

Abstract
Mechanical cues such as stiffness have been shown to influence cell gene expression, protein expression, and cell behaviors critical for tissue engineering. The mechanical cue of confinement is also a pervasive parameter affecting cells in vivo and in tissue-engineered constructs. Despite its prevalence, the mechanical cue of confinement lacks assays that provide precise control over the degree of confinement induced on cells, yield a large sample size, enable long-term culture, and enable easy visualization of cells over time. In this study, we developed a process to systematically confine cells using micropillar arrays. Using photolithography and polydimethylsiloxane (PDMS) molding, we created PDMS arrays of micropillars that were 5, 10, 20, or 50 μm in spacing and between 13 and 17 μm in height. The tops of micropillars were coated with Pluronic F127 to inhibit cell adhesion, and we observed that mesenchymal stem cells (MSCs) robustly infiltrated into the micropillar arrays. MSC and nucleus morphology were altered by narrowing the micropillar spacing, and cytoskeletal elements within MSCs appeared to become more diffuse with increasing confinement. Specifically, MSCs exhibited a ring of actin around their periphery and punctate focal adhesions. MSC migration speed was reduced by narrowing micropillar spacing, and distinct migration behaviors of MSCs emerged in the presence of micropillars. MSCs continued to proliferate within micropillar arrays after 3 weeks in culture, displaying our assay's capability for long-term studies. Our assay also has the capacity to provide adequate cell numbers for quantitative assays to investigate the effect of confinement on gene and protein expression. Through deeper understanding of cell mechanotransduction in the context of confinement, we can modify tissue-engineered constructs to be optimal for a given purpose.
Impact Statement
In this study, we developed a novel process to systematically confine cells using micropillar arrays. Our assay provides insight into cell behavior in response to mechanical confinement. Through deeper understanding of how cells sense and respond to confinement, we can fine tune tissue-engineered constructs to be optimal for a given purpose. By combining confinement with other physical cues, we can harness mechanical properties to encourage or inhibit cell migration, direct cells down a particular lineage, induce cell secretion of specific cytokines or extracellular vesicles, and ultimately direct cells to behave in a way conducive to tissue engineering.
Keywords: confinement, mechanobiology, mesenchymal stem cells
Introduction
While biological cues have long been recognized as important regulators of cell behavior, mounting evidence indicates that mechanical cues also have critical influences on cell migration, division, differentiation, and other essential cell processes.1–3 Understanding and capitalizing on the effects of these mechanical cues can promote the successful outcome of tissue engineering strategies for repair and regeneration of tissues. Relevant external mechanical cues may include stiffness, topography, shear stress, viscoelasticity, and, as we discuss specifically herein, confinement. The field is recognizing that a variety of cell types, including fibroblasts, cancer cells, and epithelial cells, undergo migrational plasticity and enlist different mechanisms for confined migration than for unconfined migration.4–6 For example, we have shown that confinement reduces the influence of microtubule polymerization on mesenchymal stem cell (MSC) migration.3
Intravital imaging has revealed that cells indeed experience physical confinement in vivo.7,8 In addition, cells integrated into tissue-engineered scaffolds likely experience physical confinement depending on the porosity and organization of the scaffold at the microscale. Three-dimensional cellular assays are becoming increasingly popular, but many laboratories also have distinct methods and devices to study cell behavior in confinement. One approach uses micropatterned lines of a particular adhesive protein or grooves, creating 1D tracks upon which cells can migrate.9–12 This technique is advantageous due to its simplicity of production, ease of live cell imaging, and potential for single-cell studies. While 1D patterning techniques can be similar to the migration of cells along extracellular matrix protein “tracks” in vivo, they do not account for the compression of the cell body and nucleus that a cell may experience in 3D in vivo microenvironments.13
To mimic in vivo cell compression, systems have been designed to confine cells vertically, in which cell migration can be markedly different than on a 2D substrate.14–16 However, in this method, cells are limited to only one axis of confinement.16 Boyden chambers are also often used for studying the effects of 3D confined migration on cell behavior.17 While Boyden chambers are a useful tool for postconfinement readouts, they do not allow for easy live visualization of cells within the chamber and do not easily allow for long-term culture in confinement.18 Hydrogels are also commonly used to confine cells in a 3D microenvironment.19,20 However, hydrogels can lack precise control over the degree of 3D confinement experienced by the cells, and it is difficult to image and accurately track cells in 3D hydrogels over time.
To address these shortcomings, we and others have analyzed cell migration through confining microchannels of various widths.21–25 While this approach offers precise control over the degree of confinement experienced and ease of imaging, it provides a relatively small sample size that is inadequate for proteomic or genetic analysis. In addition, microfluidic devices often require the introduction of a chemotactic gradient to encourage migration into small channels,21,22 which may or may not be physiologically relevant for a given tissue engineering strategy.
In this study, we have developed a novel micropillar confinement assay that allows for precise control over the degree of confinement experienced by cells, enables visualization of cells in real-time (on the order of weeks), and provides a large sample size for downstream biological assays. Our data show that MSCs alter their cell and nuclear morphology in response to confinement induced by micropillars. Furthermore, it appears that MSCs may alter their migration mode based on the degree of confinement experienced or by the mere existence of micropillars. Overall, this micropillar assay will provide new fundamental information about cellular migration and mechanobiology in response to physical confinement.
Materials and Methods
Cell culture and reagents
Bone marrow-derived human MSCs (Donor 1: 20-year-old female, Donor 2: 22-year-old male) were purchased from RoosterBio, Inc. (Frederick, MD). Experiments were performed with Donor 1 unless otherwise noted in the figure caption.
Cells were removed from liquid nitrogen and grown in RoosterBio basal media with media booster (RoosterBio, Inc.) for the first day post-thaw. Thereafter, cells were cultured in medium composed of Dulbecco's modified Eagle's medium with high glucose (ThermoFisher Scientific, Waltham, MA), 10% fetal bovine serum (FBS) (ThermoFisher Scientific), and 1% penicillin–streptomycin 10,000 U/mL (ThermoFisher Scientific). Cells were cultured and used until a population doubling level of 20 and cells were passaged at or below 80% confluency. Cells were washed with phosphate-buffered saline (PBS) (VWR, Radnor, PA), and detached with TrypLE Express Enzyme (ThermoFisher Scientific). All cells were cultured at 37°C, 50% humidity, and 5% CO2:95% air.
Micropillar device fabrication
A polydimethylsiloxane (PDMS) micropillar device with micropillars of different spacing (Fig. 1A) was prepared via photolithography, as previously described.21,22 All photolithography procedures were carried out in the University of Maryland Nanocenter FabLab. In brief, a mask was made in AutoCAD (AutoDesk, San Rafael, CA) to represent the micropillars of different spacings. A layer of SU-8 3010 negative photoresist (MicroChem, Westborough, MA) was spin coated onto a 4-inch diameter silicon wafer (University Wafer, Boston, MA). Using an EVG620 mask aligner (EVG Group, Albany, NY), the mask in the image of micropillars was placed over the wafer and exposed to ultraviolet (UV) light to crosslink. Excess SU-8 3010 was dissolved using SU-8 developer (MicroChem). Finished wafers were silanized using tridecafluoro-1,1,2,2,tetrahydrooctyl-1-trichlorosilane (OTS, 97%) (UCT, Inc., Bristol, PA) overnight in a vacuum desiccator.
FIG. 1.

Micropillar preparation. (A) Design of micropillar arrays. (B) Representative workflow of micropillar array preparation.
The resulting silicon master contained micropillars with 50, 20, 10, and 5 μm spacing and 13–17 μm height. These distances were chosen because they are representative of the dimensions of in vivo tracks and in vitro scaffold pores.26 PDMS micropillar arrays were manufactured based on previously described protocols.16,27 PDMS (Krayden, Denver, CO) was mixed at a 10:1 base:curing agent ratio, poured over the silicon master, and degassed in a vacuum desiccator. The PDMS and master were baked for at least 10 min at 100°C, and then the PDMS mold was cut and removed from the silicon master. The PDMS mold was then placed in a plasma cleaner (Harrick Plasma, Ithaca, NY) and plasma treated for 2.5 min. Finished molds were silanized using OTS (97%) overnight in a vacuum desiccator.
PDMS was mixed at a 10:1 base:curing agent ratio, poured over the PDMS mold, and degassed in a vacuum desiccator. Glass coverslips (thickness #1; Fisher Scientific, Fair Lawn, NJ) were then applied to each set of micropillars while applying gentle pressure. The PDMS and coverslips were baked overnight at 80°C, then the coverslip-PDMS micropillar constructs were carefully removed from the PDMS mold. The PDMS-coverslip construct ensured that the base PDMS layer was sufficiently thin for microscopy, but that cell would sense PDMS on all sides.
PDMS blocks were coated in 8% Pluronic F127 (Sigma-Aldrich, St. Louis, MO) solution for 1 h at room temperature, then washed with DI water. Meanwhile, micropillar arrays were plasma treated for 2.5 min. Micropillar arrays were subsequently stamped with the Pluronic F127-coated PDMS blocks, and placed into six-well plates. The plates were UV sterilized for 10 min; 20 μg/mL collagen I (Sigma-Aldrich) was added to all wells and incubated for at least 1 h at 37°C. The collagen I solution was then removed, and devices were washed twice with PBS before seeding cells at a density of 5 × 104 cells/well (Fig. 1B). Cell medium was changed every 2–3 days.
Scanning electron microscopy
All SEM images were acquired in the University of Maryland Nanocenter AimLab by Dr. Sz-Chian Liou. SEM images were acquired using Hitachi SU-70 FEG SEM to visualize pillar spacing and Tescan XEIA FEG SEM/FIB to visualize pillar height. Pillar spacing and diameter were measured in ImageJ, and height was measured using Tescan microscope software.
Immunofluorescence
The following steps were carried out at room temperature, unless otherwise specified. Cells were fixed in 3.7% formaldehyde (Fisher Scientific) for 10 min, and then washed two to three times in PBS (VWR). Cells were permeabilized with 0.5% Triton-X 100 (Sigma-Aldrich) for 5 min, washed in PBS two to three times, and blocked for nonspecific binding in 2.5% goat serum (Abcam, Cambridge, MA) for at least 1 h. Primary antibody in 1% goat serum was added to cells and incubated at 4°C overnight. Primary antibodies and dilutions used were mouse anti-α-tubulin (ThermoFisher Scientific #A11126; 1:100) and rabbit anti-phospho-paxillin (Cell Signaling Technology #2541S, Danvers, MA; 1:100).
Cells were washed two to three times in PBS, incubated in 2.5% goat serum for at least 1 h, then incubated with AlexaFluor 488 Phalloidin (ThermoFisher Scientific; 1:500), Hoechst (ThermoFisher Scientific; 1:2500), and a fluorescently labeled secondary antibody for 1 h. Secondary antibodies and dilutions used were AlexaFluor 568 goat anti-mouse (ThermoFisher Scientific #A11004; 1:200) and AlexaFluor 568 goat anti-rabbit (ThermoFisher Scientific #A11011; 1:200). Cells were washed two to three times in PBS and then imaged.
Images were acquired on an Olympus IX83 microscope (Olympus) using a 20 × objective for cell morphology measurements or using a 60 × objective for high magnification images. Images were acquired on a PerkinElmer UltraVIEW Vox confocal spinning disk microscope (PerkinElmer, Waltham, MA) using a 20 × objective to obtain z-stacks and to visualize actin, nucleus, microtubules, and focal adhesions. Use of the PerkinElmer confocal microscope was performed courtesy of the University of Maryland imaging core.
Migration experiments
Cells were seeded onto micropillar arrays and allowed to incubate overnight. The following day, medium was changed and imaging began thereafter. Images were acquired on an Olympus IX83 microscope (Olympus) using a 20 × objective. A chamber adjusted to 37°C, 50% humidity, and 5% CO2:95% air was used on the microscope stage to sustain cell viability. Images were taken at 10 min intervals overnight.
Data analysis
For cell density, images of cell nuclei were thresholded and analyzed with the ImageJ Particle Analyzer. Cell density for each image was calculated by dividing the number of nuclei in an image by the total image area. For cell morphology, cells and nuclei were manually traced in ImageJ. Area, circularity, solidity, roundness, major axis, and minor axis were extracted using ImageJ built in morphology measurements. Roundness is equated to the inverse aspect ratio. Solidity was defined as the cell area divided by the convex area.
To track cell migration in ImageJ, the cell centroid was manually identified for each frame. Only cells that stayed within frame for the duration of the time lapse were tracked. A custom MATLAB program was implemented to calculate the speed, trajectory, mean squared displacement (MSD), and turning angle for each cell based on the position of the cell centroid. To calculate cell migration parameters every 30 min, we considered the position of the cell centroid every third frame since images were originally captured every 10 min. Skewness of turning angle distributions was calculated in GraphPad.
Statistical analysis
Data for cells from at least two independent trials were pooled for statistical analysis. A one-way ANOVA with Holm–Sidak's multiple comparisons test, in the case of normally distributed data, or Kruskal–Wallis test with Dunn's multiple comparisons test, in the case of nonnormally distributed data, was performed to determine differences in data from micropillar arrays of different spacing and differences between time points. A significance level of 0.05 was used. Error bars report the standard error of the mean (SEM) or standard deviation (SD), as indicated in figure or table captions.
Results
MSCs completely infiltrate into micropillar arrays
To entrap cells into the micropillar arrays, we aimed to encourage cell infiltration between pillars via cell attachment to the sides of the micropillars and migration on the cells' own accord. The tops of the pillars were stamped with Pluronic F127, a common surfactant used to minimize cell adhesion, using a microcontact printing method described previously.27 The remaining surface area was coated in collagen type I to enable cell attachment to the micropillar sides and base (Fig. 1B).
We next investigated whether cells were completely entrapped within the pillars (for a full confinement effect), or whether they protruded above the pillar height (for only partial confinement). Our goal was for cells to only “feel” the basal PDMS plane and the walls of the micropillars. When posts were shorter, with a height of ∼11 μm, parts of the cell body protruded above the micropillars, and the cell appeared to extend over the tops of pillars in spots (Fig. 2A). Hence, we fabricated 14 μm tall pillars and confirmed that these arrays fully confined the cell body and nucleus below the pillar tops (Fig. 2B).
FIG. 2.

Micropillar specifications selection. Confocal images of MSCs within (A) 11 μm tall micropillars or (B) 14 μm tall micropillars. For both panels (i) shows the XY plane, (ii) shows the YZ plane, and (iii) shows the XZ plane. White arrows indicate cell regions atop the micropillars. Cells were fixed and stained for actin (green) and the nucleus (blue). Scale bars represents 50 μm.
Next, we verified the dimensions of the 14 μm tall micropillars using scanning electron microscopy (SEM, Fig. 3). Micropillars with spacing of 5, 10, 20, and 50 μm were indeed measured to be approximately the theorized spacing, and the micropillar diameter was ∼9 μm for all micropillar spacings (Fig. 3A–D and Table 1). Micropillars with spacing of 5, 10, 20, and 50 μm were ∼13, 14, 13, and 17 μm tall, respectively, as measured by SEM (Fig. 3E–H and Table 1). We speculate that this height discrepancy could be minimized if each micropillar array was manufactured on its own silicon wafer, such that each array has its own optimized parameters during photolithography. However, because we achieved micropillar arrays that fully confined cells without their protruding onto the micropillar tops, we moved forward with this fabrication technique.
FIG. 3.

Micropillar specifications validation. SEM images of micropillar array distribution with spacings of (A) 5 μm, (B) 10 μm, (C) 20 μm, and (D) 50 μm. SEM images of micropillar array height with spacings of (E) 5 μm, (F) 10 μm, (G) 20 μm, and (H) 50 μm. Quantification of pillar spacing and dimensions are shown in Table 1.
Table 1.
Measurements of SEM Images
| Pillar spacing (theorized) | Pillar spacing (mean ± SD) | Pillar diameter (mean ± SD) | Pillar height (mean ± SD) |
|---|---|---|---|
| 5 μm | 5.861 ± 0.21 μm | 9.160 ± 0.16 μm | 12.82 ± 0.29 μm |
| 10 μm | 10.95 ± 0.30 μm | 8.622 ± 0.19 μm | 14.27 ± 0.21 μm |
| 20 μm | 20.35 ± 0.25 μm | 9.217 ± 0.25 μm | 13.26 ± 0.20 μm |
| 50 μm | 51.57 ± 0.60 μm | 9.011 ± 0.51 μm | 16.94 ± 0.92 μm |
Cell density is consistent across micropillar spacings in short-term culture
After cells were seeded atop the micropillar arrays and incubated overnight, cells appeared to have infiltrated between the micropillars. A change in cell culture medium removed any cells remaining in suspension, but did not dislodge most cells within pillars. Cells remained within the micropillars after 48 h, ensuring cell infiltration was not transient and could withstand media changes. Despite this, we wanted to ensure that cell density was fairly constant between micropillar arrays and for cells from multiple MSC donors.
We quantified cell density within the micropillars at both 24 and 48 h post-seeding. For Donor 1 cells, at 24 h, there was no difference in the cell density between micropillar arrays (Fig. 4A). However, for Donor 1 cells, by 48 h, there were less cells within 50 μm spaced pillars than any other pillar spacing (Fig. 4B). To investigate whether this observation was donor-specific, we seeded MSCs from a second donor (Donor 2) within micropillar arrays. There was no difference in cell density between micropillar spacings at 24 or 48 h, suggesting that cell seeding density may need to be optimized for each MSC donor and carefully considered when interpreting biological outcomes from the experiments (Fig. 4C, D).
FIG. 4.

MSC density within micropillars. Donor 1 MSC density within micropillars for (A) 24 h or (B) 48 h. Donor 2 MSC density within micropillars for (C) 24 h or (D) 48 h. Dot plots report mean ± SD. n.s. = not significant. *p < 0.05, ****p < 0.0001. Each dot indicates one analyzed image, pooled from N = 2 independent experiments. MSC, mesenchymal stem cell.
Cell and nucleus morphology within micropillars is altered by micropillar spacing
Fluorescence images of cell actin demonstrated that micropillar spacing drastically affected cell and nucleus morphology (Fig. 5A–D). Qualitatively, we observed cell spreading behaviors typical of 2D substrates in the 50 μm spaced pillars, while cells in the narrow spacings displayed much more spindly, protrusive morphologies with most cells physically wrapping around micropillars.
FIG. 5.

MSC morphology within micropillars. MSC actin cytoskeleton and nucleus after 48 h in micropillar arrays with spacings of (A) 5 μm, (B) 10 μm, (C) 20 μm, or (D) 50 μm. In (A–D), cells were fixed and stained for actin (green) and the nucleus (blue). (E) Area, (F) solidity, (G) roundness, and (H) circularity of MSC body as a function of pillar spacing after 24 and 48 h within the micropillars. Dot plots report mean ± SEM. *p < 0.05, **p < 0.005, ***p < 0.0005, and ****p < 0.0001. Each dot indicates one cell, pooled from N = 2 independent experiments with n (5 μm, 24 h) = 56, n (5 μm, 48 h) = 94, n (10 μm, 24 h) = 45, n (10 μm, 48 h) = 78, n (20 μm, 24 h) = 47, n (20 μm, 48 h) = 66, n (50 μm, 24 h) = 35, and n (50 μm, 48 h) = 42.
At both 24 and 48 h, cells within arrays of 20 and 50 μm spaced pillars were significantly larger in area than those in arrays of 5 μm spaced pillars (Fig. 5E). At both time points, cells within arrays of 5 μm spaced pillars were significantly less solid than cells in any other micropillar array and significantly less circular than cells within 20 μm arrays (Fig. 5F, H), which was expected given their spindly, protrusive morphology. There was no significant difference in any cell morphology measure within any pillar spacing from 24 to 48 h (Fig. 5E–H).
Cell nuclei followed a slightly more consistent trend. Nucleus morphology was measured as above, and we additionally measured the major and minor axis. Major and minor axes indicate the longest and shortest axes, respectively, of an ellipse fit to the nucleus (Fig. 6A, B). In pillar arrays of 5 μm spacing, the fitted ellipse was typically larger than the nucleus (Fig. 6A). In pillar arrays of 10 μm spacing or wider, the nuclei were naturally ellipsoidal (Fig. 6B). At both 24 and 48 h, cell nuclei within pillar arrays of 5 μm spacing were significantly smaller in 2D area, less solid, less round, and less circular than cell nuclei in any other array (Fig. 6C–F). Interestingly, there was no significant difference in the nucleus major axis among micropillars of different spacing, but the nucleus minor axis was significantly smallest in arrays with 5 μm pillar spacing (Fig. 6G, H). Future studies should examine if cell nuclei alter their volume in response to confinement.
FIG. 6.

MSC nucleus morphology within micropillars. MSC nucleus in micropillar arrays with spacings of (A) 5 μm and (B) 10 μm. In (A, B), cells were fixed and stained for actin (green) and the nucleus (blue). (C) Area, (D) solidity, (E) roundness, (F) circularity, (G) major axis, and (H) minor axis of MSC nucleus as a function of pillar spacing after 24 and 48 h within the micropillars. Dot plots report mean ± SEM. *p < 0.05, **p < 0.005, ***p < 0.0005, and ****p < 0.0001. Each dot indicates one cell, pooled from N = 2 independent experiments with n (5 μm, 24 h) = 56, n (5 μm, 48 h) = 94, n (10 μm, 24 h) = 45, n (10 μm, 48 h) = 78, n (20 μm, 24 h) = 47, n (20 μm, 48 h) = 66, n (50 μm, 24 h) = 35, and n (50 μm, 48 h) = 42.
We have also demonstrated that cells can remain viable and integrated within the micropillar confinement array for at least 3 weeks, and likely longer. When MSCs were cultured in micropillars for 3 weeks, they formed elongated networks within the narrowest micropillar arrays (Fig. 7A, B). MSCs cultured within the widest micropillar arrays for 3 weeks were similar in appearance to a two-dimensional monolayer (Fig. 7C, D). In addition to network formation, some cells began to protrude over the pillar tops (Fig. 7A). There were fewer cells within pillar arrays of 5 μm than those of 20 or 50 μm; thus, quantitative biological readouts may need to be normalized to cell density (Fig. 7E). Such long-term culture and high cell counts may allow for the examination of cell response to confinement in situations relevant for tissue engineering applications, such as the changing secretome or differentiation status of the cells.
FIG. 7.

MSCs within micropillars for 3 weeks. MSCs in micropillar arrays with spacings of (A) 5 μm, (B) 10 μm, (C) 20 μm, or (D) 50 μm. (E) MSC density after 3 weeks. In (A–D), cells were fixed and stained for actin (green) and the nucleus (blue). Scale bars represent 50 μm. In (E) dot plots report mean ± SEM. **p < 0.005. Each dot indicates one analyzed image.
MSC cytoskeletal elements become more diffuse with increasing confinement
We next investigated whether the changes in cellular morphology as a function of micropillar array spacing were accompanied by alterations in organization of the cell cytoskeleton. Within all micropillar arrays, regardless of pillar spacing, some cells exhibited F-actin-rich protrusions wrapping entirely around the pillars (Fig. 8).
FIG. 8.

MSC cytoskeleton within micropillars. Basal plane of MSC cytoskeleton in micropillar arrays with spacing of (A) 5 μm, (B) 10 μm, (C) 20 μm, and (D) 50 μm. Maximum projection of MSC cytoskeleton in micropillar arrays with spacing of (E) 5 μm, (F) 10 μm, (G) 20 μm, and (H) 50 μm. In (A–D), cells were fixed and stained for py-paxillin (red), actin (green), and the nucleus (blue). Areas of strong signal are indicated by white arrows. In (E–H), cells were fixed and stained for 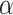 -tubulin (red), actin (green), and the nucleus (blue). Color channels were altered individually for optimal visualization. Scale bars represent 10 μm.
-tubulin (red), actin (green), and the nucleus (blue). Color channels were altered individually for optimal visualization. Scale bars represent 10 μm.
To determine whether cells were adhering to the pillars via focal adhesions, we immunostained for tyrosine-phosphorylated (py-) paxillin, a protein associated with focal adhesions. Indeed, we observed areas of high py-paxillin signal associated with the micropillars (Fig. 8A, B). MSCs within 50 μm spaced micropillars exhibited many linear, mature focal adhesions at the end of F-actin stress fibers, indicative of highly contractile cells (Fig. 8D). These linear, mature focal adhesions decreased with each subsequent decrease in pillar spacing (Fig. 8B, C). MSCs within 5 μm spaced micropillars exhibited mostly punctate py-paxillin dispersed through the cell (Fig. 8A). These results are consistent with our own previous work in microchannels.3
We also examined how microtubules were arranged within MSCs via immunostaining of α-tubulin. Microtubules were fairly well localized to regions of F-actin in confined MSCs within 5 and 10 μm spaced micropillars (Fig. 8E, F). MSCs within 20 and 50 μm spaced micropillar arrays exhibited the strongest microtubule signal surrounding the cell nucleus, while F-actin mainly resided along the cell perimeter (Fig. 8G, H). Similar to F-actin, microtubules also wrapped around individual micropillars. Together, these results provide further confirmation that the cell cytoskeleton and adherence patterns can be altered by physical confinement, which we also hypothesized could result in cell phenotypes with altered functionality in behaviors, such as cell migration, relevant to tissue engineering strategies.
MSCs migrate within micropillar arrays
Our next goal was to use phase contrast imaging to confirm that cells were capable of robust migration within the micropillars (Fig. 9). Cells displayed several interesting behaviors when migrating within the micropillars. In arrays where pillars were 5 μm or 10 μm apart, cells remained mostly elongated. MSCs typically approached a pillar, probed around both sides, and “chose” one of those two sides to migrate around (Fig. 9A). Occasionally, cells appeared to engulf a pillar entirely to migrate around it.
FIG. 9.

MSC migration phenotypes within micropillars. Representative image sequences of (A) MSC migrating in confined 5 μm pillar spacing, (B) MSC migrating in a mesenchymal mode within wide 50 μm pillar spacing, (C) MSC migrating in amoeboidal mode within wide 50 μm pillar spacing, and (D) MSC attaching to a micropillar within wide 50 μm pillar spacing (MSC of interest denoted by white arrow). Scale bars represent 25 μm.
In arrays where pillars were 20 μm or 50 μm apart, different migration patterns emerged. The cells often migrated in a mesenchymal mode, like their behavior in 2D (Fig. 9B). However, cells also migrated in a somewhat amoeboidal mode, characterized by alternating periods of fast, directional migration and slower, more random migration (Fig. 9C). Occasionally, another mode appeared where cells attached to a pillar, clung to it for some time, and then rapidly migrated to another pillar where it attached and rounded up yet again (Fig. 9D). These observations of different migration modes encouraged us to further analyze MSC migration within the micropillar arrays.
To investigate cell migration, we manually tracked each cell's movement every 10 min or every 30 min. We hypothesized that cell migration parameters, including speed and turning angle distribution, would depend on time interval chosen for analysis, where, at short timescales, cells may be more persistent, but turn more or measure slower speeds when undergoing random migration. Qualitatively, MSC trajectories within the 5 μm spaced micropillars appeared to migrate with more “straight” paths, presumably due to guidance by the constricting pillars (Fig. 10A, B). As pillar spacing was increased, cell motion appeared more random (Fig. 10A, B).
FIG. 10.

Quantitative MSC migration parameters within micropillars. Trajectories of MSCs within 5, 10, 20, or 50 μm micropillar arrays tracked every (A) 10 min or (B) 30 min. Histogram displaying relative frequencies of turning angles for cells tracked every 10 min within micropillars, and considered from (C) 0–180° where 0° corresponds to no deviation in migration path from previous time step and 180° corresponds to a complete reversal of direction; or (D) 0 to 90° where there is no distinction between forward and backward motion. Histogram displaying relative frequencies of turning angles for cells tracked every 30 min within micropillars, and considered from (E) 0–180° or (F) 0 to 90°. (G) Skewness of histograms in (C–F). Speed of MSCs as a function of pillar spacing tracked every (H) 10 min or (I) 30 min. MSD as a function of time for cells within micropillars spacing tracked every (J) 10 min or (K) 30 min. (C–K) display data for 5 μm (green), 10 μm (blue), 20 μm (red), or 50 μm (cyan). Dot plots in (H, I) report mean ± SEM, and each dot represents one cell. n.s. = not significant, *p < 0.05. Cells pooled from N = 4 independent experiments with n (5 μm) = 25, n (10 μm) = 30, n (20 μm) = 32, and n (50 μm) = 21.
We quantified the turning angle of each cell for each time point as it migrated and plotted the relative frequency of turning angles (Fig. 10C–F). The turning angle represents the angle between a cell's previous trajectory and its current trajectory. We observed that most turning angles were close to 0°, and there was a slight increase in the frequency of angles very close to 180° for cells in 5 μm micropillars (Fig. 10C). In this definition, a turning angle of 0° indicates a cell continued moving forward in a straight line, while a turning angle of 180° indicates a cell completely reversed its direction. However, we also considered straightness of cell migration without considering directionality. In this definition, we considered turning angles from 0° to 90°, by subtracting the angle from 180° if it was over 90° (Fig. 10D). For example, a previous turning angle of 175° (nearly complete reversal of migration direction) would now be represented as 5°. In this case, turning angles within all micropillar spacings were most frequently close to 0°, with 5 μm being the most extreme case (Fig. 10D).
Interestingly, different trends emerged when we analyzed cell turning angles using 30-min time steps instead of 10-min time steps (Fig. 10E-F). Specifically, we found an increase in the distribution of turning angles close to 180° (complete reversal of direction), especially for the 5 μm pillar spacing. We subsequently quantified the skewness of each histogram and found that, when considering turning angles from 0° to 90°, there was an increase in skewness with increasing confinement, which provided quantitative confirmation that more confined cells migrated in straighter paths than less confined cells (Fig. 10G).
MSCs migrated faster in 50 μm spaced pillars than 3 μm spaced pillars when tracked every 10 min, but there were no differences in speed when cells were tracked every 30 min (Fig. 10H, I). The MSD versus time of the MSCs appeared very similar for all micropillar spacings over short timescales (up to ∼200 min), but began to diverge (with no monotonic trend) between micropillar spacings at larger timescales (Fig. 10J, K). Therefore, physical confinement of MSCs in this micropillar assay affects not only cell morphology and cytoskeletal organization but also functional behaviors such as cell migration.
Discussion
We have demonstrated the potential of using a micropillar array for confining cells and studying their mechanobiology with a downstream goal of providing information that could help optimize cell culture systems for tissue engineering purposes. Our system was developed using information learned from many previous studies using micropillars in various applications. An excellent review of how micropillars have been used to evaluate cell behavior is found in Roca-Cusachs et al.28 In Table 1, we summarize studies where cells were cultured within,27,29,30 within and atop,31–34 or solely atop35–39 micropillar arrays.
Our system herein is distinct in that we use a wide range of micropillar spacings and show that cells can be maintained within micropillars over long periods of time (Table 2). In addition, cells infiltrate directly into the micropillar array, in contrast to other systems, in which cells migrate from 2D into micropillars.29,30 In fact, collagen-coated micropillar arrays can be placed within a noncell-adhesive plate to disallow cell attachment to any 2D surface. This allows one to investigate the effect of confinement on cell behavior by ensuring that no cells are attached to a 2D surface.
Table 2.
Micropillars Used in Literature
| Pillar size and spacing | Unique features | Cells | Functional assessment | Reference |
|---|---|---|---|---|
| Within pillars | ||||
| 10 μm diameter | Wide range of pillar spacings, long-term culture | human MSCs | Cell morphology, cytoskeletal arrangement, cell migration | The work herein |
| 14–16 μm height | ||||
| 5, 10, 20, 50 μm spacing | ||||
| 7.6 × 7.7 μm2 to 3.7 × 5.7 μm2 dimensions | Anisotropically stiff | human MSCs | Cell alignment | 27 |
| 15 μm height | ||||
| ∼3 μm spacing | ||||
| 10 μm diameter | Correlated invasiveness to nucleus mechanical properties | human MSCs | Invasiveness, persistence length | 29 |
| 20 μm height | ||||
| 8,10,12 μm spacing | ||||
| 10 μm diameter | Cells migrate from 2D into pillars | 3T3 fibroblasts, Hutchinson–Gilford Progeria Syndrome patient derived fibroblasts | Morphology, invasiveness, trajectory, MSD, diffusion coefficient | 30 |
| 20 μm height | ||||
| 6, 8, 12 μm spacing | ||||
| Atop and within pillars | ||||
| 5 μm diameter | Cells sit atop and deform pillars | REF52 fibroblasts | Displacement of pillars, force exerted on pillars during spreading | 31 |
| 20 μm height | ||||
| 4–12 μm spacing | ||||
| 3 μm side length | Examined effect of osteogenesis on nuclear deformation | rat MSCs, osteoblasts | Nuclear height, nuclear deformation | 32 |
| 7 μm height | ||||
| 6 μm spacing | ||||
| 2–15 μm side length | Compared pillar materials | osteosarcoma cells | Nuclear deformation | 33 |
| 6 μm height | ||||
| 2–20 μm spacing | ||||
| 5–15 μm side length | Compared pillars to grooves | rat MSCs | Proliferation, morphology, cytoskeletal elements | 34 |
| 5 μm height | ||||
| 5–15 μm spacing | ||||
| Atop pillars | ||||
| 3 μm diameter | Micropillars within microchannels | 3T3 fibroblasts, osteosarcoma cells | Traction forces | 35 |
| 9 μm height | ||||
| 6 μm spacing | ||||
| 3 μm diameter | Magnetic microposts to apply forces to cells | 3T3 fibroblasts | Traction forces, focal adhesion area | 36 |
| 10 μm height | ||||
| 9 μm spacing | ||||
| 1–2 μm diameter | Improved resolution | MDCK epithelial cells | Traction forces | 37 |
| 3–8 μm height | ||||
| 2–4 μm spacing | ||||
| 2–10 μm diameter | Origin of micropillars | Pulmonary artery smooth muscle cells, endothelial cells, 3T3 fibroblasts | Traction forces | 38 |
| 3–50 μm height | ||||
| 6 μm spacing | ||||
| 2–7 μm diameter | Stiffness gradient of pillars | C2C12 myoblasts, 3T3 fibroblasts, HeLa cells | Traction forces, cell migration | 39 |
| 12 μm height | ||||
| 10 μm spacing | ||||
An additional important feature of our system is the nondeformability of micropillars by cells. Our micropillar arrays are fabricated with a stiff polymer that has previously been measured to be ∼1.75 MPa.40 Even in the most extreme case, if cells were to reach the top of the pillar and exert a force perpendicular to the pillar, the bending stiffness, 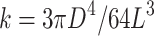 ,41 of the tallest pillar would be 629 nN/μm, assuming a pillar diameter (D) of 10 μm and a pillar height (L) of 16 μm. Cells typically exert traction forces through focal adhesions of ∼100–1000 nN.42 Therefore, the micropillars should be primarily nondeformable to the cells and induce the specified degree of confinement. Although the PDMS used here is quite stiff, it is similar to the stiffness of various tissues in vivo such as bone, cartilage, and ligament.43 To mimic other anatomical features, future studies may use gels of lower elastic modulus, thus enabling investigation into the coupled effects of confinement and stiffness.
,41 of the tallest pillar would be 629 nN/μm, assuming a pillar diameter (D) of 10 μm and a pillar height (L) of 16 μm. Cells typically exert traction forces through focal adhesions of ∼100–1000 nN.42 Therefore, the micropillars should be primarily nondeformable to the cells and induce the specified degree of confinement. Although the PDMS used here is quite stiff, it is similar to the stiffness of various tissues in vivo such as bone, cartilage, and ligament.43 To mimic other anatomical features, future studies may use gels of lower elastic modulus, thus enabling investigation into the coupled effects of confinement and stiffness.
Distinct morphological differences appeared between MSCs in 5 μm spaced pillars and wider spaced pillars. Morphology is an important indicator of MSC behavior, as morphology alone can be used to predict the long-term mineralization potential or the immunosuppressive capacity of MSCs.44,45 Future work may leverage freely available software such as CellProfiler to create high-throughput pipelines and analyze cells on a population level. Such high-throughput analysis may be applied to this micropillar confinement system to reveal subsets of distinct cells within a heterogeneous population or between different populations, such as different MSC donors.
Similar to what we saw within micropillar arrays, it has been shown previously that mesenchymal cells can spontaneously switch to an amoeboidal migration mode when they experience both vertical confinement and low adhesion.14 One of these amoeboidal migration modes is initiated by an external polarization signal, such as a piece of debris. In our case, the micropillars may act as an external polarizer, even though MSCs in this study attach well to the micropillars on three sides and show evidence of some focal adhesions. Similar to our migration assays, another group also observed cells alternate between fast, directed migration and slow, random migration when cells encountered micropillars.46
Furthermore, the cell and nuclear deformation and altered cytoskeletal organization that occur in the most confining micropillar arrays may lead to differences in cell growth, as we have shown for sarcoma cells,2 or cell fate, which is influenced by other mechanical cues that have similar impact on cell morphology.
Interestingly, we saw different trends in turning angles when cells were tracked every 10 min compared to when they were tracked every 30 min. When cell migration is tracked using a 10 min interval, the subsequent analysis is representative of changes in cell migration at short timescales. This potentially increases the histogram's skewness toward small turning angles, as cells make small probing motions. Increasing the time interval to 30 min could be more representative of cell migration over long time periods, as cells move through the micropillars. Regardless of the time interval, cells migrated straighter within more confined micropillar arrays, perhaps due to the high density of micropillars guiding their migration.
The migration patterns of MSCs we observed within micropillar arrays are in slight contrast to our previous reports of MSC behavior within microchannels.3 Herein, MSCs migrated faster in 50 μm spaced pillars than 5 μm spaced pillars when tracked every 10 min. However, when MSCs migrated through microchannels, cells within 50 μm wide channels migrated slower than or with similar speed to those in 6 μm or 10 μm wide channels, depending on MSC passage.3 We hypothesize that contact guidance has a larger influence on cell migration in microchannels than in micropillar arrays, as cells tend to follow the surface to which they are attached.47 Cells have more choice over their path within micropillar arrays, potentially slowing them in narrower spacings. Furthermore, our previous results in microchannels were in the presence of a chemotactic gradient, which in combination with contact guidance from the microchannel walls, may lead to altered trends in migration. Overall, different methods of confinement may induce different cell behaviors.
It was critical that our micropillar design yield adequate sample sizes for downstream quantitative assays to investigate the concentration of secreted factors as well as gene and protein expression. We have successfully performed an ELISA on MSC-conditioned media from micropillar arrays at various time points (data not shown). This is possible due to the capacity of our micropillar arrays to support long-term cell growth. Analysis of cell density after 3 weeks in micropillars showed that cells did indeed proliferate within the micropillar arrays, yet, there were significantly fewer cells within the 5 μm spaced micropillars. We speculate this may be due to there being less planar surface area for cell attachment and growth in the narrowest spaced micropillars. In addition, we have previously shown that increasing confinement (in microchannels) decreases the percentage of cells that successfully divide,2 which may explain cells being more sparse in 5 μm arrays.
Despite differences after 3 weeks in culture, the majority of seeded cells infiltrated into the micropillars yielding a sample size of ∼5 × 104 cells per micropillar array at 24 h. This was fairly consistent for two different MSC donors, but minor differences suggest that cell seeding parameters be optimized for each new cell type. This cell count could provide enough RNA to allow for qRT-PCR and analysis. However, an order of magnitude more cells are necessary for one western blot. This could be achieved by running multiple wells in parallel, increasing the area of the micropillar array or increasing the seeding density. However, we note that when the seeding density was increased to a very high number (5–10 × 105 cells per array), some groups of cells began to aggregate and did not infiltrate into the pillars (data not shown). The combination of our confinement assay with western blot or PCR will reveal downstream effects of mechanical confinement, making it an even more powerful tool for tissue engineering. Through deeper understanding of cell mechanotransduction in the context of confinement, we can fine tune tissue-engineered constructs to produce cell phenotypes that are optimal for a given purpose.
Conclusions
In summary, we have successfully developed and validated a PDMS micropillar array that systematically confines cells, allows for easy visualization and scale up, allows for long-term cell culture, and provides an adequate sample size for analysis of the genome, secretome, or proteome. Future modifications of the micropillar confinement assay, including fabrication of micropillar arrays with more narrow spacing and applying a top confining post to the micropillar array, would enable us to study the effects of physical confinement on cell behaviors in greater depth. This knowledge is critical to improve the design of cell-laden tissue-engineered constructs and in vivo cell therapies. By combining confinement with other physical cues, we can harness mechanical properties to encourage or inhibit cell migration, direct cells down a particular lineage, induce cell secretion of specific cytokines or extracellular vesicles, and ultimately direct cells to behave in a way conducive to tissue engineering.
Acknowledgments
The University of Maryland Imaging Core Facility is acknowledged for providing resources for confocal imaging. We acknowledge the support of the Maryland NanoCenter: its FabLab for providing photolithography resources and its AIMLab for providing SEM resources. Specifically, we thank Dr. Sz-Chian Liou for his assistance with sample preparation and acquisition of SEM images.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by a Burroughs Wellcome Career Award at the Scientific Interface (to KMS), the University of Maryland Research and Scholarship Award (to KMS), the University of Maryland Graduate School Summer Fellowship (to MTD), the Fischell Department of Bioengineering, and the University of Maryland.
Research reported in this publication was supported by the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Number F31HL145991. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health."
References
- 1. Engler A.J., Sen S., Sweeney H.L., and Discher D.E. Matrix elasticity directs stem cell lineage specification. Cell 126, 677, 2006 [DOI] [PubMed] [Google Scholar]
- 2. Moriarty R.A., and Stroka K.M. Physical confinement alters sarcoma cell cycle progression and division. Cell Cycle Taylor Francis 17, 2360, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doolin M.T., and Stroka K.M. Physical confinement alters cytoskeletal contributions towards human mesenchymal stem cell migration. Cytoskeleton 75, 103, 2018 [DOI] [PubMed] [Google Scholar]
- 4. Stroka K.M., Jiang H., Chen S.-H., et al. . Water permeation drives tumor cell migration in confined microenvironments. Cell 157, 611, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hung W.-C., Chen S.-H., Paul C.D., et al. . Distinct signaling mechanisms regulate migration in unconfined versus confined spaces. J Cell Biol 202, 807, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Petrie R.J., Koo H., and Yamada K.M. Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science (80-.) 345, 1062, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weigelin B., Bakker G.-J., and Friedl P. Intravital third harmonic generation microscopy of collective melanoma cell invasion. IntraVital 1, 32, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pittenger M.F., and Martin B.J. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res 95, 9, 2004 [DOI] [PubMed] [Google Scholar]
- 9. Chang S.S., Guo W.-H., Kim Y., and Wang Y.-L. Guidance of Cell Migration by Substrate Dimension. Biophys J 104, 313, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Doyle A.D., Wang F.W., Matsumoto K., and Yamada K.M. One-dimensional topography underlies three-dimensional fibrillar cell migration. J Cell Biol 184, 481, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim D., Han K., Gupta K., Kwon K.W., Suh K.-Y., and Levchenko A. Mechanosensitivity of fibroblast cell shape and movement to anisotropic substratum topography gradients. Biomaterials 30, 5433, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Teixeira A.I. Epithelial contact guidance on well-defined micro- and nanostructured substrates. J Cell Sci 116, 1881, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ray A., Lee O., Win Z., et al. . Anisotropic forces from spatially constrained focal adhesions mediate contact guidance directed cell migration. Nat Commun 8, 14923, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu Y.J., Le Berre M., Lautenschlaeger F., et al. . Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 160, 659, 2015 [DOI] [PubMed] [Google Scholar]
- 15. Le Berre M., Aubertin J., and Piel M. Fine control of nuclear confinement identifies a threshold deformation leading to lamina rupture and induction of specific genes. Integr Biol 4, 1406, 2012 [DOI] [PubMed] [Google Scholar]
- 16. Le Berre M., Zlotek-Zlotkiewicz E., Bonazzi D., Lautenschlaeger F., and Piel M. Methods for two-dimensional cell confinement. Methods Cell Biol 121, 213, 2014 [DOI] [PubMed] [Google Scholar]
- 17. Chen H.-C. Boyden Chamber Assay. Methods Mol Biol 294, 15, 2005 [DOI] [PubMed] [Google Scholar]
- 18. Hulkower K.I., and Herber R.L. Cell migration and invasion assays as tools for drug discovery. Pharmaceutics 3, 107, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Doyle A.D., Carvajal N., Jin A., Matsumoto K., and Yamada K.M. Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat Commun 6, 8720, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wolf K., te Lindert M., Krause M., et al. . Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol 201, 1069, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balzer E.M., Tong Z., Paul C.D., et al. . Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J 26, 4045, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tong Z., Balzer E.M., Dallas M.R., Hung W.-C., Stebe K.J., and Konstantopoulos K. Chemotaxis of Cell Populations through Confined Spaces at Single-Cell Resolution. Rao C V., editor. PLoS One 7, e29211, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koch B., Meyer A.K., Helbig L., et al. . Dimensionality of Rolled-up Nanomembranes Controls Neural Stem Cell Migration Mechanism. Nano Lett 15, 5530, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davidson P.M., Sliz J., Isermann P., Denais C., and Lammerding J. Design of a microfluidic device to quantify dynamic intra-nuclear deformation during cell migration through confining environments. Integr Biol 7, 1534, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Irimia D., and Toner M. Spontaneous migration of cancer cells under conditions of mechanical confinement. Integr Biol 1, 506, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wolf K., Alexander S., Schacht V., et al. . Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol 20, 931, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alapan Y., Younesi M., Akkus O., and Gurkan U.A. Cell-aligning substrates: anisotropically Stiff 3D micropillar niche induces extraordinary cell alignment and elongation. Adv Healthc Mater 5, 1833, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roca-Cusachs P., Conte V., and Trepat X. Quantifying forces in cell biology. Nat Cell Biol 19, 742, 2017 [DOI] [PubMed] [Google Scholar]
- 29. Spagnol S.T., Lin W.-C., Booth E.A., Ladoux B., Lazarus H.M., and Dahl K.N. Early passage dependence of mesenchymal stem cell mechanics influences cellular invasion and migration. Ann Biomed Eng 44, 2123, 2016 [DOI] [PubMed] [Google Scholar]
- 30. Booth-Gauthier E.A., Du V., Ghibaudo M., Rape A.D., Dahl K.N., and Ladoux B. Hutchinson-Gilford progeria syndrome alters nuclear shape and reduces cell motility in three dimensional model substrates. Integr Biol (United Kingdom) 5, 569, 2013 [DOI] [PubMed] [Google Scholar]
- 31. Ghibaudo M., Di Meglio J.M., Hersen P., and Ladoux B. Mechanics of cell spreading within 3D-micropatterned environments. Lab Chip 11, 805, 2011 [DOI] [PubMed] [Google Scholar]
- 32. Liu X., Liu R., Gu Y., and Ding J. Nonmonotonic self-deformation of cell nuclei on topological surfaces with micropillar array. ACS Appl Mater Interfaces 9, 18521, 2017 [DOI] [PubMed] [Google Scholar]
- 33. Badique F., Stamov D.R., Davidson P.M., et al. . Directing nuclear deformation on micropillared surfaces by substrate geometry and cytoskeleton organization. Biomaterials 34, 2991, 2013 [DOI] [PubMed] [Google Scholar]
- 34. Li Z., Gong Y., Sun S., et al. . Differential regulation of stiffness, topography, and dimension of substrates in rat mesenchymal stem cells. Biomaterials 34, 7616, 2013 [DOI] [PubMed] [Google Scholar]
- 35. Raman P.S., Paul C.D., Stroka K.M., and Konstantopoulos K. Probing cell traction forces in confined microenvironments. Lab Chip 13, 4599, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sniadecki N.J., Anguelouch A., Yang M.T., et al. . Magnetic microposts as an approach to apply forces to living cells. Proc Natl Acad Sci USA 104, 14553, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. du Roure O., Saez A., Buguin A., et al. . Force mapping in epithelial cell migration. Proc Natl Acad Sci USA 102, 2390, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tan J.L., Tien J., Pirone D.M., Gray D.S., Bhadriraju K., and Chen C.S. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci USA 100, 1484, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee S., Hong J., and Lee J. Cell motility regulation on a stepped micro pillar array device (SMPAD) with a discrete stiffness gradient. Soft Matter 12, 2325, 2016 [DOI] [PubMed] [Google Scholar]
- 40. Johnston I.D., McCluskey D.K., Tan C.K.L., and Tracey M.C. Mechanical characterization of bulk Sylgard 184 for microfluidics and microengineering. J Micromech Microeng 24, 035017, 2014 [Google Scholar]
- 41. Schoen I., Hu W., Klitzsch E., and Vogel V. Contribution of Substrate Warping to Pillar Deflection. Nano Lett 10, 1823, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fu J., Wang Y., Yang M.T., et al. . Mechanical regulation of cell function with geometrically modulated elastomeric substrates. Nat Methods 7, 733, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang T.W., Wu C.H., Liau J.J., and Cheng C.K. The effect of graft strength on knee laxity and the in-situ forces of grafts after posterior cruciate ligament reconstruction. IFMBE Proc 25, 809, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marklein R.A., Lo Surdo J.L., Bellayr I.H., Godil S.A., Puri R.K., and Bauer S.R. High content imaging of early morphological signatures predicts long term mineralization capacity of human mesenchymal stem cells upon osteogenic induction. Stem Cells 34, 935, 2016 [DOI] [PubMed] [Google Scholar]
- 45. Klinker M.W., Marklein R.A., Lo Surdo J.L., Wei C.-H., and Bauer S.R. Morphological features of IFN-γ–stimulated mesenchymal stromal cells predict overall immunosuppressive capacity. Proc Natl Acad Sci USA 114, E2598, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arcizet D., Capito S., Gorelashvili M., et al. . Contact-controlled amoeboid motility induces dynamic cell trapping in 3D-microstructured surfaces. Soft Matter 8, 1473, 2012 [Google Scholar]
- 47. Paul C.D., Shea D.J., Mahoney M.R., et al. . Interplay of the physical microenvironment, contact guidance, and intracellular signaling in cell decision making. FASEB J 30, 2161, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]