

Abstract
Background
The structural remodeling of atrial architecture, especially increased amounts of fibrosis, is a critical substrate to atrial fibrillation (AF). Doxycycline (Doxy) has recently been shown to exert protective effects against fibrogenic response. This study investigated whether doxycycline (Doxy) can sufficiently ameliorate the fibrosis-induced changes of atrial conduction and AF vulnerability in a chronic intermittent hypoxia (CIH) rat model.
Material/Methods
Sixty rats were randomized into 3 groups: Control, CIH, and CIH with Doxy treatment (DOXY) group. CIH rats were exposed to CIH (6 h/d) and Doxy-treated rats were treated with Doxy during processing CIH. After 6 weeks, echocardiographic and hemodynamic parameters were measured. Isolated atrial epicardial activation mapping and heart electrophysiology were performed. The extent of atrial interstitial fibrosis were estimated by Masson’s trichrome staining. The expression levels of TGF-β1 and downstream factors were determined by real-Time PCR, immunohistochemistry, and Western blot analysis.
Results
Compared to Control rats, the CIH rats showed significant atrial interstitial fibrosis, longer inter-atrial conduction time, and elevated conduction inhomogeneity and AF inducibility, and the expression of TGF-β1, TGF-βRI, TGF-βRII, P-Smad2/3, α-SMA, CTGF, and Collagen I were significantly increased, whereas the velocity of atrial conduction and the expression of miR-30c were dramatically decreased. All of these changes were significantly improved by Doxy treatment.
Conclusions
The findings suggested that Doxy can profoundly mitigate atrial fibrosis, conduction inhomogeneity as well as high AF inducibility secondary to fibrosis in a CIH rat model through suppressing the TGF-β1 signaling pathway.
MeSH Keywords: Atrial Fibrillation; Doxycycline; Receptors, Transforming Growth Factor beta
Background
Atrial fibrillation (AF), the most common sustained arrhythmia, has become one of the severe challenges in the field of global cardiovascular diseases, and can lead to many clinical complications such as stroke and heart failure [1]. Unfortunately, there is still no a comprehensive and clear understanding of the specific pathogenesis of AF. Clinical and experimental evidence suggest that fibrosis activates reentry circuits in the atrium by increasing inhomogeneous electrical conduction, which is considered to be a crucial contributor to AF [2–4].
Although the underlying pathophysiological mechanisms leading to cardiac fibrosis are complex, transforming growth factor-β1 (TGF-β1) has a close correlation with AF [1,5]. In a variety of organ systems, such as lung, liver, and heart, TGF-β1 and its related factors are critical inflammatory, pro-fibrotic cytokines that contribute to the production of extracellular matrix, and the overproduction of extracellular matrix in atrial tissues indicates the formation of structural reconstruction [6,7]. Indeed, the majority of experimental studies in animals have demonstrated that upregulated expression of TGF-β1 can lead to atrial-specific fibrosis and increase AF vulnerability [7,8]. TGF-β1 regulates its effects via interactions of transmembrane type I receptor (TGF-βRI) and type II receptor (TGF-βRII). Once bound to TGF-β1, TGF-βRII recruits and phosphorylates TGF-βRI, and this triggers phosphorylation of Smads, which can then manipulate the expression of fibrosis marker proteins, including CTGF, α-SMA, and Collagen I [10–12]. A previous study [13] reported that miR-30c, an microRNA participating in the anti-fibrotic process, is a downstream factor of TGF-β1, and found that TGF-β1 can suppress miR-30c level, which in turn negatively regulates the expression of its target gene TGF-βRII and thereby amplifies the biological effects of the TGF-β1 signaling pathway.
Doxycycline (Doxy) is a long-acting semisynthetic tetracycline antibiotic clinically used for several decades to treat infectious diseases [14]. Besides its well-known anti-inflammatory properties, Doxy has been found to improve the fibrogenic response. Hua et al. [15] reported that Doxy can markedly improve pulmonary interstitial fibrosis induced by paraquat via inhibiting the TGF-β1 signal transduction pathway.
Recently, chronic intermittent hypoxia (CIH) has been frequently used to explore the pathogenesis of AF because of its role in causing atrial interstitial fibrosis [16,17]. It has been unclear whether Doxy can also attenuate CIH-induced atrial interstitial fibrosis, atrial conduction, and AF susceptibility by interfering with TGF-β1 signaling pathway; therefore, we performed the present study to test this hypothesis by establishing a CIH-induced AF rat model.
Material and Methods
Experimental animals and protocol
This study was approved by the Experimental Animal Administration Committee of Tianjin Medical University.
Sixty Sprague-Dawley male rats (8 weeks old, weight 180–200 g), purchased from the BEIJING HFK BIOSCIENCE CO. (Beijing, China). Rats in the DOXY group were administered Doxy (30 mg/kg/d, according to animal pharmacology data) by oral gavage prior to exposure to CIH (6 h/d) for 6 consecutive weeks.
During the exposure periods, CIH and Doxy-treated rats were placed inside a plexiglas chamber connected to supplies of pure O2 and N2, which was applied to create the intermittent hypoxia condition. In the CIH protocol, the O2 concentration in the chamber was gradually reduced from 21% to 8% by flushing with 100% nitrogen for 160 s, held at 8% for 50 s, and then returned to 21% by flushing with O2 for 90 s. Each complete cycle lasted approximately 300 s, and the O2 concentration inside the chamber was alternately switched between 8% and 21%. Within 24 h after the last exposure, from each group, 10 rats were randomized to perform experiments including echocardiography, hemodynamic examination, and histological and molecular biological studies. The remaining 30 rats underwent isolated atrial epicardial activation mapping and heart electrophysiology.
Echocardiographic assessment
After 6 weeks of CIH, rats were anesthetized with isoflurane (1 L/min) and transthoracic echocardiography was performed. The interventricular septum thickness, left atrial diameter, left ventricular end-diastolic and end-systolic diameters, mean pulmonary artery pressure, and left ventricular ejection fraction were determined.
Hemodynamic assessment and sample collection
After echocardiography, a Millar pressure cannula was carefully inserted into the right carotid artery to record the aortic systolic and diastolic blood pressure of rats. Subsequently, the rats were instantly euthanized and left atrial (LA) tissues were rapidly separated. Samples for histological assessment were soaked in neutral buffered 10% formalin solution, and samples for molecular biological experiments were quickly stored in a liquid nitrogen tank.
Histological studies
5-micron tissue sections were collected and corresponding staining was performed. Masson’s trichrome staining was used to estimate the extent of interstitial fibrosis. The left atrial interstitial collagen volume fraction (LACVF) was calculated as [blue area/(red area+blue area)]×100%. Immunohistochemistry staining was applied to identify the protein expression levels of TGF-β1 and related downstream factors, with the mean optical density (MOD) was defined as integral optical density (IOD)/the area of brown granules. Blood vessels were excluded from quantitative analysis.
Real-time PCR
For detection of mRNAs, 1 μg of total RNA extracted from LA was reverse transcribed into cDNA, after which relative quantification was conducted using the 7500 Real-Time PCR Detection System. For detection of miR-30c, the cDNA was added to a miR-30c stem-loop. The expression levels of mRNAs and miR-30c were normalized to GAPDH and U6, respectively. Data analysis was performed by 2−ΔΔCt method and was repeated 3 times for each specimen. The primers used are shown in Table 1.
Table 1.
Primers used for quantitative RT-PCR.
| Target gene | Primer sequence (5′-3′) | Tm (°C) |
|---|---|---|
| U6 | F: CTCGCTTCGGCAGCACA | 56.00 |
| R: AACGCTTCACGAATTTGCGT | 53.35 | |
| miR-30c stem-loop | GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACGACGAGAGTCG | 77.61 |
| miR-30c forward primer | GCGGTGTAAACATCCTACA | 56.00 |
| Universal reverse primer | GTATCCAGTGCAGGGTCCGAGGT | 63.77 |
| GAPDH | F: AGTGCCAGCCTCGTCTCATA | 57.45 |
| R: ACCAGCTTCCCATTCTCAGC | 57.45 | |
| TGF-β1 | F: CGCTTCTGCTCCCACTCC | 60.00 |
| R: TGTTGCGGTCCACCATTA | 54.00 | |
| TGF-βRI | F: ACTTCCCAACTACAGAAAAGCA | 56.00 |
| R: ATGACAGTGCGGTTATGGCA | 56.00 | |
| TGF-βRII | F: CCCAAGTCGGTTAACAGCGAT | 57.80 |
| R: TGGCAATGACAGCTATGGCA | 55.40 | |
| α-SMA | F: GGAGATGGCGTGACTCACAA | 57.45 |
| R: CGCTCAGCAGTAGTCACGAA | 57.45 | |
| CTGF | F: CTTCCCGAGAAGGGTCAAGC | 60.39 |
| R: TTCCAGTCGGTAGGCAGCTA | 60.32 | |
| CollagenI | F: CCCAGCGGTGGTTATGACTT | 57.45 |
| R: TCGATCCAGTACTCTCCGCT | 57.45 |
Tm (°C) – melting temperature; F – forward; R – reverse
Western blotting
Total protein was extracted by RIPA lysis buffer (KeyGEN Biotech, Nanjing) and then underwent Western blot analysis, as described elsewhere [18]. β-actin served as the loading control.
Surface ECGs, isolated atrial epicardial activation mapping, and heart electrophysiology
The remaining 30 rats underwent isolated atrial epicardial activation mapping and heart electrophysiology assessment after surface ECGs. Briefly, the beating heart isolated from an anesthetized rat was carefully but rapidly removed and connected to a Langendorff perfusion system full of warmed Tyrode’s solution (37°C) saturated with a mixture of 95% O2 and 5% CO2 gas mixture.
With the spontaneously and normally beating of heart, a 36-electrode microelectrode array arranged in a 6×6 configuration (PA03606060101, Multielectrode Probe array) was positioned on the epicardial surface of the left atrium for recording multiple activation signals. Activation waveforms acquired were amplified by a filter amplifier (EMS64-USB-1003, 64 channels) and transmitted to the connected computer. All activation times were digitized and then used to draw activation maps. The activation times were determined as the point of maximal negative slope of activation waveforms. Inhomogeneity index, absolute inhomogeneity, and conduction velocity were calculated using the Electric Mapping Scope System (MappingLab, UK).
At the end of activation mapping, 3 pairs of electrodes were connected to the epicardial surface of the high right atrium, high left atrium, and right ventricular apex, respectively. The atrial effective refractory period (AERP) and intermediary atrial conduction time (IACT) were measured by our electrophysiological recording and measurement program. AF vulnerability was evaluated by burst pacing for 3 s (10 times). The occurrence of AF was determined as rapid, irregular atrial, and ventricular responses lasting for more than 1000 ms. Between these stimulation procedures, there were 30-s recovery periods.
Statistical analysis
All results are expressed as mean±SD and analyzed by one-way analysis of variance (ANOVA) after Bonferroni correction. SPSS 22.0 software was applied for data analysis. The statistically significant p value was set at 0.05.
Results
Echocardiographic and hemodynamic characteristics
Table 2 indicates the findings of transthoracic echocardiography and hemodynamics.
Table 2.
Hemodynamic and echocardiographic studies.
| Data | Control group(n=10) | CIH group(n=10) | Doxy group (n=10) | P value |
|---|---|---|---|---|
| SBP, mm Hg | 147.9±19.7 | 163.7±15.7 | 152.3±17.6 | 0.162 |
| DBP, mm Hg | 115.9±18.3 | 126.5±13.1 | 120.7±16.3 | 0.342 |
| MBP, mm Hg | 131.6±18.4 | 144.2±13.4 | 136.6±15.1 | 0.242 |
| Pulse Pressure, mm Hg | 32.0±7.9 | 37.2±4.6 | 31.6±6.8 | 0.169 |
| LAD (mm) | 3.63±0.33 | 3.85±0.20 | 3.77±0.26 | 0.162 |
| LVESD (mm) | 4.34±0.40 | 4.19±0.40 | 4.38±0.41 | 0.547 |
| LVEDD (mm) | 6.85±0.47 | 6.98±0.43 | 7.26±0.35 | 0.150 |
| IVS (mm) | 1.75±0.11 | 1.64±0.26 | 1.74±0.16 | 0.305 |
| mPAP (mm Hg) | 65.51±2.66 | 67.88±1.65* | 66.40±1.41 | 0.035 |
| LVEF (%) | 64.77±5.73 | 69.12±4.48 | 67.85±9.18 | 0.236 |
| Heart weight ratio (1/1000) | 3.07±0.07 | 3.24±0.24 | 3.16±0.22 | 0.168 |
Values are expressed as mean±SD. SBP – systolic blood pressure; DBP – diastolic blood pressure; MBP – mean blood pressure; LAD – left atrial diameter; LVESD – left ventricular end-systolic dimension; LVEDD – left ventricular end-diastolic dimension; IVS – interventricular septum; mPAP – mean pulmonary artery pressure; LVEF – left ventricular ejection fraction;
p<0.05 vs. Control group.
Compared with the Control group, mean pulmonary artery pressure (mPAP) was significantly increased in the CIH group (67.88±1.65 vs. 65.51±2.66, P <0.05), which was improved by Doxy treatment, but the difference was not significant. No significant differences in LAD, LVESD, LVEDD, IVS, LVEF, heart weight ratio, and the hemodynamic parameters were observed among the 3 groups.
LA interstitial fibrosis
Figure 1 showed an obvious broader geographic areas of atrial interstitial collagen deposition in the CIH group than in the Control group, which was markedly improved by Doxy treatment. Left atrial interstitial collagen volume fraction (LACVF) among the Control, CIH, and DOXY groups were 1.70±0.92%, 5.44±2.94%, and 2.85±1.72%, respectively (P<0.01).
Figure 1.

Representative photomicrographs of left atrial sections subjected to Masson’s trichrome staining in three groups (original magnification ×400). Control group (A); CIH group (B); DOXY group (C); The illustration of the LACVF (D). *** p<0.001 vs. Control(CON) group. ## p<0.01 vs. CIH group. NS – no significance.
Comparison of immunohistochemistry
The CIH group showed more positive particles deposition in left atrium than in the Control group in TGF-β1, TGF-βRII, p-Smad2/3, α-SMA, CTGF, and Collagen I. Similarly, the MOD of these factors were also prominently higher in the CIH group compared to the Control group, and these changes were mitigated by Doxy treatment (Figure 2).
Figure 2.
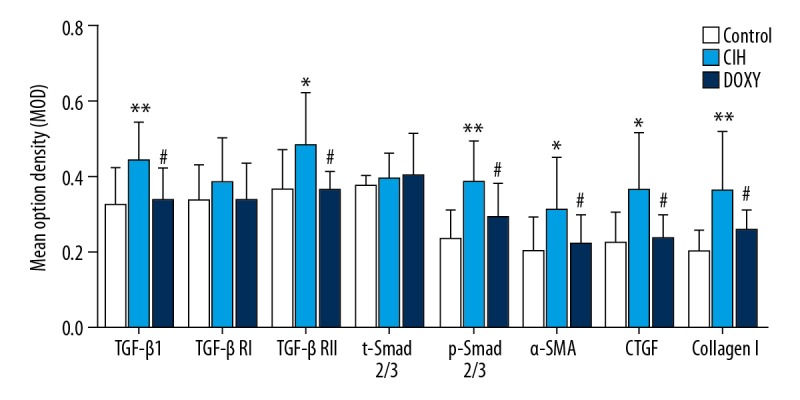
Mean optical density of target proteins (TGF-β1, TGF-βRI, TGF-βRII, Smad2/3, p-Smad2/3, α-SMA, CTGF, CollagenI) in three groups. * p<0.05, ** p< 0.01 vs. Control (CON) group. # p<0.05 vs. CIH group.
Expressions of TGFβ1 signaling factors and fibrosis marker proteins
Compared to the Control rats, the CIH rats exhibited increased mRNA and protein expression levels of TGF-β1, TGF-βRI, TGF-βRII, and the ratio of the phosphorylated (p) form versus total (t) Smad2/3, and decreased expression level of miR-30c. These alterations were ameliorated in Doxy-treated rats (Figures 3A–3D, 4A–4D). Similarly, we detected notably increased mRNA and protein expression levels of the fibrosis marker proteins CTGF, α-SMA, and Collagen I in the CIH group, and the expression levels of these factors were markedly ameliorated in the DOXY group (Figures 3E–3G, 4E–4G).
Figure 3.

mRNA expression levels of TGF-β1 (A), miR-30c (B), TGF-βRI (C), TGF-βRII (D), α-SMA (E), CTGF (F), CollagenI (G) in three groups.* p<0.05, ** p<0.01, and *** p<0.001 vs. Control group (CON). # p<0.05, ## p< 0.01, and ### p< 0.001 vs. CIH group. NS – no significance.
Figure 4.

Protein expression levels of TGF-β1 (A), TGF-βRI (B), TGF-βRII (C), p-Smad2/3 and Smad2/3 (D), α-SMA (E), CTGF (F), CollagenI (G) in three groups. * p<0.05, ** p<0.01, and *** p< 0.001 vs. Control group (CON). # p<0.05, ## p<0.01, and ### p<0.001 vs. CIH group. NS – no significance.
Surface ECGs, electrophysiology, and activation mapping parameters
The specific values of the surface ECGs and isolated heart electrophysiological parameters are shown in Table 3. The Control group had a notable difference from the CIH group in P-wave duration (14.13±1.78 vs. 18.13±6.22, P<0.05). Electrophysiological results exhibited longer inter-atrial conduction time (IACT) in the CIH group than in the Control group after pacing stimulation at basic cycle lengths of 150, 200, and 250 ms. These changes were clearly reversed by Doxy treatment. Representative diagrammatic sketch of AF induction was shown in Figure 5G. Furthermore, the CIH group had a higher induction ratio of AF than in the Control group, and Doxy treatment greatly reduced the occurrence of AF. AF inducibility among the 3 groups was 21.00±15.95%, 35.00±12.69%, and 20.00±11.55%, respectively (P<0.05) (Figure 5H). However, other parameters from surface ECGs and isolated heart electrophysiology showed no statistically significant differences among the 3 groups.
Table 3.
Surface ECGs and electrophysiological parameters.
| Control group (n=10) | CIH group (n=10) | Doxy group (n=10) | P value | |
|---|---|---|---|---|
| Surface ECG parameters, ms | ||||
| HR, beats per min | 413.98±26.39 | 421.07±17.30 | 415.72±21.54 | 0.757 |
| PR interval (ms) | 43.63±2.89 | 41.89±2.53 | 44.37±3.35 | 0.174 |
| P-wave duration (ms) | 14.13±1.78 | 18.13±6.22* | 15.37±2.09 | 0.084 |
| QRS duration (ms) | 17.33±2.14 | 17.13±1.70 | 17.07±2.53 | 0.962 |
| QT interval (ms) | 61.83±8.29 | 66.14±9.00 | 69.85±17.83 | 0.369 |
| QTc interval (ms) | 161.30±19.95 | 175.92±23.48 | 181.06.32±47.93 | 0.391 |
| Tpeak Tend Interval (ms) | 33.11±5.59 | 36.53±7.10 | 38.99±18.46 | 0.546 |
| Electrophysiological parameters | ||||
| IACT | ||||
| 150 ms | 23.90±2.38 | 27.00±2.94 * | 23.00±2.05## | 0.003 |
| 200 ms | 23.40±2.84 | 26.40±3.53 * | 22.70±2.31## | 0.021 |
| 250 ms | 23.00±2.45 | 27.10±3.14 ** | 22.30±2.50## | 0.001 |
| P value | 0.736 | 0.871 | 0.793 | |
| HRAERP | ||||
| 150 ms | 49.80±3.46 | 51.40±1.90 | 49.40±4.01 | 0.359 |
| 200 ms | 51.00±3.16 | 50.20±3.33 | 51.60±2.95 | 0.614 |
| 250 ms | 51.80±3.05 | 49.60±2.95 | 51.60±3.10 | 0.219 |
| P value | 0.391 | 0.354 | 0.262 | |
| HLAERP | ||||
| 150 ms | 52.20±3.71 | 50.80±2.53 | 51.60±3.50 | 0.638 |
| 200 ms | 52.00±2.49 | 51.40±3.13 | 52.60±2.32 | 0.610 |
| 250 ms | 51.20±3.29 | 51.80±6.00 | 51.00±4.03 | 0.921 |
| P value | 0.763 | 0.865 | 0.567 | |
| AF inducibility (burst pacing) | ||||
| 50 ms | 2.10/10±1.60/10 (21.00%±15.95%) | 3.50/10±1.27/10 (35.00%±12.69)* | 2.00/10±1.15/10 (20.00%±11.55)# | 0.034 |
Values are expressed as mean±SD. IACT – inter-atrial conduction time; HLAERP – high left atrium effective refractory period; HRAERP – high right atrium effective refractory period.
p<0.05,
p<0.01 vs. Control group (CON).
p<0.05,
p<0.01 vs. CIH group.
Figure 5.

Abridged general view of atrial epicardial activation mapping and AF inducibility. Representative maps of spontaneous left atrial epicardial activation among Control (A), CIH (B) and DOXY group (C). From dark red to dark blue, scale bar indicates total time from first activated locus to last activated locus within one heartbeat. Comparison of inhomogeneity index in 3 groups (D). Comparison of absolute inhomogeneity between 3 groups (E). Comparison of mean conduction velocity in 3 groups (F). Representative diagrammatic sketch of AF induction (G). Comparison of AF inducibility in three groups (H). HRA – high right atrium; HLA – high left atrium; RV – right ventricular. * p<0.05 vs. Control (CON) group. # p<0.05 vs. CIH group. NS – no significance. n=10 per group.
Representative maps of three groups were shown in Figure 5A–5C. Atrial activation mapping showed that, compared with the Control group, the atrial electrical conduction inhomogeneity index and absolute inhomogeneity were remarkably increased in the CIH group, whereas the mean conduction velocity was significantly decreased. These changes were improved by Doxy treatment (Figures 5D–5F).
Discussion
In this study, we assessed the protective functions of Doxy on atrial fibrosis, atrial conduction, and AF vulnerability using a CIH rat model. The main findings were: (1) 6 weeks of CIH can significantly increase LA interstitial fibrosis; (2) profoundly enhanced atrial fibrosis contributed to some electrophysiological abnormalities, including increased atrial electrical conduction inhomogeneity index, absolute inhomogeneity, inter-atrial conduction time, AF vulnerability, and decreased mean velocity of left atrial conduction; and (3) Doxy can significantly improve CIH-induced fibrosis and electrophysiological properties changes in atrium secondary to fibrosis via inhibition of the TGF-β1/Smad2/3 signaling pathway.
AF is a commonly encountered cardiac arrhythmia in clinical practice, with very high mortality and disability rates [19]. It is clear that atrial electrophysiological and structural alterations participate in the initiation and maintenance of AF [20,21]. Of the atrial structural changes, fibrosis is regarded as particularly critical to creating the AF substrate, with fibrosis likely promoting the formation of intra-atrial reentry circuits by increasing non-uniform anisotropy and local electrical conduction heterogeneities [4,22]. Our electrophysiological results demonstrated that severe interstitial fibrosis contributed to increased conduction inhomogeneity and decreased conduction velocity of the left atrium in CIH rats, and these changes are essential to the development and maintenance of atrial conduction reentry circuits and progression to AF.
P-wave indices such as duration, variance, and dispersion are reliable noninvasive marker of intra-atrial and inter-atrial conduction [23]. Previous studies [24,25] demonstrated that prolonged P-wave duration is not a totally benign condition. Indeed, P-wave prolongation is an indicator of AF in patients after bypass surgery, and people with an prolonged P-wave duration are more likely to experience paroxysmal AF and progress toward persistent AF [26,27]. We observed that CIH caused remarkably prolonged P-wave duration and some abnormalities in atrial conduction, as well as higher AF inducibility. These findings were similar to published results showing that P-wave prolongation might be a reliable indicator for AF incidence.
TGFβ1, a pro-fibrotic cytokine, has been proved to be involved in formation and deposition of extracellular matrix components, contributing to tissue and organ fibrosis [6]. Additionally, TGF-β1 a pivotal fibrotic signaling ligands in a number of different animal models of atrial remodeling established by rapid atrial pacing, angiotensin II stimulation, aortic constriction, and CIH [28]. When TGF-β1 binds to its heterodimeric receptor complex, TGF-βRI and TGF-βRII, the downstream canonical Smads signaling cascades are activated. The signaling molecules Smad2/3 are more dedicated downstream effectors of the classical TGF-β1 pathway in the initiation and progression of cardiac fibrosis [29–31]. Hence, we used Smad2/3 activation as an indicator of the TGF-β1 pathway. Our results indicated that activation of the TGF-β1/Smad2/3 pathway plays a critical role in CIH-induced atrial fibrosis and atrial conduction abnormalities caused by fibrosis, which might be the key molecular mechanism by which Doxy exerts a remarkable therapeutic efficacy on atrial interstitial fibrosis.
MicroRNAs (miRs) are small, endogenous, 20–23 nucleotide noncoding RNAs which can downregulate gene expression by combining with the 3′untranslated region of the target mRNA [32]. Recently, miRs were found to take part in many biological and disease processes, including fibrosis [33,34]. miR-30c, identified as an anti-fibrotic microRNA, is involved in the atrial fibrogenic response. A recent study [13] showed that TGF-β1 can also mediate its pro-fibrotic function by repressing the expression level of miR-30c, which in turn upregulates the expression level of the target gene TGF-βRII and further amplifies the effects of the TGF-β1 pathway. In accordance with the above-mentioned study, the expression level of miR-30c was negatively related to TGF-β1 and was downregulated in our CIH rats, which was accompanied by a remarkably increased expression level of TGF-βRII.
Doxy, a major tetracycline antibiotic, is widely used in the treatment of infectious disorders in humans and animals. Recently, Doxy was found to have an anti-fibrotic role by reducing the production of extracellular matrix. As outlined above, Fujita et al. [35] indicated that Doxy significantly alleviated bleomycin-induced pulmonary interstitial fibrosis in mice. A related study [36] found that Doxy can also ameliorate cardiac fibrosis and hypertrophy caused by isoproterenol in rats. Moreover, Doxy was confirmed to reduce the production of matrix metalloprotein 9 by blocking upregulation of the TGF-β1/Smad pathway in human corneal epithelial cells [37].
In the present study, we found that atrial interstitial fibrosis can be alleviated by Doxy treatment in rats exposed to CIH for 6 weeks (5.44±2.94% vs. 2.85±1.72%). To further determine the specific molecular mechanisms by which Doxy interferes with the process of fibrogenesis, we assessed the expression levels of TGF-β1 pathway-related factors in our 3 groups, showing that the TGF-β1/Smad2/3 pathway was highly activated and the expression levels of its downstream fibrosis marker proteins were also significantly elevated in the CIH group; those changes were somewhat inhibited by Doxy treatment.
Our results show that the TGF-β1 pathway participates in the course of CIH-induced atrial interstitial fibrosis and is crucial in this pathological process. Doxy might be a promising drug for use in preventing and treating atrial fibrosis, as well as the consequent electrophysiological abnormalities by mediating the TGF-β1/Smad2/3 pathway. Further investigations are needed to better understand its beneficial effects on atrial remodeling.
Limitations
Firstly, we did not examine the serum levels of TGF-β1and fibrotic markers among the 3 groups, but the expression levels of those factors in atrial tissue should be more telling. Secondly, our experiment failed to directly validate the targeted relationships among TGF-β1, miR-30c, and TGF-βRII. However, relevant conclusions were verified by using neonatal cardiac fibroblasts separated from the atria of Sprague-Dawley rats, which is consistent with our animal species and organ specificity.
Conclusions
Fibrosis-induced alternations in atrial conduction and AF susceptibility in CIH rats can be mitigated by Doxy, which was closely associated with the TGF-β1/Smad2/3 signaling pathway.
Footnotes
Source of support: This work was supported by the National Natural Science Foundation of China (81570304)
References
- 1.Benjamin EJ, Wolf PA, D’Agostino RB, et al. Impact of atrial fibrillation on the risk of death: Rhe Framingham Heart Study. Circulation. 1998;98(10):946–52. doi: 10.1161/01.cir.98.10.946. [DOI] [PubMed] [Google Scholar]
- 2.Pellman J, Sheikh F. Atrial fibrillation: Mechanisms, therapeutics, and future directions. Compr Physiol. 2015;5(2):649–65. doi: 10.1002/cphy.c140047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vagos M, van Herck IGM, Sundnes J, et al. Computational modeling of electrophysiology and pharmacotherapy of atrial fibrillation: Recent advances and future challenges. Front Physiol. 2018;9:1221–50. doi: 10.3389/fphys.2018.01221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckstein J, Verheule S, de Groot NM, et al. Mechanisms of perpetuation of atrial fibrillation in chronically dilated atria. Prog Biophys Mol Biol. 2008;97(2–3):435–51. doi: 10.1016/j.pbiomolbio.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 5.Rahmutula D, Marcus GM, Wilson EE, et al. Molecular basis of selective atrial fibrosis due to overexpression of transforming growth factor-β1. Cardiovasc Res. 2013;99(4):769–79. doi: 10.1093/cvr/cvt074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lijnen P, Petrov V. Transforming growth factor-beta 1-induced collagen production in cultures of cardiac fibroblasts is the result of the appearance of myofibroblasts. Methods Find Exp Clin Pharmacol. 2002;24(6):333–44. doi: 10.1358/mf.2002.24.6.693065. [DOI] [PubMed] [Google Scholar]
- 7.Dixon IMC, Reid NL, Ju H. Angiotensin II and TGF-β in the development of cardiac fibrosis, myocyte hypertrophy, and heart failure. Heart Fail Rev. 1997;2(2):107–16. [Google Scholar]
- 8.Polejaeva IA, Ranjan R, Davies CJ, et al. Increased susceptibility to atrial fibrillation secondary to atrial fibrosis in transgenic goats expressing transforming growth factor-β1. J Cardiovasc Electrophysiol. 2016;27(10):1220–29. doi: 10.1111/jce.13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi EK, Chang PC, Lee YS, et al. Triggered firing and atrial fibrillation in transgenic mice with selective atrial fibrosis induced by overexpression of TGF-β1. Circ J. 2012;76(6):1354–62. doi: 10.1253/circj.cj-11-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vander Ark A, Cao J, Li X. TGF-β receptors: In and beyond TGF-β signaling. Cell Signal. 2018;52:112–20. doi: 10.1016/j.cellsig.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Heldin CH, Moustakas A. Role of Smads in TGF beta signaling. Cell Tissue Res. 2012;347:21–36. doi: 10.1007/s00441-011-1190-x. [DOI] [PubMed] [Google Scholar]
- 12.Khalil H, Kanisicak O, Prasad V, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770–83. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu J, Wu H, Chen S, et al. MicroRNA-30c suppresses the pro-fibrogenic effects of cardiac fibroblasts induced by TGF-β1 and prevents atrial fibrosis by targeting TGFβRII. J Cell Mol Med. 2018;22:3045–57. doi: 10.1111/jcmm.13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henehan M, Montuno M, De Benedetto A. Doxycycline as an anti-inflammatory agent: updates in dermatology. J Eur Acad Dermatol Venereol. 2017;31:1800–8. doi: 10.1111/jdv.14345. [DOI] [PubMed] [Google Scholar]
- 15.Hua XF, Li XH, Li MM, et al. Doxycycline attenuates paraquat-induced pulmonary fibrosis by downregulating the TGF-β signaling pathway. J Thorac Dis. 2017;9:4376–86. doi: 10.21037/jtd.2017.10.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei Q, Bian Y, Yu F, et al. Chronic intermittent hypoxia induces cardiac inflammation and dysfunction in a rat obstructive sleep apnea model. J Biomed Res. 2016;30:490–95. doi: 10.7555/JBR.30.20160110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Zhang L, Liu H, et al. Cardiac sympathetic denervation suppresses atrial fibrillation and blood pressure in a chronic intermittent hypoxia rat model of obstructive sleep apnea. J Am Heart Assoc. 2019;19(8):e010254. doi: 10.1161/JAHA.118.010254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang K, Ma Z, Wang W, et al. Beneficial effects of tolvaptan on atrial remodeling induced by chronic intermittent hypoxia in rats. Cardiovasc Ther. 2018;36:e12466. doi: 10.1111/1755-5922.12466. [DOI] [PubMed] [Google Scholar]
- 19.Chugh SS, Havmoeller R, Narayanan K, et al. Worldwide epidemiology of atrial fibrillation: A Global Burden of Disease 2010 Study. Circulation. 2014;129:837–47. doi: 10.1161/CIRCULATIONAHA.113.005119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pathak R, Lau DH, Mahajan R, et al. Structural and functional remodeling of the left atrium: Clinical and therapeutic implications for atrial fibrillation. J Atr Fibrillation. 2013;6:986. doi: 10.4022/jafib.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kistler PM, Sanders P, Dodic M, et al. Atrial electrical and structural abnormalities in an ovine model of chronic blood pressure elevation after prenatal corticosteroid exposure: Implications for development of atrial fibrillation. Eur Heart J. 2006;27:3045–56. doi: 10.1093/eurheartj/ehl360. [DOI] [PubMed] [Google Scholar]
- 22.Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: Mechanisms and implications. Circ Arrhythm Electrophysiol. 2008;1:62–73. doi: 10.1161/CIRCEP.107.754564. [DOI] [PubMed] [Google Scholar]
- 23.Magnani JW, Williamson MA, Ellinor PT, et al. P wave indices: Current status and future directions in epidemiology, clinical and research applications. Circ Arrhythm Electrophysiol. 2009;2:72–79. doi: 10.1161/CIRCEP.108.806828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magnani JW, Johnson VM, Sullivan LM, et al. P wave duration and risk of longitudinal atrial fibrillation risk in persons ≥60 years old (from the Framingham Heart Study) Am J Cardiol. 2011;107:917–21. doi: 10.1016/j.amjcard.2010.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kishima H, Mine T, Takahashi S, et al. The impact of left atrial pressure on filtered P-wave duration in patients with atrial fibrillation. Heart Vessels. 2016;31(11):1848–54. doi: 10.1007/s00380-015-0789-3. [DOI] [PubMed] [Google Scholar]
- 26.Zaman AG, Alamgir F, Richens T, et al. The role of signal averaged P wave duration and serum magnesium as a combined predictor of atrial fibrillation after elective coronary artery bypass surgery. Heart. 1997;77:527–31. doi: 10.1136/hrt.77.6.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aytemir K, Amasyali B, Kose S, et al. Maximum P-wave duration and P-wave dispersion predict recurrence of paroxysmal atrial fibrillation in patients with Wolff-Parkinson-White syndrome after successful radiofrequency catheter ablation. J Interv Card Electrophysiol. 2004;11:21–27. doi: 10.1023/B:JICE.0000035925.90831.80. [DOI] [PubMed] [Google Scholar]
- 28.Nishida K, Michael G, Dobrev D, et al. Animal models for atrial fibrillation: Clinical insights and scientific opportunities. Europace. 2010;12:160–72. doi: 10.1093/europace/eup328. [DOI] [PubMed] [Google Scholar]
- 29.Qiu H, Liu W, Lan T, et al. Salvianolate reduces atrial fibrillation through suppressing atrial interstitial fibrosis by inhibiting TGF-β1/Smad2/3 and TXNIP/NLRP3 inflammasome signaling pathways in post-MI rats. Phytomedicine. 2018;51:255–65. doi: 10.1016/j.phymed.2018.09.238. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Zhuo X, Liu W, et al. Resveratrol inhibits high glucose induced collagen upregulation in cardiac fibroblasts through regulating TGF-β1-Smad3 signaling pathway. Chem Biol Interact. 2015;227:45–52. doi: 10.1016/j.cbi.2014.12.031. [DOI] [PubMed] [Google Scholar]
- 31.Lei B, Hitomi H, Mori T, et al. Effect of efonidipine on TGF-β1-induced cardiac fibrosis through Smad2-dependent pathway in rat cardiac fibroblasts. J Pharmacol Sci. 2011;117(2):98–105. doi: 10.1254/jphs.11065fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ventura A, Jacks T. MicroRNAs and cancer: Short RNAs go a long way. Cell. 2009;136:586–91. doi: 10.1016/j.cell.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang D, Cui Y, Li B, et al. miR-155 regulates high glucose-induced cardiac fibrosis via the TGF-β signaling pathway. Mol Biosyst. 2016;13(1):215–24. doi: 10.1039/c6mb00649c. [DOI] [PubMed] [Google Scholar]
- 34.Abonnenc M, Nabeebaccus AA, Mayr U, et al. Extracellular matrix secretion by cardiac fibroblasts: Role of microRNA-29b and microRNA-30c. Circ Res. 2013;113:1138–47. doi: 10.1161/CIRCRESAHA.113.302400. [DOI] [PubMed] [Google Scholar]
- 35.Fujita M, Ye Q, Ouchi H, et al. Doxycycline attenuated pulmonary fibrosis induced by bleomycin in mice. Antimicrob Agents Chemother. 2006;50:739–43. doi: 10.1128/AAC.50.2.739-743.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Errami M, Tassa AT, Galindo CL, et al. Carbamazepine alone and in combination with doxycycline attenuates isoproterenol-induced cardiac hypertrophy. Heart Int. 2010;5:e7. doi: 10.4081/hi.2010.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim HS, Luo L, Pflugfelder SC, et al. Doxycycline inhibits TGF-beta1-induced MMP-9 via Smad and MAPK pathways in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2005;46:840–48. doi: 10.1167/iovs.04-0929. [DOI] [PubMed] [Google Scholar]