALL)].6,7,36,50 In the large GRAALL experience, the application of the pediatric French ALL programs was more difficult in patients aged >45 and >55 years, causing significantly more induction and consolidation deaths than in younger patients receiving the same therapy.6,7 Among the most notable examples given the large patient number, the more favorable age range (up to 45 and 55 years, respectively) and length of follow up (5-year outcomes available) are the NOPHO and the GMALL data, with an EFS of 73% (87% in SR patients) in the NOPHO trial,46 and OS and CR duration rates of 84% and 74% for SR patients treated in the intensive Pegylated-Asparaginase (Peg-ASP) cohort in the GMALL trial,49 respectively. Outcome was improved in T-ALL patients in some studies (DFCI, NILG, RALL).36,47,48,50
Improved drug regimens
The general lay-out of modern pediatric-type regimens for AYAs consists of a four or five-drug CR induction phase [V-corticosteroids (CS)-anthracycline-l-asparaginase/Peg-ASP, with or without fractionated C], along with an early intrathecal (IT) prophylaxis. Fractionated C in induction or preinduction is frequently used but was not found advantageous in a randomized GRAALL trial.7 Patients achieving CR receive a complex postremission sequence with six to eight rotational multi-agent chemotherapy cycles, variously denominated (intensification, consolidation, cytoreduction), comprising systemic MTX and HD cytarabine courses, also useful to optimize the central nervous system (CNS) prophylaxis together with further IT injections, more Peg-ASP, and a reinduction course (or delayed intensification), which was demonstrated highly effective in prior BFM studies.56 The total duration of intensive therapy may exceed 6 months and approach 1 year, followed by long-term low-dose maintenance for 2–3 years. Some typical components of pediatric protocols deserve special attention in view of their characteristics and related toxicity issues (Peg-ASP, MTX, and CS).
Peg-ASP in pediatric-based regimens
As shown above, treatment results were significantly improved in GMALL trials in SR patients treated with a Peg-ASP-containing protocol,44,49 as well as in DFCI studies,47,48 reporting OS rates of greater than 70% with programs based again on an intensive use of l-asparaginase or Peg-ASP,48 and in UKALL,43 NOPHO46 and US Intergroup45 trials. Peg-ASP is a unique anti-ALL drug that hydrolyzes serum asparagine which is essential to ALL cells for protein synthesis and proliferation.57 This drug is a core component of current pediatric regimens because it provides longer and better asparagine depletion than the native compound from Escherichia coli. A single Peg-ASP injection at 2000–2500 IU/m2 can warrant an effective serum activity (⩾0.1 IU/ml) for ⩾14 days and up to 30+ days).57,58 A DFCI study reported excellent results with Peg-ASP monotherapy consolidation (2500 IU/m2 q14d × 30 weeks initially, reduced to 2000 IU/m2 q21d over the same time period because of toxicity).48 To exert sufficient therapeutic activity the number of Peg-ASP doses should be equal to or greater than four.58
Peg-ASP related toxicity: prevention and management
Despite its central role in the management of ALL in AYAs and more in general in adult ALL, Peg-ASP can cause severe toxicity in the form of allergic reactions (less frequently than with the native form), coagulopathy (antithrombin III or fibrinogen deficiency), thrombosis, hyperglycemia, hypertriglyceridemia, pancreatitis, and severe liver toxicity, this latter more frequent and more severe in adults and obese patients than in children.47–49,58–60 Therefore, drug toxicity should be carefully monitored, while to avoid excess toxicity drug schedule and dosing should take into account patient’s age (higher risk >45–55 years), body mass index (BMI, higher toxicity with BMI >30), and liver steatosis (higher risk of hepatotoxicity if detected on ultrasound scan).49 Coagulopathy and thrombosis can be prevented by the periodic infusion of antithrombin III and fibrinogen concentrates as needed, and by subcutaneous low molecular weight heparin, at least until the platelet count remains >30 × 109/L. This kind of antithrombotic prophylaxis is recommended in some treatment protocols and is likely to represent a sensible choice (without contraindications) in intensive Peg-ASP regimens, though no general consensus exists as yet. The use of l-carnitine and vitamin B was occasionally found to ameliorate severe liver injury by Peg-ASP with direct bilirubin >3 mg/day/l,61,62 and may be considered along with other established measures to prevent or reduce serious adverse events by Peg-ASP.63 The recommended pediatric dosage of Peg-ASP is 2500 IU/m2. This can be difficult to maintain in older AYAs and adults, requiring a reduction to 1500–2000 IU/m2 or less when risk factors for drug-related toxicity are detected at baseline, especially obesity and liver steatosis, or when severe toxicity develops despite initial dose reduction. The most recent Gruppo Italiano Malattie Ematologiche dell’Adulto (GIMEMA) trial (ClinicalTrials.gov identifier: NCT03367299), following a prior experience with a Peg-ASP-containing program for adult patients with Ph– ALL in the 18–65 age range,64 provided empirical guidelines to Peg-ASP dosing, combining baseline risk factors (age, BMI, hepatosteatosis) with organ-specific grade 3–4 drug toxicity during first or prior Peg-ASP exposure (Table 5). With or without Peg-ASP dosage reduction guided by risk factors or toxicity, the assessment of serum drug activity may be informative about silent drug inactivation, which is, however, less common than in pediatric ALL and using the pegylated product. In this case a shift to Erwinia asparaginase is known to provide therapeutic drug levels. In the GMALL study, the outcome of AYA/adult patients who receive full dose Peg-ASP was improved,44,49 but many of those who had age-/risk-adapted Peg-ASP at 1000 IU/m2 exhibited sustained drug levels ⩾0.1 IU/ml for 14 days (77% in induction and 96% in consolidation); even with 500 IU/m2, the therapeutic drug level was maintained for 7 days in 86% and 92% of the patients during induction and consolidation, respectively (lasting for 14 days in 25% and 59%, respectively).65 Therefore, even a lower drug concentration can exert some therapeutic benefit in patients at risk of excess toxicity. Another pediatric study introducing a more sensitive laboratory method could confirm a complete serum asparagine depletion obtained with a drug activity >0.02 IU/ml,66 sensibly lower than the standard 0.1 IU/ml threshold. This study could sustain the use of a lower and safer Peg-ASP dosing in AYA/adult ALL.
Table 5.
Operative algorithm based on patient age, BMI, and toxicities related to prior drug exposure used for the administration of Peg-ASP during chemotherapy courses no. 1, 2, 5, and 6 in GIMEMA trial LAL 2317 for adult Ph– ALL (age range 18–65 years; ClinicalTrials.gov identifier: NCT03367299). G denotes grade of toxicity according to the Common Toxicity Criteria scale.
| Age group (years) | Cycle no. | Risk factorsa | Peg-ASP-related G3-4 toxicity observed at prior cycle/exposureb,c | Peg-ASP IU/m2 (max cumulative) |
|---|---|---|---|---|
| ⩽55 | 1 | No | – | 1500 (3000) |
| Yes | – | 1000 (2000) | ||
| 2, 5, 6 | No | No | 2000 (3750) | |
| Yes | 1000 (2000) | |||
| Yes | No | 1500 (3000) | ||
| Yes | 500 (1000) | |||
| >55 | 1 | No | – | 1000 (2000) |
| Yes | – | 500 (1000) | ||
| 2, 5, 6 | No | No | 1000 (2000) | |
| Yes | 500 (1000) | |||
| Yes | No | 1000 (2000) | ||
| Yes | No Peg-ASPd |
Pretreatment risk factors for Peg-ASP-related toxicity: hepatosteatosis (ultrasound scan), BMI >30.
Hepatic, pancreatic, coagulation/thrombosis.
Must be reduced to less than G2 before next Peg-ASP dosing; G4 pancreatitis causes permanent Peg-ASP discontinuation.
In subsequent cycle Peg-ASP will be restarted at a dose of 500 IU/m2; if G3-4 toxicity occurs again, PEG-ASP will be permanently discontinued.
ALL, acute lymphoblastic leukemia; BMI, body mass index; Peg-ASP, pegylated asparaginase; Ph–, Philadelphia chromosome-negative B-ALL.
Methotrexate in pediatric-based regimens
The antimetabolite MTX is another essential drug of ALL therapy that is usually administered as HD infusion, typically at intermediate dosage of 1–1.5 g/m2 over 24 h (followed by folinic acid rescue), either alone or together with either cytarabine at 1–3 g/m2 or Peg-ASP, for three to six blocks or more. Higher MTX doses between 3 and 5 g/m2 have been used in HR patients and T-ALL (Table 4), and may have contributed to above average results in some series.36 A randomized trial demonstrated an improved outcome for patients treated with MTX 3 versus 0.5 g/m2; however, the lower dose is nonstandard for adult ALL.52 The US Intergroup trial in AYAs used a lower MTX dose (100 mg/m2) with weekly dose adaptions (Capizzi style).45 This approach was previously tested in two phase III COG trials including AYAs (patient age 1–30), proving superior to HD MTX 5 g/m2 in T-ALL, while, on the contrary, HD MTX was better than Capizzi MTX in B-ALL.67,68 Of note, patients in the Capizzi MTX arm received two more Peg-ASP doses compared with the HD MTX arm. HD MTX 5 g/m2 was used for the first time in adult T-ALL in a NILG trial,69 with very good results and low toxicity score. Altogether, the use of MTX at doses higher than 1.5 g/m2 may be preferable in B-ALL, while the place of the lower Capizzi MTX schedule should be further investigated in T-ALL.
Corticosteroids in pediatric-based regimens
CS represent another highly effective ALL drug class, administered during prephase (when they allow to classify patients according to their prednisone sensitivity, in either ‘good or poor prednisone responder’ patients), induction chemotherapy, and, most of the times in a pulsed fashion, during consolidation courses. Apart from a strong antileukemic activity, CS may exert considerable short- and long-term toxicity (metabolism and diabetes, fluid retention/hypertension, gastritis/peptic ulcer, insomnia, osteoporosis/osteonecrosis) as well as mask the clinical signs of early infectious complications during induction chemotherapy and favor the spread invasive fungal infections. Among the different compounds available, Dex seems more active as an antileukemic agent than prednisone/prednisolone, at both the systemic and CNS levels, as indicated by the results of a large European pediatric randomized trial.70 In this study, Dex-treated patients had significantly less relapses, particularly in extramedullary sites, in CNS (p < 0.0001), and in the cohort of T-ALL, but suffered from higher incidence of induction death (2.5% versus 0.9%, p = 0.00013) often ascribable to infections, with higher incidence of fungal infections. Selecting a more appropriate Dex schedule is therefore necessary, along with the administration of an effective antimicrobial and antifungal prophylaxis. A GMALL induction study on 843 adult patients reported a lower early infectious rate, from 30% and 33% to 14% (p < 0.0001), with an associated early death rate varying from 16% to 8% and 5%, respectively, lowering Dex from 40 mg/m2 days 1–3 and 10 mg/m2 days 4–17 (cumulative dose 260 mg/m2) to 10 mg/m2 days 1–6 and 11–16 (cumulative dose 120 mg/m2) and 10 mg/m2 days 1–5 and 11–14 (cumulative dose 90 mg/m2), respectively, together with the addition of granulocyte colony-stimulating factor from as early as day 4 of the intensive induction schedule to shorten the duration of absolute severe granulocytopenia.71
Risk- and MRD-oriented therapy
Many of the studies detailed in Tables 3 and 4 and others had a risk-oriented design, which meant, above all, the assessment of MRD for final risk stratification and the allogeneic HCT choice for patients with HR/MRD-positive ALL. All these trials reporting MRD-based results confirmed the leading prognostic significance of this parameter (UKALL, GMALL, MDACC, US Intergroup, GRAALL, PETHEMA, NILG) and the benefit provided by an allogeneic HCT to these patients (GMALL, MDACC, NOPHO, GRAALL, PETHEMA, NILG), despite the outcome of MRD positive patients being globally poor in intention-to-treat- and meta-analyses.28–31 However, on analyzing trial details, this finding can be seen in relation to the combined effects of pretransplantation relapse (rating 40% or higher in some studies), nonrelapse mortality and post-transplantation relapse, which is more frequent in MRD-positive patients. Nevertheless, when feasible, allogeneic HCT is preferable to standard intensive, pediatric-based chemotherapy in MRD-positive patients (usually defined HR, Table 2), to reduce the risk of relapse and thereby increasing their survival from ⩽25% without HCT to approximately 45–55% (GMALL, NILG, GRAALL trials, reviewed by Bassan and colleagues29; plus several other studies totaling 1299 HCT patients with known MRD status, reviewed by Bassan and Spinelli72). Most notably, adopting the Simon-Makuch statistics to eliminate the time-dependent bias of pretransplantation relapse, the GRAALL study demonstrated a significant prognostic improvement with allogeneic HCT for the patients with postinduction MRD levels ⩾10−4 (p = 0.04)73 or 10−3 (p = 0.002).74 The most difficult category to treat consists of the patients who still harbor MRD ⩾ 10−3 following intensive induction-consolidation.75 Here too, a reduction of MRD prior to allogeneic HCT would be highly desirable to enhance the likelihood of a successful HCT, as demonstrated in MRD-directed phase II studies with blinatumomab, a bispecific CD3 × CD19 product engaging cytotoxic normal T cells against CD19+ B-precursor ALL cells.76,77 This type of immunotherapy acted as successful bridge to allogeneic HCT, and improved the outcome of study patients compared with the historical MRD positive cohort. A companion study with the T-targeting agent nelarabine is being conducted by the GMALL in MRD positive T-ALL patients.37 An open question is represented by the clinical management of patients who, despite achieving MRD negativity, had other HR features at diagnosis; in this respect there is not yet a clear consensus, although individual trial recommendations should usually be used (see Table 2 and related sections).
Management of specific ALL subsets: Ph+ ALL, Ph–like ALL and early thymic precursor ALL
Ph+ ALL
The BCR/ABL1 rearrangement, derived from the t(9;22) translocation, alias Ph chromosome, can be detected in about 20–30% of adult cases with ALL; its incidence increases with age, representing the most frequent of ALL in the elderly population, and is therefore a relatively rare event in AYAs (<20%).22,23 The outcome of Ph+ ALL patients, historically poor, has changed drastically since the introduction of tyrosine-kinase inhibitors (TKIs) of first-, second-, and, more recently, third-generation TKIs. TKIs are administered alone or in combination with chemotherapy, followed by consolidation and allogeneic HCT. With these strategies, survival rates are very close to those documented in Ph– ALL (Table 6, inclusive of extensive study references), and can reach an outstanding rate close to 80% using the more effective TKI ponatinib in conjunction with chemotherapy.78
Table 6.
Front-line treatments including TKI used in adult/AYA Ph+ ALL (study reference indicated).
| TKI used | Study group and reference | No. of patients | Median age (range), years | CR (%) | DFS (%) | OS (%) | Allo-HCT (%) |
|---|---|---|---|---|---|---|---|
| Intensified treatment | |||||||
| Im 600 mg | GMALL, Wassmann B, et al. Blood 2006; 108: 1469–1477. | 92 | Alternatinga: 46 (21–65) Concurrentb: 41 (19–63) |
95 | 36 alternating cycles at 2 years; 43 concurrent cycles at 2 years |
77 | |
| Im 600 mg | GRAALL*, Delannoy A, et al. Leukemia 2006; 20: 1526–1532. | 30 | 65.8 (58–78) | 72 | 58 at 1 year (RFS) | 66 at 1 year | NA |
| Im 600 mg | JALSG, Yanada M, et al. JCO 2006; 24: 460–466. | 80 | 45 (15–64) | 96 | 76 at 1 year | 61 | |
| Im 600 mg | GRAALL, de Labarthe A, et al. Blood 2007; 109: 1408–1413. | 45 | 45 (16–59) | 96 | 51 at 18 months | 65 at 18 months | 48 |
| Im 400 mg | PETHEMA, Ribera JM, et al. Haematologica 2010; 95: 87–95. | 30 | 44 (18–62) | 90 | 30 at 4 years | 30 at 4 years | 53 |
| Im 600 mg | NILG, Bassan R, et al. JCO 2010; 28: 3644–3652. | 59 | 45 (20.4–66) | 92 | 39 at 5 years | 38 at 5 years | 57 |
| Im 400 mg | Thyagu S, et al. BJH 2012; 158: 506–514. | 32 | 46 (18–60) | 94 | NA | 53 at 3 years | 50 |
| Im 600/800 mg | GRAALL, Tanguy-Schmidt A, et al. Biol Blood Marrow Transplant 2013; 19: 150–155. | 45 | 45 (16–59) | 96 | 44 at 4 years | 52 at 4 years | 53 |
| Im 600 mg | NCRI/ECOG, Fielding AK, et al. Blood 2014; 123: 843–850. | 175 | 42 (16–64) | 92 | 50 at 4 years (RFS) |
38 at 4 years | 46 |
| Im 400/800 mg | MDACC, Daver N, et al. Haematologica 2015; 100: 653–661. | 45 | 51 (17–84) | 93 | 43 at 5 years | 43 at 5 years | 30 |
| Das 50 mg BID or 100 daily | MDACC, Ravandi F, et al. Blood 2010; 116: 2070–2777. | 35 | 52 (21–77) | 94 | 60 at 2 years | 64 at 2 years | 12 |
| Nil 400 mg BID | KAALL WP, Kim DY, et
al. Blood 2015; 126: 746–756. |
50 | 44.5 (18–71) | 91 | NA | 66 at 2 years | 91 |
| Das 100 mg | Yoon JH, et al. Ann of Oncol 2016; 27: 1081–1088. | 51 | 46 (19–64) | 94 | 52 at 4 years | 51 at 4 years | 76 |
| Pon. 45 mg | MDACC, Jabbour E, et al. Lancet Oncol 2015; 16: 1547–1555. | 37 | 51 (27–75) | 100 | NA | 86 at 1 year | 24 |
| De-intensified treatment | |||||||
| Im 600 mg | PETHEMA, Ribera JM, et al. BJH 2012; 159: 78–81. | 29 | 38 (n.a.) | 100 | NA | 63 at 2 years (EFS) | 90 |
| Das 70 mg BID | GIMEMA. Foà R, et al. Blood 2011; 118: 6521–6528. | 53 | 53.6 (23.8–76.5) | 100 | 51 at 20 months | 69 at 20 months | – |
| Im 600 mg | GIMEMA, Chiaretti S, et al. Haematologica 2016; 101: 1544–1552. | 49 | 45.9 (16.9–59.7) | 100 | 50 at 36 months | 69 at 36 months | – |
| Das 140 mg daily | GIMEMA, Chiaretti S, et al. Blood 2015; abstract 81. | 60 | 41.9 (18.7–59.1) | 97 | 47 at 5 years | 56 at 5 years | |
| Im 800 mg | GRAALL, Chalandon Y, et al. Blood 2015; 125: 3711–3719 | 268 | 47 (18–59) | 98 | NA | 45 at 5 years | 63 |
ALL, acute lymphoblastic leukemia; AYA, adolescents and young adults; CR, complete remission; DFS/RFS/EFS, disease- or relapse- or event-free survival; OS, overall survival; NA, not available; Ph+, Philadelphia chromosome-positive B-ALL.; TKI, tyrosine kinase inhibitor (Das, dasatinib; Im, imatinib; Nil, nilotinib; Pon, ponatinib), daily dosage reported.
Alternating to chemotherapy.
Concurrent to chemotherapy.
As a general principle, induction treatment must be based on TKI, and the burden of chemotherapy can be reduced drastically to minimize toxicity: in this respect, Chalandon and colleagues clearly showed in the phase III GRAALL trial how chemotherapy deintensification led to higher CR and slightly higher survival rates. The GIMEMA has, for several years, carried out trials based on a chemotherapy-free induction with TKI, corticosteroids, and IT CNS prophylaxis, achieving CR rates of 97–100% without induction deaths.
Consolidation is usually based on chemotherapy including high-dose chemotherapy. With novel drugs available, namely monoclonal antibodies, chemotherapy might be abrogated also in this phase. Indeed, although data are preliminary because of the short follow up, the GIMEMA experience indicated that postinduction blinatumomab along with dasatinib is highly effective, with remarkably high 1-year OS and DFS rates (94.8% and 87.8%, respectively)79: in this trial, final treatment after blinatumomab was according to investigator’s choice, consisting of either dasatinib maintenance or allogeneic HCT. The results of similar chemotherapy-free, and possibly transplant-free, programs such as the MDACC’s ponatinib-blinatumomab study are awaited with interest.
At present, also in light of the smaller incidence of Ph+ ALL in AYAs, treatment does not differ between older adults and AYAs.80 Despite the improvement described above, there are still open issues. In fact, it is becoming clear that Ph+ ALL patients can be further stratified at diagnosis on the basis of additional genomic lesions. The cases harboring additional genomic lesions, particularly IKZF1 and CDKN2A/B and PAX5 deletions have a poorer outcome, which is not greatly improved by HCT: for these patients, alternative strategies are required.81,82 Furthermore, during follow up, a set of patients can acquire deleterious TK domain mutations (see exhaustive reviews by Soverini and colleagues83,84) or show MRD persistence. Thus, it is ever more frequently debated whether all Ph+ ALL patients should be allocated to HCT, or if HCT should be reserved for HR patients on the basis of the biological features described above. Moreover, in a recent chemotherapy-dasatinib combination COG trial including AYAs (age range 1–30 years), outcome was comparable among nontransplant and transplant patients, the latter identified through expression of HR features or availability of a related HCT donor.80
Ph–like ALL
As mentioned above, the outcome of AYA patients is poorer than children because of intrinsic biological features. One adverse subset is represented by the Ph–like (or BCR-ABL1-like) subgroup, which accounts for about 20% of B-ALL overall and is detected exclusively in cases lacking BCR-ABL1, KMT2A-based, and TCF3-PBX1 rearrangements.27,85–88 This subset was initially recognized by means of gene expression profile, revealing a transcriptional profile similar to that of BCR-ABL1+ patients. Later, with the integration of CNA, and DNA- and RNA-sequencing, the genetic basis of this subset, as well as its heterogeneity, were unraveled. Overall, the genetic scenario of Ph–like ALL is characterized by cytokine receptor deregulation or TK mutations and rearrangements. The first lesion to be described was CRLF2 deregulation. CRLF2 encodes a member of the type I cytokine receptor family involved in B-cell development. CRLF2 overexpression is sustained by a cryptic chromosomal translocation that juxtaposes CRLF2 to the immunoglobulin heavy chain locus (IGH),89 interstitial deletion of the PAR1 region centromeric to CRLF2,90 and, rarely, elevated CRLF2 expression is sustained by F232C mutation.91 In AYAs, CRLF2 overexpression is detected in 50–60% of Ph–like cases,85–88,92 with P2RY8/CRLF2 prevailing in children and IGH/CRLF2 more frequent in AYAs and adults.85,86,92,93 CRLF2 overexpression is usually coupled with other mutations, the most frequent affecting JAK/STAT pathway members (JAK2, JAK1, IL7R, and CRLF2).85–87,92–95 More rarely, another cytokine receptor rearranged in Ph–like ALL is the erythropoietin receptor (EPOR, 4%).85,92,94 Regarding TKs, the most frequent classes involved are ABL-class and JAK/STAT genes. ABL-class genes are relatively often detected (10% of AYA group) and include rearrangements of ABL1, ABL2, CSF1R, and PDGFRB with multiple partner genes. Similarly, JAK2 rearrangements can recognize several partner genes, comprising EBF1, ETV6, PAX5, and BCR (7–8% overall).85,92,94
From a clinical standpoint, Ph–like patients are often young male adults presenting with hyperleukocytosis, who display an inferior response to induction therapy, higher incidence of relapse and lower survival than other B-precursor Ph– ALL patients.45,85–87,95–97 Only a SJH report showed that pediatric patients treated with intensive therapies including HCT had a survival similar to non-Ph–like patients.92
Given their dismal outcome, these patients should be recognized promptly at diagnosis, for targeted treatment including TKIs and other agents. However, because of the plethora of associated genetic lesions, the optimal therapeutic approach to Ph–like ALL is not yet defined, and different alternative approaches have been proposed97–100: the first is on the basis of the underlying lesion, including dasatinib for cases with ABL class genes, and with JAK2 inhibitors, particularly ruxolitinib, for cases with JAK/STAT pathway lesions. However, this approach is not applicable in all treatment centers. Several clinical trials are ongoing to test the efficacy and safety of these approaches. Furthermore, regarding ruxolitinib, preliminary results from MDACC on nine R/R Ph–like patients did not show significant responses, and the study was prematurely closed (E. Jabbour, personal communication, 2019). A second approach could be the use of the pan-TKI ponatinib, as suggested by Chiaretti and colleagues,88 and tested in a French patient.101 Third, the role of antibody constructs, namely blinatumomab and inotuzumab, must be assessed. Presently, the best therapeutic approach is to treat such difficult cases upfront with a combination of intensified therapy and a targeted therapy, followed, also according to disease response, by allogeneic HCT.
ETP ALL
While B-lineage subsets are predominant in ALL, about 20–25% of AYA patients have T-ALL, which in children is no longer considered a HR subset due to the progress obtained with modern intensive regimens.1,2 Within T-ALL, about 15% of the cases share the peculiar diagnostic features of early T-cell precursor (ETP) ALL, consisting of a mixed early T-cell/myeloid immunophenotype lacking the pan T-cell antigen CD5 (weakly expressed on <75% of blast cell in the ‘near-ETP’ subset), co-expressing early myeloid antigens, and displaying higher genomic instability and different gene expression profile, closer to myeloid stem cells, compared with classical T-ALL.102 These patients fared significantly less well than standard T-ALL patients. However, a more recent COG trial documented and improved outcome for ETP ALL, not significantly inferior to non-ETP ALL patients.103 The topic of ETP ALL is less well known in adult and AYA patients. A review of MDACC results using Hyper-CVAD chemotherapy confirmed the inferior outcome of this subset (3-year OS about 30%, p = 0.037).104 Instead, the GRAALL, using pediatric-inspired therapy with risk/MRD-based stratification for HCT allocation, reported an improved outcome (5-year OS 59.6% versus 66.5% in non-ETP ALL patients, p = 0.33). In this study,105 ETP ALL patients were more likely to express high levels of postinduction MRD at the two study time-points (p < 0.001 and p = 0.005) and were more frequently offered an allogeneic HCT than non-ETP ALL patients (48.9% versus 28.3%, p = 0.008), which conferred a survival advantage.
Allogeneic HCT in AYA patients
Allogeneic HCT is still the most effective consolidation treatment for HR patients in whom the relapse risk is significantly higher than after chemotherapy, despite the risk of transplant-related mortality (TRM, ranging from 10 to 30%).106–110 Taking into consideration advances in chemotherapy (pediatric-style protocols) as well as new immunotherapy approaches, the advantage of one approach over the other is becoming less clear and defining the exact indications of HCT in AYA ALL in CR1 is increasingly difficult and should be regarded as matter for prospective clinical studies.
Critical issues of allogeneic HCT versus modern intensive AYA chemotherapy
In the pre-MRD era, the large prospective UKALL XII/ECOG E2993 study,111 together with the meta-analysis by Gupta and colleagues summarizing several HCT-based trials,112 evidenced that the benefit of transplantation over chemotherapy was restricted to AYAs <35 years with Ph– ALL (5-year OS 62% versus 52%; RFS 55% versus 45%, respectively). The main limitation of all these studies was the heterogeneous definition of HR ALL, which did not include MRD and the new genetic characterization, and considered only adult-type chemotherapy. The improved results of chemotherapy in AYA patients using pediatric-inspired protocols, combined with an MRD/risk-oriented treatment strategy, re-opened the debate on the value of HCT. In a retrospective comparative analysis from the DFCI Consortium and the Center for International Blood and Marrow Transplant Research,113 4-year OS was significantly improved in nontransplant Ph–negative AYA patients treated with the DFCI pediatric protocol (73% versus 45%, p < 0.001), essentially due to lower treatment mortality (6% versus 37%, p < 0.0001), while 4-year relapse incidence was almost identical (23% versus 24%). Age >30 years was confirmed to be an independent risk factor for TRM. Of note, the two patients cohorts were not fully comparable for the prevalence of HR features in HCT group, and, in addition, the TRM rate was higher than the figure currently expected in this age group, perhaps in relation to the limited use of antithymocyte globulin for the prevention of graft-versus-host disease, aggravating the risk of TRM.
Current place of allogeneic HCT in AYAs
Several studies demonstrated postinduction MRD and ALL genetics to represent the most important prognostic markers, as discussed above. With the limit of patient selection because not originally intended as a MRD-oriented trial, the GRAALL experience with 522 Ph– HR patients (aged 15–55 years, hence including a large proportion of AYA patients) showed no differences in RFS/OS between donor (HCT) and no-donor (no HCT) patient cohorts.74 However, HR patients expressing MRD positivity (>10−3) after 6 weeks of chemotherapy and KMT2A-rearranged or IKZF1-deleted ALL benefited from allogeneic HCT (5-year OS 70% versus 35%; p < 0.002).25,74 Other studies focused exclusively on AYA populations,106,108,109 although mostly retrospective and heterogeneous concerning the status at transplantation (CR1 and CR2), the definition of HR (with or without new genetic markers), and MRD evaluation, indirectly highlighted the advantage of HCT (OS ranging from 40% to 70%) over no transplant approaches in suboptimal responders (MRD positive, adverse genetic subsets). In view of these uncertainties, it is preferable to adhere to the transplantation policy of specific treatment protocols, which usually reserve this treatment modality in first CR to very HR patients with high postinduction MRD or highly adverse genetic ALL variants (see section Risk stratification). For refractory and relapsed ALL patients, an allogeneic HCT still represents the main curative option, with up to 40% of long survivors.
Improving HCT results
Because of its adverse impact on post-transplantation outcome, achieving MRD negativity with new targeted therapies before HCT may be crucial to bring survival above 50%, compared with less when the transplant is performed in molecular failure.28–34,37,72–77 In new AYA trials employing inotuzumab ozogamicin (such as the ongoing US Intergroup AYA trial 041501; ClinicalTrials.gov identifier: NCT03150693), dual alkylator HCT conditioning must be avoided due to the associated risk of hepatic veno-occlusive disease.114
Allogeneic HCT in Ph+ ALL
As for Ph– ALL, the role of HCT in Ph+ ALL patients is changing in relation to the efficacy of TKI associations with low-dose chemotherapy and new immunotherapeutic approaches.83 MRD status and genomic characterization at diagnosis are key factors in this decision-making process,78,83,115,116 especially with the most effective combinations tested upfront to date, such as ponatinib-chemotherapy78 and dasatinib-blinatumomab,79 which are providing excellent early results without HCT in MRD negative patients. However, apart from these very recent examples, most studies evidenced a better outcome for transplanted patients (3–5 year EFS 50–69%), in comparison with TKI-based regimens without transplantation (3–5 year EFS 30–46%),83 although the younger the patient, the better the outcome, even with transplant-free regimens in patients lacking HR features.80,83
Concluding remarks and future directions
The most useful approach to the frontline management of AYA ALL is summarized in Figure 3. It must be emphasized that any effort should be devoted to curing the disease upfront, since survival is still largely unsatisfactory with any new treatment so far tested in patients who display primary resistance or develop a recurrence, unless they belong to the minority of younger patients that suffer from an isolated late marrow relapse >24 months from the date of initial CR.32,117 Based on a large body of evidence, the initial diagnostic work up should aim to identify the ALL subsets that have clear prognostic and therapeutic relevance (i.e. Ph+ ALL, Ph–like ALL, KMT2A-rearranged ALL, ETP ALL, etc.) and include the generation of patient-specific molecular probes (or corresponding leukemia-specific immunophenotypes) for MRD analysis. The patients should be enrolled into pediatric-inspired national or institutional trials of modern design, including a postremission strategy orientated by clinical risk class, genetics, and MRD. In Ph+ ALL, the concurrent use of TKI therapy is, of course, mandatory. It is also essential to develop new trials with specific therapeutic elements for discrete ALL entities, and test new risk stratification systems integrating MRD with the most important genetic abnormalities to identify more precisely the patient subsets that benefit (or not) from any given therapeutic intervention. As reviewed extensively elsewhere, trials with new agents targeting different ALL subsets and molecular variants were performed successfully in advanced ALL, and are flourishing in frontline studies.118–120 Among them, it is worth mentioning several North-American and European immunotherapy studies with rituximab (targeting CD20 antigen),53–55 blinatumomab (bispecific CD19 × CD3 T-cell engager antibody), and inotuzumab ozogamicin (anti-CD22 drug-antibody conjugate) for CD20, CD19, and CD22-positive, Ph+, and Ph– ALL, respectively; a variety of TKI-based trials for Ph-like ALL83; and the evaluation of several other small molecules (such as BCL2 inhibitors in KMT2A-rearranged ALL, ETP ALL, etc.) and of CAR T cells, at present mainly in relapsed/refractory states and MRD+ ALL.121,122 These new approaches, once confirmed safe and effective, would be transferred upfront, leading to significant improvements not only in AYAs, but also in highly difficult conditions, as demonstrated for the first time in elderly ALL and Ph+ ALL, even employing low-dose chemotherapy or without any chemotherapy.83,123,124 The reduction of severe toxicity due to intensive chemotherapy or allogeneic HCT, that causes both therapy-related deaths and significant short- and long-term morbidity, is a major concern in AYA’s therapy, and is evaluated in new targeted agent trials. This progressive therapeutic shift could be facilitated and strengthened by molecular and drug response screening programs for the identification of actionable targets, and the confirmation of expected or unexpected drug vulnerabilities. These new assays have already led to optimal therapeutic choices in patients refractory to standard treatments,125,126 and deserve to be tested in chemotherapy-naïve patients. This global challenge could enlarge our therapeutic horizon, and increase the curability of AYA patients with ALL.
Figure 3.

Current and future status of ALL therapy in AYA patients. The essential steps are a correct risk stratification (genetics, MRD), the use of an institutional/national pediatric-based protocol containing Peg-ASP among other elements, enriched whenever possible with targeting agents (Ph+ ALL: additional TKI therapy; B-ALL: monoclonal antibodies if CD20+, CD19+, CD22+), and with a prospective risk-oriented allogeneic HCT policy according to study protocol. Further improvements, under evaluation in ongoing trials, may be possible with the intensification of immunotherapy, the introduction of other targeting agents (as suggested by molecular profiling data), and the optimization of drug therapy (as suggested by drug sensitivity screening). New trials will have to evaluate novel drug combinations and sequences, demonstrating therapeutic progress with manageable toxicity, finally allowing depotentiation of intensive chemotherapy and reducing the need for allogeneic HCT.
ALL, acute lymphoblastic leukemia; AYA, adolescents and young adults; HCT, hematopoietic cell transplantation; HR, high risk; MRD, minimal (or measurable) residual disease; Peg-ASP, pegylated-asparaginase; Ph–, Philadelphia chromosome-negative B-ALL; Ph+, Ph chromosome-positive B-ALL; TKI, tyrosine kinase inhibitor.
Footnotes
Funding: The author(s) received no financial support for the research, authorship, and publication of this article.
Conflict of interest statement: F. Carobolante, C. Skert: none; R. Bassan: fees, travel and accommodation expenses from Amgen, Pfizer, Shire, Servier, Incyte for consultancies and participation into boards and symposia; S. Chiaretti: fees, travel and accommodation expenses from Amgen, Pfizer, Shire, Incyte for consultancies and participation into boards and symposia.
ORCID iD: Renato Bassan  https://orcid.org/0000-0001-8214-2894
https://orcid.org/0000-0001-8214-2894
Contributor Information
Francesca Carobolante, UOC Ematologia, Ospedale dell’Angelo, Venezia, Mestre, Italy.
Sabina Chiaretti, Division of Hematology, ‘Sapienza’ University, Rome, Italy.
Cristina Skert, UOC Ematologia, Ospedale dell’Angelo, Venezia, Mestre, Italy.
Renato Bassan, UOC Ematologia, Ospedale dell’Angelo, Via Paccagnella 11, Venezia, Mestre, 30174, Italy.
References
- 1. Pui CH, Evans WE. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin Hematol 2013; 50: 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med 2015; 15: 1541–1552. [DOI] [PubMed] [Google Scholar]
- 3. Siegel SE, Stock W, Johnson RH, et al. Pediatric-inspired treatment regimens for adolescents and young adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: a review. JAMA Oncol 2018; 4: 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Curran E, Stock W. How I treat acute lymphoblastic leukemia in older adolescents and young adults. Blood 2015; 125: 3702–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boissel N, Baruchel A. Acute lymphoblastic leukemia in adolescent and young adults: treat as adults or as children? Blood 2018; 132: 351–361. [DOI] [PubMed] [Google Scholar]
- 6. Huguet F, Leguay T, Raffoux E, et al. Pediatric-inspired therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: the GRAALL-2003 study. J Clin Oncol 2009; 27: 911–918. [DOI] [PubMed] [Google Scholar]
- 7. Huguet F, Chevret S, Leguay T, et al. Intensified therapy of acute lymphoblastic leukemia in adults: report of the randomized GRAALL-2005 clinical trial. J Clin Oncol 2018; 36: 2514–2523. [DOI] [PubMed] [Google Scholar]
- 8. Rytting ME, Jabbour EJ, Jorgensen JL, et al. Final results of a single institution experience with a pediatric-based regimen, the augmented Berlin-Frankfurt-Münster, in adolescents and young adults with acute lymphoblastic leukemia, and comparison to the hyper-CVAD regimen. Am J Hematol 2016; 91: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kantarjian H, Thomas D, O’Brien S, et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer 2004; 101: 2788–2801. [DOI] [PubMed] [Google Scholar]
- 10. Siegel SE, Advani A, Seibel N, et al. Treatment of young adults with Philadelphia-negative acute lymphoblastic leukemia and lymphoblastic lymphoma: Hyper-CVAD vs. pediatric-inspired regimens. Am J Hematol 2018; 93: 1254–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muffly L, Alvarez E, Lichtensztajn D, et al. Patterns of care and outcomes in adolescent and young adult acute lymphoblastic leukemia: a population-based study. Blood Adv 2018; 2: 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gupta S, Pole JD, Baxter NN, et al. The effect of adopting pediatric protocols in adolescents and young adults with acute lymphoblastic leukemia in pediatric vs adult centers: an IMPACT Cohort study. Cancer Med 2019; 8: 2095–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bleyer A, Tai E, Siegel S. Role of clinical trials in survival progress of American adolescents and young adults with cancer-and lack thereof. Pediatr Blood Cancer 2018; 65: e27074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hough R, Sandhu S, Khan M, et al. Are survival and mortality rates associated with recruitment to clinical trials in teenage and young adult patients with acute lymphoblastic leukaemia? A retrospective observational analysis in England. BMJ Open 2017; 7: e017052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boissel N, Auclerc MF, Lhéritier V, et al. Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol 2003; 21: 774–780. [DOI] [PubMed] [Google Scholar]
- 16. Stock W, La M, Sanford B, et al. What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of children’s cancer group and cancer and leukemia group B studies. Blood 2008; 112: 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ribera JM, Oriol A, Sanz MA, et al. Comparison of the results of the treatment of adolescents and young adults with standard-risk acute lymphoblastic leukemia with the Programa Español de Tratamiento en Hematología pediatric-based protocol ALL-96. J Clin Oncol 2008; 26: 1843–1849. [DOI] [PubMed] [Google Scholar]
- 18. Clarkson B, Ellis S, Little C, et al. Acute lymphoblastic leukemia in adults. Semin Oncol 1985; 12: 160–179. [PubMed] [Google Scholar]
- 19. Bassan R, Lerede T, Barbui T. Institutional performance and dose intensity as prognostic factors in adult ALL. Leukemia 1995; 9: 933–934. [PubMed] [Google Scholar]
- 20. Rytting ME, Jabbour EJ, O’Brien SM, et al. Acute lymphoblastic leukemia in adolescents and young adults. Cancer 2017; 123: 2398–2403. [DOI] [PubMed] [Google Scholar]
- 21. Giebel S, Marks DI, Boissel N, et al. Hematopoietic stem cell transplantation for adults with Philadelphia chromosome-negative acute lymphoblastic leukemia in first remission: a position statement of the European working group for adult acute lymphoblastic leukemia (EWALL) and the acute leukemia working party of the European society for blood and marrow transplantation (EBMT). Bone Marrow Transplant 2019; 54: 798–809. [DOI] [PubMed] [Google Scholar]
- 22. Moorman AV. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica 2016; 101: 407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chiaretti S, Vitale A, Cazzaniga G, et al. Clinico-biological features of 5202 patients with acute lymphoblastic leukemia enrolled in the Italian AIEOP and GIMEMA protocols and stratified in age cohorts. Haematologica 2013; 98: 1702–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev 2012; 26: 123–135. [DOI] [PubMed] [Google Scholar]
- 25. Lafage-Pochitaloff M, Baranger L, Hunault M, et al. Impact of cytogenetic abnormalities in adults with Ph-negative B-cell precursor acute lymphoblastic leukemia. Blood 2017; 130: 1832–1844. [DOI] [PubMed] [Google Scholar]
- 26. Hamadeh L, Enshaei A, Schwab C, et al. Validation of the United Kingdom copy-number alteration classifier in 3239 children with B-cell precursor ALL. Blood Adv 2019; 3: 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herold T, Baldus CD, Gökbuget N. Ph-like acute lymphoblastic leukemia in older adults. N Engl J Med 2014; 371: 2235. [DOI] [PubMed] [Google Scholar]
- 28. Berry DA, Zhou S, Higley H, et al. Association of minimal residual disease with clinical outcome in pediatric and adult acute lymphoblastic leukemia: a meta-analysis. JAMA Oncol 2017; 3: e170580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bassan R, Intermesoli T, Scattolin A, et al. Minimal residual disease assessment and risk-based therapy in acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk 2017; 17S: S2–S9. [DOI] [PubMed] [Google Scholar]
- 30. Brüggemann M, Kotrova M. Minimal residual disease in adult ALL: technical aspects and implications for correct clinical interpretation. Blood Adv 2017; 1: 2456–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bassan R, Brüggemann M, Radcliffe HS, et al. A systematic literature review and meta-analysis of minimal residual disease as a prognostic indicator in adult B-cell acute lymphoblastic leukemia. Haematologica 2019; 104: 2028–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoelzer D, Bassan R, Dombret H, et al. Acute lymphoblastic leukaemia in adult patients: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2016; 27(Suppl. 5): v69–v82. [DOI] [PubMed] [Google Scholar]
- 33. Short NJ, Jabbour E, Albitar M, et al. Recommendations for the assessment and management of measurable residual disease in adults with acute lymphoblastic leukemia: a consensus of North American experts. Am J Hematol 2019; 94: 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gökbuget N, Kneba M, Raff T, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood 2012; 120: 1868–1876. [DOI] [PubMed] [Google Scholar]
- 35. Beldjord K, Chevret S, Asnafi V, et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood 2014; 123: 3739–3749. [DOI] [PubMed] [Google Scholar]
- 36. Bassan R, Masciulli A, Intermesoli T, et al. Final results of Northern Italy leukemia group (NILG) trial 10/07 combining pediatric-type therapy with minimal residual disease study and risk-oriented hematopoietic cell transplantation in adult acute lymphoblastic leukemia (ALL). Blood 2016; 128: abstract 176. [Google Scholar]
- 37. Goekbuget N, Bruggemann M, Beck J, et al. Evaluation of minimal residual disease (MRD) and MRD-based treatment decisions in Ph/BCR-ABL-negative adult acute lymphoblastic leukemia (ALL): experience from the German multicenter study group for adult ALL (GMALL). Blood 2017; 130(Suppl. 1): abstract 139. [Google Scholar]
- 38. Moorman AV, Kirkwood A, Enshael A, et al. Clinical efficacy of a novel validated prognostic index for trial design in adult acute lymphoblastic leukaemia. HemaSphere 2019; 3(S1): 748: abstract S1621. [Google Scholar]
- 39. O’Connor D, Enshaei A, Bartram J, et al. Genotype-specific minimal residual disease interpretation improves stratification in pediatric acute lymphoblastic leukemia. J Clin Oncol 2018; 36: 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Petit A, Trinquand A, Chevret S, et al. Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood 2018; 131: 289–300. [DOI] [PubMed] [Google Scholar]
- 41. EU Clinical Trials Register. ALLTogether Protocol_Version 1.0. A treatment study protocol of the ALLTogether Consortium for children and young adults (1-45 years of age) with newly diagnosed acute lymphoblastic leukemia. EUDRACT number: 2018-001795-38. [Google Scholar]
- 42. Hayakawa F, Sakura T, Yujiri T, et al. Markedly improved outcomes and acceptable toxicity in adolescents and young adults with acute lymphoblastic leukemia following treatment with a pediatric protocol: a phase II study by the Japan adult leukemia study group. Blood Cancer J 2014; 4: e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hough R, Rowntree C, Goulden N, et al. Efficacy and toxicity of a pediatric protocol in teenagers and young adults with Philadelphia chromosome negative acute lymphoblastic leukaemia: results from UKALL 2003. Br J Haematol 2016; 172: 439–451. [DOI] [PubMed] [Google Scholar]
- 44. Goekbuget N, Beck J, Brandt K, et al. Significant improvement of outcome in adolescents and young adults (AYAs) aged 15-35 years with acute lymphoblastic leukemia (ALL) with a pediatric derived adult ALL protocol; results of 1529 AYAs in 2 consecutive trials of the German multicenter study group for adult ALL (GMALL). Blood 2013; 122: abstract 839. [Google Scholar]
- 45. Stock W, Luger SM, Advani AS, et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: results of CALGB 10403. Blood 2019; 133: 1548–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Toft N, Birgens H, Abrahamsson J, et al. Results of NOPHO ALL2008 treatment for patients aged 1-45 years with acute lymphoblastic leukemia. Leukemia 2018; 32: 606–615. [DOI] [PubMed] [Google Scholar]
- 47. DeAngelo DJ, Stevenson KE, Dahlberg SE, et al. Long-term outcome of a pediatric-inspired regimen used for adults aged 18-50 years with newly diagnosed acute lymphoblastic leukemia. Leukemia 2015; 29: 526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. DeAngelo DJ, Stevenson K, Neuberg DS, et al. A multicenter phase II study using a dose intensified pegylated-asparaginase pediatric regimen in adults with untreated acute lymphoblastic leukemia: a DFCI ALL consortium trial. Blood 2015; 126: abstract 80. [Google Scholar]
- 49. Goekbuget N, Baumann A, Beck J, et al. PEG-asparaginase intensification in adult acute lymphoblastic leukemia (ALL): significant improvement of outcome with moderate increase of liver toxicity in the German multicenter study group for adult ALL (GMALL) study 07/2003. Blood 2010; 116: abstract 494. [Google Scholar]
- 50. Parovichnikova EN, Troitskaya V, Sokolov AN, et al. Non-intensive but constant and exhausting action on the leukemic clone is a reasonable and effective treatment approach in adult acute lymphoblastic leukemia: results of the Russian acute lymphobastic leukemia (RALL) study group. Blood 2014; 124: abstract 3662. [Google Scholar]
- 51. Ribera JM, Morgades M, Montesinos P, et al. Comparison of efficacy and safety of two types of E. coli Asparaginase (Native or Pegylated) for treatment of adult patients with high-risk (HR), Philadelphia (Ph) chromosome-negative ALL included in the prospective MRD-oriented protocol ALL-HR-11 from the Spanish Pethema group. Blood 2016; 128: abstract 180. [Google Scholar]
- 52. Sakura T, Hayakawa F, Sugiura I, et al. High-dose methotrexate therapy significantly improved survival of adult acute lymphoblastic leukemia: a phase III study by JALSG. Leukemia 2018; 32: 826–832. [DOI] [PubMed] [Google Scholar]
- 53. Thomas DA, O’Brien S, Faderl S, et al. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol 2010; 28: 3880–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hoelzer D, Gökbuget N. Chemoimmunotherapy in acute lymphoblastic leukemia. Blood Rev 2012; 26: 25–32. [DOI] [PubMed] [Google Scholar]
- 55. Maury S, Chevret S, Thomas X, et al. Rituximab in B-lineage adult acute lymphoblastic leukemia. N Engl J Med 2016; 375: 1044–1053. [DOI] [PubMed] [Google Scholar]
- 56. Möricke A, Zimmermann M, Reiter A, et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 2010; 24: 265–284. [DOI] [PubMed] [Google Scholar]
- 57. Marini BL, Perissinotti AJ, Bixby DL, et al. Catalyzing improvements in ALL therapy with asparaginase. Blood Rev 2017; 31: 328–338. [DOI] [PubMed] [Google Scholar]
- 58. Douer D, Aldoss I, Lunning MA, et al. Pharmacokinetics-based integration of multiple doses of intravenous pegaspargase in a pediatric regimen for adults with newly diagnosed acute lymphoblastic leukemia. J Clin Oncol 2014; 32: 905–911. [DOI] [PubMed] [Google Scholar]
- 59. Aldoss I, Douer D, Behrendt CE, et al. Toxicity profile of repeated doses of PEG-asparaginase incorporated into a pediatric-type regimen for adult acute lymphoblastic leukemia. Eur J Haematol 2016; 96: 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Burke PW, Aldoss I, Lunning MA, et al. Pegaspargase-related high-grade hepatotoxicity in a pediatric-inspired adult acute lymphoblastic leukemia regimen does not predict recurrent hepatotoxicity with subsequent doses. Leuk Res 2018; 66: 49–56. [DOI] [PubMed] [Google Scholar]
- 61. Roesmann A, Afify M, Panse J, et al. l-Carnitine ameliorates l-asparaginase-induced acute liver toxicity in steatotic rat livers. Chemotherapy 2013; 59: 167–175. [DOI] [PubMed] [Google Scholar]
- 62. Rausch CR, Paul S, Marx KR, et al. l-Carnitine and vitamin B complex for the treatment of pegasparaginase-induced hyperbilirubinemia. Clin Lymphoma Myeloma Leuk 2018; 18: e191–e195. [DOI] [PubMed] [Google Scholar]
- 63. Stock W, Douer D, DeAngelo DJ, et al. Prevention and management of asparaginase/pegasparaginase-associated toxicities in adults and older adolescents: recommendations of an expert panel. Leuk Lymphoma 2011; 52: 2237–2253. [DOI] [PubMed] [Google Scholar]
- 64. Bassan R, Chiaretti S, Paoloni F, et al. First results of new GIMEMA trial LAL1913 for adult patients with Philadelphia-negative acute lymphoblastic leukemia (Ph- ALL). HemaSphere 2018; 2(s1): 408: abstract PS919. [Google Scholar]
- 65. Lanvers-Kaminsky C, Niemann A, Eveslage M, et al. Asparaginase activities during intensified treatment with pegylated E. coli asparaginase in adults with newly-diagnosed acute lymphoblastic leukemia. Leuk Lymphoma 2020; 61: 138–145. [DOI] [PubMed] [Google Scholar]
- 66. Schore RL, Devidas M, Bleyer A, et al. Plasma asparaginase activity and depletion in acute lymphoblastic leukemia patients treated with pegaspargase on children’s oncology group AALL074P4. Leuk Lymphoma 2019; 60: 1740–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Larsen EC, Devidas M, Chen S, et al. Dexamethasone and high-dose methotrexate improve outcome for children and young adults with high-risk B-acute lymphoblastic leukemia: a report from children’s oncology group study AALL0232. J Clin Oncol 2016; 34: 2380–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Winter SS, Dunsmore KP, Devidas M. Improved survival for children and young adults with T-lineage acute lymphoblastic leukemia: results from the children’s oncology group AALL0434 methotrexate randomization. J Clin Oncol 2018; 36: 2926–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bassan R, Masciulli A, Spinelli O, et al. New first line chemotherapy program with lineage-targeted methotrexate infusions is feasible and improves the early minimal residual disease response and survival in acute T-lymphoblastic leukemia. Haematologica 2011; 96(Suppl. 2): 238: abstract 0557.20952517 [Google Scholar]
- 70. Möricke A, Zimmermann M, Valsecchi MG, et al. Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 2016; 127: 2101–2112. [DOI] [PubMed] [Google Scholar]
- 71. Goekbuget N, Baur KH, Beck J, et al. Dexamethasone dose and schedule significantly influences remission rate and toxicity of induction therapy in adult acute lymphoblastic leukemia (ALL): results of the GMALL pilot trial 06/99. Blood 2005; 106: abstract 1832. [Google Scholar]
- 72. Bassan R, Spinelli O. Minimal residual disease monitoring in adult ALL to determine therapy. Curr Hematol Malig Rep 2015; 10: 86–95. [DOI] [PubMed] [Google Scholar]
- 73. Dhédin N, Huynh A, Maury S, et al. Allogeneic hematopoietic stem cell transplantation (HSCT) in adults with Philadelphia chromosome (Ph)-negative acute lymphoblastic leukemia (ALL): results from the group for research on adult ALL (GRAALL). Blood 2013; 122: abstract 552. [Google Scholar]
- 74. Dhédin N, Huynh A, Maury S, et al. Role of allogeneic stem cell transplantation in adult patients with Ph-negative acute lymphoblastic leukemia. Blood 2015; 125: 2486–2496. [DOI] [PubMed] [Google Scholar]
- 75. Gökbuget N, Dombret H, Giebel S, et al. Minimal residual disease level predicts outcome in adults with Ph-negative B-precursor acute lymphoblastic leukemia. Hematology 2019; 24: 337–348. [DOI] [PubMed] [Google Scholar]
- 76. Gökbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018; 131: 1522–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gökbuget N, Zugmaier G, Klinger M, et al. Long-term relapse-free survival in a phase 2 study of blinatumomab for the treatment of patients with minimal residual disease in B-lineage acute lymphoblastic leukemia. Haematologica 2017; 102: e132–e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jabbour E, Kantarjian H, Ravandi F, et al. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: a single-centre, phase 2 study. Lancet Oncol 2015; 16: 1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chiaretti S, Bassan R, Vitale A, et al. A Dasatinib-Blinatumomab combination for the front-line treatment of adult Ph+ ALL patients. Preliminary results of the GIMEMA LAL2116 D-ALBA TRIAL; on behalf of GIMEMA acute leukemia working party. HemaSphere 2019; 3(S1): 746: abstract S1617. [Google Scholar]
- 80. Slayton WB, Schultz KR, Kairalla JA, et al. Dasatinib plus intensive chemotherapy in children, adolescents, and young adults with Philadelphia chromosome-positive acute lymphoblastic leukemia: results of children’s oncology group trial AALL0622. J Clin Oncol 2018; 36: 2306–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pfeifer H, Raum K, Markovic S, et al. Genomic CDKN2A/2B deletions in adult Ph+ ALL are adverse despite allogeneic stem cell transplantation. Blood 2018; 131: 1464–1475. [DOI] [PubMed] [Google Scholar]
- 82. Fedullo AL, Messina M, Elia L, et al. Prognostic implications of additional genomic lesions in adult Philadelphia chromosome-positive acute lymphoblastic leukemia. Haematologica 2019; 104: 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Soverini S, Bassan R, Lion T. Treatment and monitoring of Philadelphia chromosome-positive leukemia patients: recent advances and remaining challenges. J Hematol Oncol 2019; 12: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Soverini S, Vitale A, Poerio A, et al. Philadelphia-positive acute lymphoblastic leukemia patients already harbor BCR-ABL kinase domain mutations at low levels at the time of diagnosis. Haematologica 2011; 96: 552–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Roberts KG, Gu Z, Payne-Turner D, et al. High frequency and poor outcome of Philadelphia chromosome-like acute lymphoblastic leukemia in adults. J Clin Oncol 2017; 35: 394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jain N, Roberts KG, Jabbour E, Patel K, et al. Ph-like acute lymphoblastic leukemia: a high-risk subtype in adults. Blood 2017; 129: 572–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Herold T, Schneider S, Metzeler KH, et al. Adults with Philadelphia chromosome-like acute lymphoblastic leukemia frequently have IGH-CRLF2 and JAK2 mutations, persistence of minimal residual disease and poor prognosis. Haematologica 2017; 102: 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chiaretti S, Messina M, Grammatico S, et al. Rapid identification of BCR/ABL1-like acute lymphoblastic leukaemia patients using a predictive statistical model based on quantitative real time-polymerase chain reaction: clinical, prognostic and therapeutic implications. Br J Haematol 2018; 181: 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Moorman AV, Schwab C, Ensor HM, et al. IGH@ translocations, CRLF2 deregulation, and microdeletions in adolescents and adults with acute lymphoblastic leukemia. J Clin Oncol 2012; 30: 3100–3108. [DOI] [PubMed] [Google Scholar]
- 90. Cario G, Zimmermann M, Romey R, et al. Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood 2010; 115: 5393–5397. [DOI] [PubMed] [Google Scholar]
- 91. Yoda A, Yoda Y, Chiaretti S, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 2010; 107: 252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 2014; 371: 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Russell LJ, Jones L, Enshaei A, et al. Characterisation of the genomic landscape of CRLF2-rearranged acute lymphoblastic leukemia. Genes Chromosomes Cancer 2017; 56: 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Reshmi SC, Harvey RC, Roberts KG, et al. Targetable kinase gene fusions in high-risk B-ALL: a study from the children’s oncology group. Blood 2017; 129: 3352–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009; 10: 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mullighan CG, Zhang J, Harvey RC, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 2009; 106: 9414–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chiaretti S, Messina M, Foà R. BCR/ABL1-like acute lymphoblastic leukemia: how to diagnose and treat? Cancer 2019; 125: 194–204. [DOI] [PubMed] [Google Scholar]
- 98. Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood 2017; 130: 2064–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Maese L, Tasian SK, Raetz EA. How is the Ph-like signature being incorporated into ALL therapy? Best Pract Res Clin Haematol 2017; 30: 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Boer JM, den Boer ML. BCR.-ABL1-like acute lymphoblastic leukaemia: from bench to bedside. Eur J Cancer 2017; 82: 203–2018. [DOI] [PubMed] [Google Scholar]
- 101. Collette Y, Prébet T, Goubard A, et al. Drug response profiling can predict response to ponatinib in a patient with t(1;9)(q24;q34)-associated B-cell acute lymphoblastic leukemia. Blood Cancer J 2015; 5: e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 2009; 10: 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wood BL, Winter SS, Dunsmore KP, et al. T-lymphoblastic leukemia (T-ALL) shows excellent outcome, lack of significance of the early thymic precursor (ETP) immunophenotype, and validation of the prognostic value of end-induction minimal residual disease (MRD) in children’s oncology group (COG) study AALL0434. Blood 2014; 124: abstract 1. [Google Scholar]
- 104. Jain N, Lamb AV, O’Brien S, et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high risk subtype. Blood 2016; 127: 1863–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bond J, Graux C, Lhermitte L, et al. Early response-based therapy stratification improves survival in adult early thymic precursor acute lymphoblastic leukemia: a group for research on adult acute lymphoblastic leukemia study. J Clin Oncol 2017; 35: 2683–2691. [DOI] [PubMed] [Google Scholar]
- 106. Hangai M, Urayama KY, Tanaka J, et al. Allogeneic stem cell transplantation for acute lymphoblastic leukemia in adolescents and young adults. Biol Blood Marrow Transplant 2019; 25: 1597–1602. [DOI] [PubMed] [Google Scholar]
- 107. Giebel S, Labopin M, Socié M, et al. Improving results of allogeneic hematopoietic cell transplantation for adults with acute lymphoblastic leukemia in first complete remission: an analysis from the acute leukemia working party of the European society for blood and marrow transplantation. Haematologica 2017; 102: 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wood WA, Lee SJ, Brazauskas R, et al. Survival improvements in adolescents and young adults after myeloablative allogeneic transplantation for acute lymphoblastic leukemia. Biol Blood Marrow Transplant 2014; 20: 929–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Muffly L, Li Q, Alvarez E, et al. Hematopoietic cell transplantation in young adult acute lymphoblastic leukemia: a United States population-level analysis. J Adolesc Young Adult Oncol 2019; 8: 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Vignon M, Andreoli A, Dhedin N, et al. Graft-Versus-Host disease in adolescent and young adults (15-24 years old) after allogeneic hematopoietic stem cell transplantation for acute leukemia in first complete remission. J Adolesc Young Adult Oncol 2019; 6: 299–306. [DOI] [PubMed] [Google Scholar]
- 111. Goldstone AH, Richards SM, Lazarus HM, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the international ALL Trial (MRC UKALL XII/ECOG E2993). Blood 2008; 111: 1827–1833. [DOI] [PubMed] [Google Scholar]
- 112. Gupta V, Richards S, Rowe J, et al. Allogeneic, but not autologous, hematopoietic cell transplantation improves survival only among younger adults with acute lymphoblastic leukemia in first remission: an individual patient data meta-analysis. Blood 2013; 121: 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Seftel MD, Neuberg D, Zhang MJ, et al. Pediatric-inspired therapy compared to allografting for Philadelphia chromosome-negative adult ALL in first complete remission. Am J Hematol 2016; 91: 322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kantarjian HM, DeAngelo DJ, Advani AS, et al. Hepatic adverse event profile of inotuzumab ozogamicin in adult patients with relapsed or refractory acute lymphoblastic leukaemia: results from the open-label, randomised, phase 3 INO-VATE study. Lancet Haematol 2017; 4: e387–e398. [DOI] [PubMed] [Google Scholar]
- 115. Short NJ, Jabbour E, Sasaki K, et al. Impact of complete molecular response on survival in patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood 2016; 128: 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lussana F, Intermesoli T, Gianni F, et al. Achieving molecular remission before allogeneic stem cell transplantation in adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: impact on relapse and long-term outcome. Biol Blood Marrow Transplant 2016; 22: 1983–1987. [DOI] [PubMed] [Google Scholar]
- 117. Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J Clin Oncol 2011; 29: 532–543. [DOI] [PubMed] [Google Scholar]
- 118. Wolach O, Amitai I, DeAngelo DJ. Current challenges and opportunities in treating adult patients with Philadelphia-negative acute lymphoblastic leukaemia. Br J Haematol 2017; 179: 705–723. [DOI] [PubMed] [Google Scholar]
- 119. Jabbour E, Pui CH, Kantarjian H. Progress and innovations in the management of adult acute lymphoblastic leukemia. JAMA Oncol 2018; 4: 1413–1420. [DOI] [PubMed] [Google Scholar]
- 120. Bassan R, Bourquin JP, DeAngelo DJ, et al. New approaches to the management of adult acute lymphoblastic leukemia. J Clin Oncol 2018; 36: 3504–3519. [DOI] [PubMed] [Google Scholar]
- 121. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018; 378: 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Park JH, Rivière I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med 2018; 378: 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kantarjian H, Ravandi F, Short NJ, et al. Inotuzumab ozogamicin in combination with low-intensity chemotherapy for older patients with Philadelphia chromosome-negative acute lymphoblastic leukaemia: a single-arm, phase 2 study. Lancet Oncol 2018; 19: 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Advani AS, Moseley A, O’Dwyer KM, et al. Results of SWOG 1318: a phase 2 trial of blinatumomab followed by pomp (prednisone, vincristine, methotrexate, 6-mercaptopurine) maintenance in elderly patients with newly diagnosed Philadelphia chromosome negative B-cell acute lymphoblastic leukemia. Blood 2018; 132: abstract 33. [Google Scholar]
- 125. Frismantas V, Dobay MP, Rinaldi A, et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood 2017; 129: e26–e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Snijder B, Vladimer GI, Krall N, et al. Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: interim results from a single-arm, open-label, pilot study. Lancet Haematol 2017; 4: e595–e606. [DOI] [PMC free article] [PubMed] [Google Scholar]