

Abstract
Highly efficient gene delivery vehicles are pursued to progress gene therapy. In this study, we developed the cell‐penetrating peptide‐labelled and degradable gene carriers for efficient external gene transfection. The cationic carriers were prepared by coupling low‐molecular‐weight polyethylenimine (PEI800) with 4ʹ4‐dithiodibutyric acid (DA), and HIV‐1 Trans‐Activator of Transcription (TAT) was conjugated to the carriers as a penetrating peptide. The resulted PEI‐DA‐TAT was able to condense plasmid DNA (pDNA) into a complex with a hydrodynamic size of around 150 nm under a neutral condition. PEI‐DA‐TAT showed negligible cytotoxicity to both Hela and HEK 293 cells with the cell viability of more than 80% beyond the carrier concentration of 50 μg/mL. This new carrier displayed better performance in regard to DNA transfection efficiency in comparison with the carriers of non‐TAT labelled PEI‐DA, commercial PEI25K and low‐molecular‐weight PEI (PEI800). The transfection efficiency of PEI‐DA‐TAT was increased by 8% compared with PEI‐DA and PEI25K. The experimental findings suggested that the developed PEI‐DA‐TAT is a promising carrier for efficient DNA delivery with low cytotoxicity for gene therapy.
Keywords: Cell‐penetrating peptide, Disulphide bond, HIV‐1 TAT, Polyethylenimine, Nanogel
Abbreviations
- FITC
fluorescein isothiocyanate
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- PEI
polyethylenimine
- TAT
trans‐activator of transcription
1. Introduction
A surging interest in the development of highly efficient gene delivery vehicles has been attracted with the ultimate goals for their use in clinical gene therapy. It has been demonstrated that therapeutic nucleic acids delivered into cells can treat various diseases including cancers 1 and genetic diseases 2. The development of efficient gene delivery vehicles is always required to assist to transport therapeutic genes into cells by overcoming several barriers in the eukaryotic cells including cellular uptake, endosome escape and nuclear transport. Viral and non‐viral gene carriers have been explored to date to overcome these barriers 3, 4. However, immunological and inflammatory issues have restricted the application of viral gene carriers, even though they exhibit higher transfection efficiency than non‐viral carriers 4. Therefore, non‐viral gene carriers such as lipid‐based gene carriers and polymer‐based gene carriers have been investigated broadly 5. However, the major drawbacks of non‐viral gene carriers are (i) some of the non‐viral carriers are not degradable and the toxic components could be accumulated in the body; (ii) non‐viral carriers often show lower DNA transfection efficiency than virus carriers 6.
Branched polyethylenimine with a molecular weight of 25K Da (BPEI25K), a cationic non‐viral gene carrier, has been widely investigated for its excellent gene protection from intracellular enzyme degradation and high transfection efficiency associated with the ‘proton sponge’ effect 7. The ionic strength in the endosome is increased by the influx of chloride ions due to the pumping of the protons to remain the charge neutrality. This is considered to be one of the reasons for the osmotic swelling and physical rupture of the endosome leading to endosomal escape of the carrier/DNA complexes 8. However, it shows high cytotoxicity owing to its high cationic charge density. Such cytotoxicity prevents PEI from its wide application in gene therapy 9. On the other hand, it has been reported that low molecular weight BPEI (LWM PEI) such as PEI800 has a low cytotoxicity while its transfection efficiency is low 10.
To harness the low cytotoxicity of LMW PEI, strategies have been adopted, including modifying LMW PEI with polyethylene glycol (PEG) 11, targeting ligands or hydrophobic groups 12 and cross‐linking LMW PEI with different linkages 13. Among these strategies, cross‐linking LMW PEI with intracellular sensitive degradable linkage can be more promising to achieve low cytotoxicity and high transfection efficiency 14. For example, disulphide bonds that can be cleaved by glutathione (GSH) in the cytosol or the nucleus have been used to crosslink PEI 15. The degradation of disulphide bonds is triggered after the escape of carriers from the endolysosomal compartment 16. Other reducible PEI‐based gene carriers have been prepared by different linkers with disulphide bonds such as N,Nʹ‐cystamine bisacrylamide (CBA) 17, 18, dithiobis(succinimidyl propionate) (DSP) 19, and dimethyl‐3,3’‐dithiobis(propionimidate) (DTBP) 19. However, the lower DNA transfection efficient performance of gene delivery by these degradable carriers still needs to be solved due to various limitations 19, 20
In order to improve the cellular uptake ability of gene carriers, several functional materials can be conjugated to the carriers. One of the strategies is employing cell‐penetrating peptides (CPPs), to facilitate the translocation of macromolecules like proteins and polymers through the biological membrane in mammal cells 21, 22. However, the mechanism for internalization of the CPPs is still not clear. Researchers have proposed several different hypotheses, such as micropinocytosis 23 and lipid raft mediated cellular uptake 24, 25. HIV‐1 Trans‐Activator of Transcription peptide (HIV‐1 TAT) is one of the most popular CPPs, since it represents a protein transduction domain and a nuclear localization sequence (NLS) 26, 27. In literature, HIV‐1 TAT has been grafted to different carriers to improve delivery performance. Lee et al. 28 reported a hybrid‐synthesized drug carrier through chemical conjugation of doxorubicin (DOX) with TAT to the chitosan backbone. The anticancer efficiency of the resulted carrier was enhanced and the efficiency of cell internalization was improved compared with free DOX. Significant inhibition of tumour growth in CT26 xenograft‐bearing mice after treating by the TAT‐modified carrier/DOX complex was also found. Moreover, Li et al reported an efficient drug delivery system by conjugating TAT with magnetic mesoporous silica nanoparticles (MMSN) with a Fe3O4 nanocore 29. DNA‐toxin anticancer drugs were delivered by the MMSN‐TAT in vitro and in vivo. In addition to the modification of drug carriers, TAT peptide was also used to improve the cellular uptake ability of polymer based gene carriers. For example, TAT was conjugated to liposomes to deliver external gene into H9C2 cells and BT20 cells in vitro 30. The transfection efficiency of TAT conjugated liposomes was significantly increased compared with commercial Lipofectin®. Even though TAT has been applied to modify different carriers, there is no report so far on TAT‐labelled intracellular degradable PEI‐based carriers for gene delivery with high transfection efficiency and low cytotoxicity.
In this study, HIV‐1 TAT labelled intracellular degradable PEI (PEI‐DA‐TAT) was synthesized by cross‐linking LMW PEI (PEI800) with 4′4‐dithiodibutyric acid (DA) and then conjugated with HIV‐1 TAT. We hypothesize that the transfection efficiency of PEI‐DA‐TAT should be improved since TAT could improve the cellular uptake ability of the gene carrier. The cytotoxicity of PEI‐DA‐TAT should be low since the disulphide bonds would be cleaved after being exposed to the reductive environment of cells. PEI‐DA‐TAT was evaluated for its gene binding ability, cytotoxicity, cellular uptake and gene transfection efficiency in Hela and HEK 293 cells.
2. Materials and methods
2.1. Materials
PEI800 and PEI25K (MW = 0.8 and 25 kDa), EDC (1‐ethyl‐3‐(3‐dimethy‐laminopropyl) carbodiimide), NHS (N‐hydroxysulfosuccinimide) were purchased from Sigma‐Aldrich (St.Louis, MO). HIV‐1 TAT peptide (Tyr‐Gly‐Arg‐Lys‐Lys‐Arg‐Arg‐GLn‐Arg‐Arg‐Arg) and FITC labelled HIV‐1 TAT (FITC‐TAT) were purchased from GenicBio (Shanghai, China). QIAGEN Maxi kit was obtained from Qiagen (Boncaster, Australia). Plasma membrane and nuclear labelling kit (Alexa Fluor 594 and Hoechst33258), nucleic acid stains (YOYO‐1), Fetal Bovine Serum (FBS), trypsin–EDTA, penicillin–streptomycin (PS) mixture, kanamycin, phosphate buffered saline (PBS) and TAE (Tris–acetate) were purchased from Life Technologies (Mulgrave, Australia). Gel red, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bro‐mide (MTT), 1,4‐dithio‐DL‐threitol (DTT) and other chemicals/solvents were purchased from Sigma‐Aldrich (St. Louis, MO, USA).
2.2. Preparation of plasmid DNA
The pEGFP‐N1 plasmid expressing the enhanced green fluores‐cent protein (EGFP) was prepared in Escherichia coli DH5α strain and further extracted using a QIAGEN Maxi kit. The integrity and purity of plasmid DNA (pDNA) were analyzed using 0.8 % agarose gel electrophoresis and the DNA concentration was determined using a Jasco UV–vis spectrophotometer (Tokyo, Japan) at a fixed wavelength of 260 nm. The pDNA was further labelled by fluorescent dye (YOYO‐1) at a ratio of 1 molecular dye to 100 molar nucleic acid base pairs for the cellular uptake study.
2.3. Synthesis of PEI‐DA and PEI‐DA‐TAT
The synthesis of the PEI‐DA is described in Scheme 1. PEI‐DA was synthesized by the reaction of PEI800 and 4ʹ4‐dithiodibutyric acid (DA) using carbodiimide chemistry. Briefly, NHS/EDC (86.32 mg/143.78 mg) and DA (89.37 mg) were added to 6 ml DMSO at room temperature and the mixture was stirred for 1 h. PEI800 (100 mg, 100 mg/mL) was then added to the mixture. The pH of the reaction mixture was adjusted to 7 by adding NaOH (0.1 M) or HCl (0.1 M). The mixture was further reacted for 16 h at room temperature. After the reaction, the product was dialyzed against deionized water (pH 7) for 3 days to remove the unreacted molecules. The obtained PEI‐DA polymer was lyophilized and characterized.
Scheme 1.
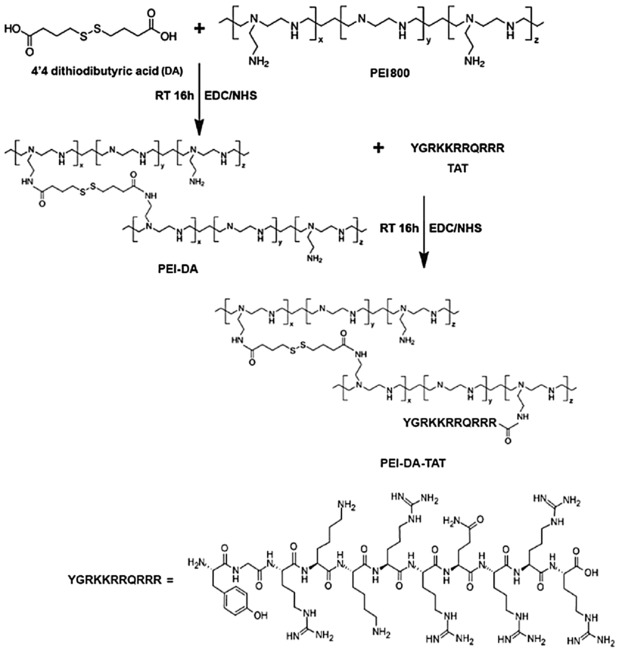
Schematic description on the synthesis of PEI‐DA and PEI‐DA‐TAT
Then the HIV‐1 TAT (Tyr‐Gly‐Arg‐Lys‐Lys‐Arg‐Arg‐GLn‐Arg‐Arg‐Arg) peptide was conjugated to PEI‐DA using an NHS/EDC coupling system to produce PEI‐DA‐TAT. Briefly, 3 mg TAT peptides were added to 5 mL MilliQ water at room temperature. Then, 234.4 and 390.4 mg of EDC and NHS were added to the solution separately. The mixture was stirred at room temperature for 1 h and then PEI‐DA (30 mg) was added to the mixture. The reaction was stirred for 16 h, and the pH of the reaction mixture was adjusted to 7.0 by NaOH (0.1 M) or HCl (0.1 M). The mixture solution was dialyzed against deionized water (pH 7.0) for 3 days to get rid of unreacted molecules. The obtained PEI‐DA‐TAT polymers were lyophilized and characterized.
To confirm the successful conjugation and the quantification of TAT to PEI‐DA, FITC‐labelled TAT was conjugated to PEI‐DA to prepare the PEI‐DA‐TAT‐FITC. Briefly, 0.01 % (w/w) FITC was grafted with TAT peptide (30 mg). Then, 3 mg of FITC labelled TAT peptides were reacted with 30 mg PEI‐DA at the same condition described above in the presence of EDC/NHS. The PEI800, PEI‐DA, PEI‐DA‐TAT‐FITC (concentration of 1 mg/mL) and FITC‐TAT from 0.1–0.6 mg/mL were evaluated with UV‐vis at the wavelength of 495 nm and room temperature to assure the conjugation of TAT peptide to PEI‐DA.
2.4. Characterization of PEI‐DA and PEI‐DA‐TAT
The Fourier transform infrared spectrometer (FTIR) spectra of PEI800, DA, HIV‐1 TAT peptide, PEI‐DA and PEI‐DA‐TAT were monitored using a Thermo NICOLET 6700 Fourier transform infrared spectrometer at room temperature under reflection mode. The 1H‐NMR spectra of PEI‐DA was measured by using a 600 MHz Bruker NMR in D2O.
The buffering capacity of PEI800, PEI25K, PEI‐DA and PEI‐DA‐TAT was measured by acid–base titration. Briefly, 50 mL of sample solution in the concentration of 0.05 mg/mL including PEI‐DA and PEI‐DA‐TAT was adjusted to pH 3 with HCl and then a diluted NaOH was gradually added until the solution pH reached 11. After the addition of NaOH, the pH was measured by a microprocessor pH meter. PEI800 and PEI25K were tested as positive controls. 50 ml of 150 mM NaCl was titrated as a negative control 31.
Particle sizes and zeta potentials of a gene carrier are related to the efficiency of its cellular uptake ability and transfection efficiency. To achieve optimal N/P ratio of carriers to pDNA, various carriers/pDNA complexes were prepared at different N/P ratios (the ratio of the number of nitrogen residues of PEI per DNA phosphate) from 0.5 to 50 at pH 7. Particle sizes and zeta potentials were examined by a Malvern Nano‐ZS 90 laser particle size analyzer equipped with ZET5104 cell at room temperature. For particle size analysis, the cumulate method was used to convert intensity–intensity autocorrelation functions to apparent particle sizes according to the Stokes–Einstein relationship 32. The Smuloschowski model was used to convert electrophoresis mobility to zeta potentials. Fifteen parallel runs were carried out for each measurement and the final data were obtained based on statistical analysis.
2.5. DNA binding ability of PEI‐DA and PEI‐DA‐TAT
To evaluate the DNA binding ability of gene carriers of PEI‐DA and PEI‐DA‐TAT, different carriers/pDNA complexes were prepared by mixing PEI‐DA and PEI‐DA‐TAT with pDNA (0.2 μg/mL) at various N/P ratios ranging from 0.5 to 50. PEI800 and PEI25K were also measured as references. The complexes were diluted to 6 μl and incubated at room temperature for 30 min. Subsequently, the samples were loaded onto the 0.8 wt. % agarose gels containing GelRed™ with Tris‐acetate (TAE) running buffer at 80 V for 90 min. The resulted pDNA migration patterns were read under UV irradiation (G‐BOX, SYNGENE).
2.6. Degradability of PEI‐DA and PEI‐DA‐TAT
To confirm the degradability of PEI‐DA and PEI‐DA‐TAT, PEI‐DA/pDNA and PEI‐DA‐TAT/pDNA complexes was prepared at the N/P ratio of 10, and 20 mM or 50 mM DTT was mixed with the complexes and incubated at 37°C for 30 min. After the incubation, the DTT treated PEI‐DA/pDNA and PEI‐DA‐TAT/pDNA complexes were loaded onto the 0.8 wt. % agarose gel containing GelRed™ with Tris‐acetate (TAE) running buffer at 80 V for 90 min to check the migration of the pDNA. PEI800/pDNA, PEI25K/pDNA, PEI‐DA/pDNA and PEI‐DA‐TAT/pDNA complexes without DTT treatment were used as references. Naked pDNA was also applied as the negative control. The migration image of pDNA was read under UV irradiation (G‐BOX, SYNGENE).
2.7. Evaluation of cytotoxicity of synthesized gene carriers
Hela and HEK 293 cells were cultured in DMEM medium supplied with 10% FBS in 96‐well plates (200 μL/well) at a cell density of 1.0×105 cells/mL for cytotoxicity measurement in different carriers. Cells were further incubated at 37°C in a humidified 5% CO2‐containing incubator for 24 h. After that the medium was replaced with 200 μL fresh medium. Then, different testing carriers (PEI800, PEI25K, PEI‐DA and PEI‐DA‐TAT) were added at the concentrations from 0.5 to 50 μg/mL , respectively. Cells were then incubated for 24 h before 10 μL of MTT (5 mg/mL in PBS) was added to each well to measure cell viability 33. The growth medium was replaced by 150 μL of dimethyl sulfoxide (DMSO) to ensure complete solubilization of the formed form‐azan crystals after incubating at 37°C in a humidified 5% CO2‐containing incubator for another 4 h. Eventually, the absorbance was determined using the Biotek Microplate Reader (Biotek, USA) at a wavelength of 595 nm 34. The cell viability was calculated with the absorbance data and mock cells without treating with carriers was used for 100% cell viability.
2.8. Gene transfection of PEI‐DA and PEI‐DA‐TAT
Hela and HEK 293 cells were seeded in 24‐well plates and cultured in complete DMEM supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified 5% CO2‐containing incubator for 24 h. After the confluent percentage of the cell culture reached 80%, the medium was replaced with 200 μL fresh medium in the absence of FBS. Meanwhile, the different carriers/pDNA complexes prepared by incubating individually PEI800, PEI25K, PEI‐DA and PEI‐DA‐TAT with pDNA at the N/P ratio of 10 at room temperature for 30 min were added to each well. The cultured medium was replaced by 1 mL fresh complete culture medium with 10% FBS after incubating for 6 h and the cells were further incubated for another 42 h 34.
2.9. Green fluorescent protein (GFP) expression and gene transfection efficiency
To evaluate the transfection efficiency of PEI‐DA and PEI‐TAT in gene delivery application, the GFP expression level was evaluated by flow cytometry. The green fluorescence intensity was detected directly by a FACSCalibur flow cytometer (Becton Dickinson), and the transfection efficiency was calculated by the percentage of positive cells, using non‐transfection cells (mock cells) as the blank control. Briefly, Hela and HEK 293 cells transfected with pEGFP‐N1 by different carriers including (PEI800, PEI25K, PEI‐DA and PEI‐DA‐TAT) at the N/P ratio of 10 were harvested from trypsin digestion after 48 h post‐transfection. Naked DNA without carriers was used as negative control. Cells were washed with PBS buffer, and centrifuged at 1000 rpm for 5 min. The cells were stained by propidium iodide (400 μL, 0.5 μg/mL) in 1x PBS to exclude the dead cells. Approximately, 2×104 cells were analyzed at the flow rate of 200–500 cells per second. CellQuest3.3 software was used for data analysis 35.
2.10. Cellular uptake by confocal laser scanning microscopy
Hela cells at a concentration of 2×105 cells/well were cultured in 6‐well plates loaded with cover‐glass slides for 24 h. 4 μg YOYO‐1 labelled pDNA was prepared according to the instruction from Life Technologies, and then was loaded onto different gene carriers (PEI800 PEI25K, PEI‐DA and PEI‐DA‐TAT) at the N/P ratio of 10 to form carriers/pDNA complexes. Naked DNA was applied as negative control. Cells were incubated with carriers/pDNA complexes for another 4 h before the cells were washed with PBS three times to remove the complexes. After that the cells were fixed with 4% formaldehyde. The cell membrane and nucleus were separately stained by 100 μL of Alexa Fluor 594 (5 μg/mL) and 100 μL of Hoechst 33258 (2 μM) for 15 min at 37 ºC. The cells were further washed with PBS three times to remove the dye and incubated in 1 mL PBS. The cells were kept at room temperature for further analysis. The fluorescence images were observed from a confocal laser scanning microscope (Leica Confocal 1P/FCS) equipped with a 405 nm diodelaser for Hoechst33258, a 488 nm argon ion laser for YOYO‐1 and a 561 diode laser for Alexa Fluor 594. The high magnification images were obtained with a 63x objective. Optical sections were averaged 8 times to reduce noise, and the images were processed using the Leica Confocal software 34.
2.11. Statistical analysis
Data obtained from our experiments were represented as mean ± SE (standard error). Statistical analysis of the numerical variables was performed using a two‐sample, two‐tailed t‐test. A value of p < 0.05 is considered to be significant 36.
3. Results and discussion
3.1. Synthesis and characterization of PEI‐DA and PEI‐DA‐TAT
Comparing with PEI25K, low molecular weight PEI (LMW PEI) such as PEI800 is less toxic to cell growth. However, the transfection efficiency of LMW PEI is low. In order to improve the transfection efficiency of LMW PEI, disulphide bonds were introduced to LMW PEI by the reaction of DA with PEI800 using an EDC/NHS coupling system. Carboxyl groups on DA were reacted with amino groups on PEI800. The feed ratio of the carboxyl groups on DA to the amino groups on PEI800 was controlled at 1:10 to optimise the formation of cross‐linked PEI800 network. As shown in Supporting Information Fig. S1, the FTIR spectrum of PEI‐DA is compared with PEI800 and DA to confirm the successful synthesis of PEI‐DA. PEI800 shows the characteristic bands at 1664 cm−1 and 1041 cm−1, indicating the NH2 vibration and the C‐N stretching, respectively. For the spectrum of DA, the specific signal of 890 cm−1 is contributed to the disulphide bonds. The appearance of the signal at 1639 cm−1 and 1544 cm−1 in the spectrum of PEI‐DA are ascribed to C = O stretching (amide I band) and N‐H deformation (amide II band). This confirms the formation of amide bonds by the reaction between the carboxyl groups on DA and the amino groups on PEI800. Meanwhile, the signal at 879 cm−1 in the PEI‐DA is the evidence of the successful introduction of the disulphide bonds after the reaction 16. In addition, 1H‐NMR was also used for a further confirmation of the chemical structure of PEI‐DA. As shown in Supporting Information Fig. S2, the chemical shifts located within the range of 2.4 to 2.6 ppm (close to the linker) and 3.3 to 3.6 ppm (far from the linker) represent the H‐atom of PEI800. The presence of the chemical shifts for the H atoms of DA are located at δ 2.0, 2.8 and 3.6 ppm. HIV‐1 TAT peptide was further conjugated to PEI‐DA by EDC/NHS coupling to prepare the PEI‐DA‐TAT. After purification, successful synthesis of PEI‐DA‐TAT was confirmed by the FTIR spectra (Supporting Information Fig. S1). PEI‐DA‐TAT shows a similar FTIR result as non‐TAT labelled PEI‐DA due to the low feeding ratio of TAT. In order to verify the success in conjugation of the TAT peptide, FITC labelled PEI‐DA‐TAT were also synthesized and characterized by UV‐vis at a wavelength of 495 nm. PEI‐DA, PEI800 and FITC labelled TAT peptide were measured as the references. The conjugation ratio of TAT on PEI‐DA‐TAT is around 73.3% according to the data from the UV‐vis measurements (Supporting Information Fig. S5).
Buffering capacity of gene carriers is normally considered to play an important role in cellular uptake and endosomal escape. It has been reported that the buffering capacity is related to the ‘proton sponge’ effect of the PEI based carriers which is proposed to be main reason for their endosomal escape. And the endosomal escape of the carrier/DNA complexes plays an important role to effect the final transfection efficiency 8, 37. The buffering capacity of cationic polymers is mainly contributed by the density of the positive charges on the polymers 38. The buffering capacities of PEI‐DA and PEI‐DA‐TAT were measured by back titration from pH 3 to 11 using diluted NaOH. The buffer capacities of PEI25K, PEI800 and PEI‐DA were evaluated as references. As shown in Supporting Information Fig. S3, PEI‐DA shows a broader buffering capacity than that of LMW PEI (PEI800) in the range of pH from 5 to 7 corresponding to the pH change in early endosome, and is comparable to the buffering capacity with commercial PEI (PEI25K). PEI‐DA‐TAT shows the similar buffering capacity as PEI‐DA. This indicates that conjugation of small amount of TAT peptides has no significant change in the buffering capacity. The increased buffering capacity of PEI‐DA‐TAT and PEI‐DA compared with LMW PEI may be contributed from their different chain conformations. Buffering capacity is considered to be related to the endosomal escape ability of gene carriers 8, 37. The good buffering capacity of PEI‐DA‐TAT indicates it should be a more suitable carrier for gene delivery, since it can facilitate gene escape from the late endosome/lysosome and maintain the integral structure for gene delivery leading to highly efficient gene transfection 38.
The entry of incompatible molecules into cells depends on the size of polymers and the elasticity of cell membrane 39. Particles in different sizes penetrate cell membrane via various pathways, including endosome‐mediated endocytosis; clathrin‐mediated endocytosis; caveolae‐mediated endocytosis and micropinocytosis 21. A well‐controlled particle size of polymer/gene complex is essential for gene delivery. It is reported that the optimal size for gene delivery to cells ranges from 100 to 200 nm 40. PEI‐DA/pDNA complexes samples were prepared by mixing PEI‐DA with pDNA at various N/P ratios from 0.5 to 50. The particle sizes of PEI‐DA/pDNA and PEI‐DA‐TAT/pDNA complexes were measured by dynamic light scattering (DLS). As shown in Fig. 1A, the general trend of the particle sizes is that the size of carrier/pDNA complexes prepared from PEI25K, PEI‐DA, PEI‐DA‐TAT reduces with the increase of the N/P ratios, except for PEI800/pDNA complexes, which has a size of more than 350 nm even at the N/P ratio of 50. The large size of PEI800/pDNA complexes at any N/P ratio is attributed by the poor condensing ability of PEI800 due to its chain conformation. DNA complex binding by PEI‐DA‐TAT shows similar particle sizes to the PEI‐DA/DNA complex from the N/P ratio of 0.5 to 50. At the N/P ratio of 10, PEI25K, PEI‐DA, PEI‐DA‐TAT have a similar size of around 150 nm after complexing with pDNA, which falls in the optimal range for gene delivery. Beyond the N/P ratio of 10, these four vehicles are able to condense pDNA to a smaller size. However, the change in the particle size is less significant between the smaller N/P ratios from 0.5 to 5. The particle size of the carrier/pDNA complexes prepared by PEI‐DA, PEI‐DA‐TAT indicates that conjugation of the TAT peptide has no influence on the size of carrier/pDNA complexes and the size of the complexes is mainly depends on the molecular weight of the carriers. The particle size results of PEI‐DA/pDNA and PEI‐DA‐TAT/pDNA complexes suggest that the synthesized carriers could potentially be the suitable carriers for the gene delivery.
Figure 1.
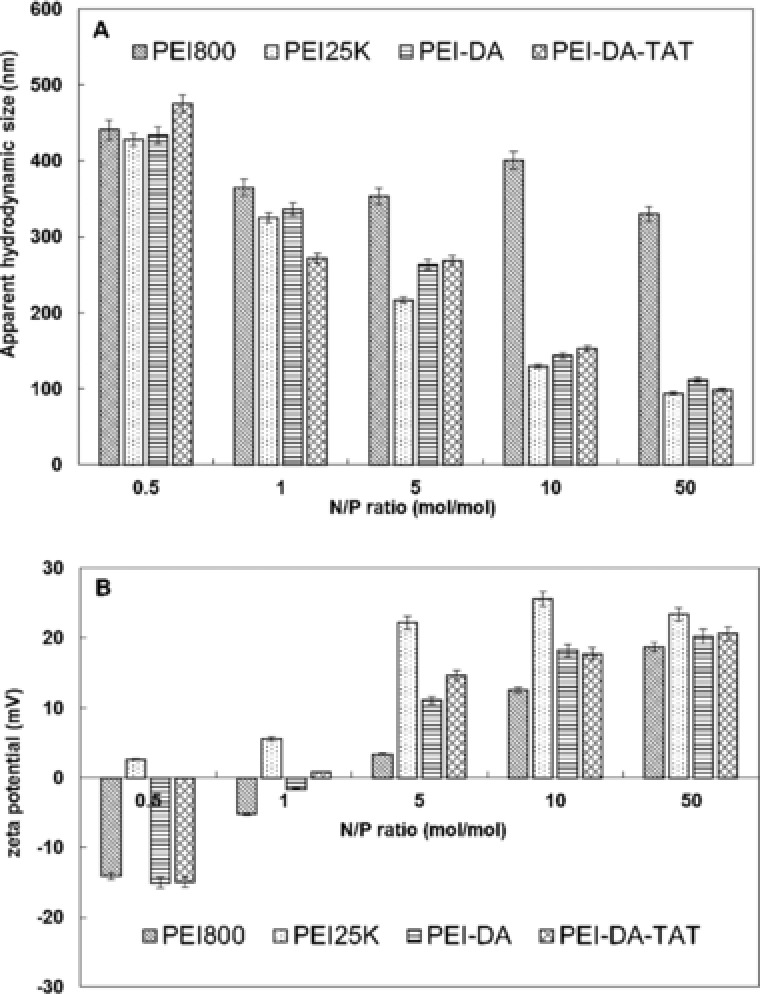
(A) Apparent hydrodynamic sizes and (B) zeta potentials of complexes prepared by mixing PEI800, PEI25K, PEI‐DA and PEI‐DA‐TAT with pDNA at different N/P ratios at pH 7.0 and 25°C. For all measurements, the pDNA concentration was fixed at 5 μg/mL. The experimental data are represented as mean ± SE (standard error, n = 3).
Zeta potential is another crucial factor to affect cellular uptake and transfection efficiency of gene carriers. It is believed that particles with a positively charged surface can implement a better translocation through cell membrane due to the interaction with the negatively charged proteoglycans on the surface of cells 41. The zeta potentials of PEI‐DA/pDNA and PEI‐DA‐TAT/pDNA complexes prepared at pH 7 but different N/P ratios were evaluated. As shown in Fig. 1B, the zeta potential of PEI800/pDNA complexes increased from –14.1 mV at the N/P ratio of 0.5 to 18.7 mV at the N/P ratio of 50. However, when compared with PEI‐DA, PEI‐DA‐TAT and PEI25K at each N/P ratio, the zeta potentials of PEI800/pDNA complexes are all lower than the rest. This confirms that the ability of PEI800 to retard pDNA is poorer than other carriers. The zeta potential of PEI‐DA/pDNA complexes is negative (–15.02 mV) at the initial N/P ratio of 0.5, and then increases with an increase of the N/P ratio from –1.64 mV (at the N/P ratio of 1) to 20.21 mV (at the N/P ratio of 50). This result confirms the gel electrophoresis result of PEI‐DA (Fig. 2C) that PEI‐DA is able to fully retard pDNA at the N/P ratio between 1 and 5. Zeta potentials of PEI‐DA‐TAT change from negative at –16 mV to positive around 20 mV with an increase of the N/P ratios. The zeta potential change becomes insignificant as the N/P ratio increases beyond 10. This could be owing to the sufficient condensation of the pDNA by PEI‐DA‐TAT. In contrast, PEI25K maintains a positive value for zeta potential at the N/P ratio of 0.5. Branched PEI (PEI25K) has been proven to have an excellent pDNA condensing ability because of its high positive charge density 9, which explains its positive zeta potential even in a low N/P ratio. The zeta potential of TAT‐labelled PEI‐DA is similar to that of non‐TAT‐labelled PEI‐DA at all N/P ratios, and this indicates that the conjugation of TAT has no significant on the surface charge of the carrier/pDNA complexes. It can be concluded that PEI‐DA‐TAT is able to condense pDNA into small particles with a positive charge for cellular uptake at and beyond an N/P ratio of 10. The above results of particle sizes and zeta potentials of PEI‐DA/pDNA complexes demonstrates that after crosslinking PEI800 with DA, the gene binding ability is improved due to the change of the chain conformation.
Figure 2.
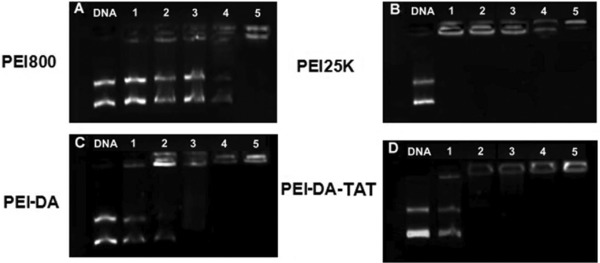
Evaluation of nucleic acid binding and protection capability of (A) PEI800, (B) PEI25K, (C) PEI‐DA and (D) PEI‐DA‐TAT at different N/P ratios (N/P ratios of 0.5, 1, 5, 10 and 50 from lanes 1–5). The left end lane, naked DNA, was used as a control. Exposure time: 400 ms.
3.2. Gene binding ability of PEI‐DA and PEI‐DA‐TAT
Gene binding ability is the prerequisite for an efficient gene delivery system. PEI‐DA and PEI‐DA‐TAT are expected be used to condense and protect the DNA from intracellular enzyme attack and improve the cellular uptake. The gene binding ability of PEI800, PEI25K, PEI‐DA and PEI‐DA‐TAT were performed at different N/P ratios by gel electrophoresis using naked pDNA as a negative control. As shown in Fig. 2, non‐TAT labelled degradable PEI based gene carrier, PEI‐DA shows better gene binding ability (Fig. 2C) than LMW PEI (PEI800) (Fig. 2A), but slightly lower than PEI25K (Fig. 2B). PEI‐DA can fully retard pDNA beyond the N/P ratio of 5 (Fig. 2C, lane 3). The increase in the gene binding ability of PEI‐DA can be contributed to the increase in the change of the chain conformation after crosslinking PEI800. Furthermore, TAT labelled degradable carrier, PEI‐DA‐TAT, can completely bind pDNA beyond the N/P ratio of 1 (Fig. 2D, lane 2). HIV‐1 TAT peptide is a positively charged peptide and it is able to facilitate the electrostatic interaction with the negatively charged pDNA 42. This property of TAT could explain the increase of the gene binding ability of PEI‐DA‐TAT (Fig. 2C) compared with PEI‐DA (Fig. 2D). The gel electrophoresis image indicates that the gene binding ability of PEI‐DA‐TAT is improved compared with PEI‐DA and PEI800. This improved gene condensing ability suggests that it may improve the cellular uptake efficiency and protect pDNA from intracellular enzymatic attacks 43.
3.3. Degradability of PEI‐DA and PEI‐DA‐TAT
To verify the degradability of PEI‐DA and PEI‐DA‐TAT, different concentrations of DTT (20 and 50 mM) were added to the gene carriers for half an hour. After then, gel electrophoresis of the treated PEI‐DA/pDNA and the treated PEI‐DA‐TAT/pDNA complexes was conducted (Fig. 3). As references (shown in Fig. 3, lanes 1 to 4) DNA migration in gel is easily observed in lane 1, which means that PEI800 shows poor gene binding ability compared with PEI25K, PEI‐DA and PEI‐DA‐TAT (lanes 2–4) at the N/P ratio of 10. Slight DNA migration is also observed in lane 5 and lane 6, indicating that PEI‐DA is degraded partially after treating with DTT. Furthermore, PEI‐DA treated with lower concentration of DTT (20 mM, lane 5) shows stronger gene binding ability than that being treated with higher dosage of DTT (50 mM, lane 6). Stronger binding ability represents less degradation of the disulfide bonds in the PEI‐DA. PEI‐DA‐TAT shows similar results to PEI‐DA after treating with DTT (lanes 4, 7 and 8). This demonstrates that the concentration of DTT exert a positive influence on the degradation of PEI‐DA and PEI‐DA‐TAT. And the similar results of PEI‐DA and PEI‐DA‐TAT can be attributed to the different chain conformation of PEI‐DA and PEI‐DA‐TAT after treated by DTT. The reduced DNA binding ability of PEI‐DA and PEI‐DA‐TAT after treating with DTT confirms the degradation of the disulphide bonds in the synthesized carriers.
Figure 3.

Evaluation of biodegradability of PEI‐DA and PEI‐DA‐TAT at the N/P ratio of 10. Naked DNA was used as reference. Lane 1–4: PEI800, PEI25K, PEI‐DA and PEI‐DA‐TAT. Lanes 5 and 6: PEI‐DA treated with 20 mM and 50 mM DTT separately. Lanes 7 and 8: PEI‐DA‐TAT treated with 20 mM and 50 mM DTT separately. Exposure time: 500 ms.
3.4. Cytotoxicity of PEI‐DA and PEI‐DA‐TAT
PEI25K has shown an excellent performance in regarding to condensation of pDNA (Fig. 2), the particle size (Fig. 1A) and the surface charge of the gene/carrier complexes (Fig. 1B). However, cytotoxicity is the major hindrance of PEI25K in gene delivery application 44. The cytotoxicity of synthesized PEI‐DA and PEI‐DA‐TAT was evaluated against Hela and HEK 293 cells. The cell viability was measured by the 3‐(4,5‐dimethy lthiazol‐2‐yl)‐2,5‐diphenyltetrazoliumbro‐mide (MTT) assay at various polymer concentrations (0.5 to 50 μg/mL). Figure 4 shows the cell viability results for Hela cells (A) and HEK 293 cells (B) after incubation of cells with polymer solution for 24 h. It can be seen that PEI800 is non‐toxic to both cell types even at the highest concentration of 50 μg/mL. This justifies our choice of this LMW PEI as the initial material. Similarly, PEI‐DA is also found to have negligible cytotoxicity to either Hela cells or HEK 293 cells at the range of testing concentrations with the cell viability above 85%. And PEI‐DA‐TAT also shows low cytotoxicity in both Hela and HEK 293 cells (cell viability ranged from 80–100%). This suggests that cross‐linking with DA and conjugation with TAT peptide would not change the property of low cytotoxicity of PEI800. This result also confirms that the positively charged TAT is non‐toxic to cells reported by Baoum et al. as well 42. In contrast, commercial PEI (PEI25K) shows a strong toxicity to both Hela and HEK 293 cells and the cell viabilities are lower than 30 % in both Hela and HEK 293 cells at a polymer concentration of 50 μg/mL. The low cytotoxicity of PEI‐DA‐TAT compared with commercial PEI (PEI25K) suggests that PEI‐DA‐TAT could be employed as a safe carrier for gene in the mammalian cells.
Figure 4.
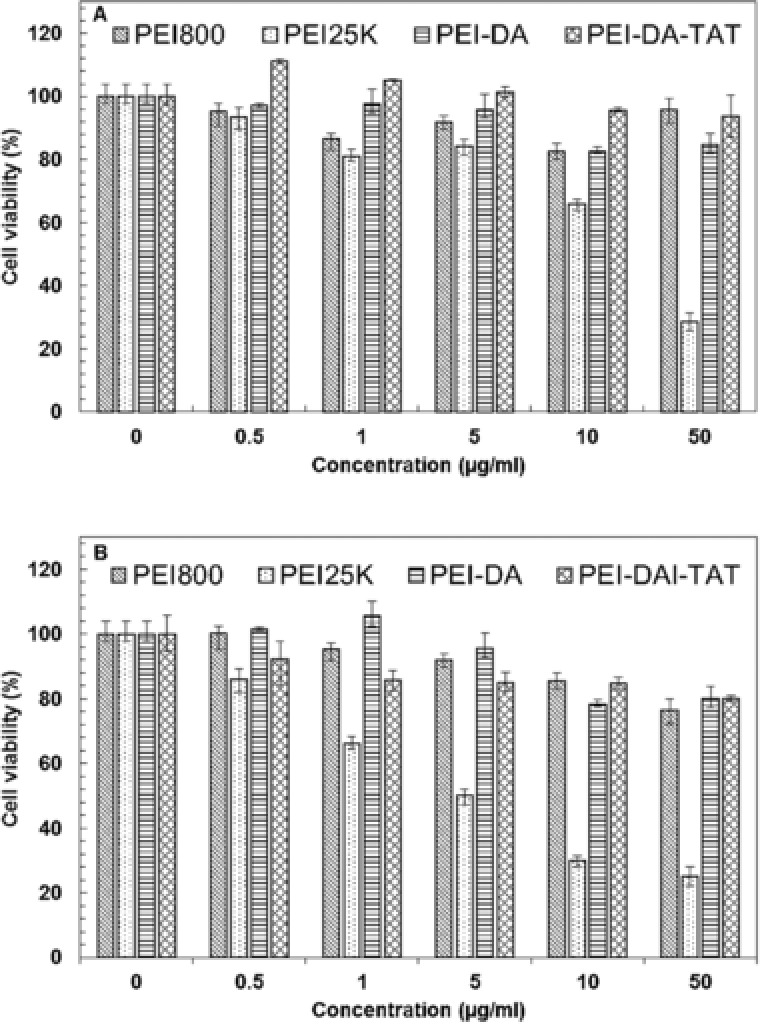
Cell viability MTT assay of (A) Hela cells and (B) HEK 293 cells after exposing to PEI800, PEI25K, PEI‐DA and PEI‐DA‐TAT at different concentrations. The absorbance was read at 570 nm using a microplate reader. The experimental data are represented as mean ± SE (standard error, n = 3).
3.5. Gene delivery of PEI‐DA and PEI‐DA‐TAT/pDNA complexes
To evaluate the DNA cellular uptake after being delivered by PEI‐DA and PEI‐DA‐TAT, pDNA dyed with YOYO‐1 (with green fluorescence) was transfected into Hela cells at the N/P ratio of 10. The cell membrane was dyed with Alexa Fluor 594 (Red) and the nucleus was dyed with Hoechst 33258 (Blue) to distinguish the distribution of pDNA. The images of DNA cellular uptake were taken under a confocal microscope after 6 h transfection before GFP protein was expressed inside the cells. Cell membrane was stained by Alexa Fluor 594 to localize the position of the pDNA after delivering it with different carriers. However, the membrane image was spray‐out in whole cell area. The reasons of spray‐out of the red signal in the image could be as follows: (i) nuclear membrane and the endoplasmic reticulum were also stained by Alexa Fluor 594; (ii) the exposure time for the image might be too long; (iii) the cells were over‐labelled with Alexa Fluor 594 which leads to a nonspecific stain. As shown in Fig. 5, green fluorescence signals representing pDNA samples are found in both cytoplasm and nucleus of Hela cells when pDNA is delivered by PEI25K, PEI‐DA and PEI‐DA‐TAT (Fig. 5, Row 2) and none is detected in cells delivered with naked DNA. Relatively weak green fluorescent signal was detected when DNA was delivered by PEI800 (Fig. 5, Row 2). PEI800 has less positive charge density than other carriers, and it causes the large size of the PEI800/pDNA complexes. This prevents the entry through the cell membrane and leads to lower DNA uptake and observably weak green fluorescence DNA signals. The naked DNA has a poor cell membrane penetration due to its negative charge and large hydrodynamic size. Naked DNA may also be easily digested by the enzymes inside the cells into small fragments which are easily ‘wash out’ from the cells, therefore the cells do not exhibit green fluorescence signals transfected by naked DNA. It is noted that when PEI25K is employed, the majority of green fluorescence signal is located in the cytoplasm in the Hela cells and a very small amount of green fluorescence signals located in the nucleus (as shown in Supporting Information Fig. S6). It could be assumed that the PEI25K/pDNA complexes escaping from late endosomes may not be able to transport DNA to the nucleus due to the lack of the capability to penetrate the nucleus barrier and/or its large hydrodynamic size 45. As comparison, transfected cells delivered by PEI‐DA appears to have distinguished green fluorescence signals located in the nucleus, and PEI‐DA‐TAT performs better than PEI‐DA regarding DNA uptake in nucleus (as shown in Supporting Information Fig. S6). The DNA cellular uptake ability of gene carriers is one of the most important factors to influence the gene transfection efficiency 46. With more pDNA translocated in the nucleus of the cells when delivered by PEI‐DA and PEI‐DA‐TAT, PEI‐DA and PEI‐DA‐TAT are expected to perform efficient gene transfection. The gene transfection efficiency of PEI‐DA and PEI‐DA‐TAT was quantified using flow cytometry based on the amount of expressed GFP. The measurements were conducted through transfecting PEI‐DA and PEI‐DA‐TAT complexed with pEGFP‐N1 plasmid at the N/P ratio of 10 in Hela and HEK 293 cells. pEGFP‐N1 plasmid delivered via PEI800 and PEI25K were used as references and naked pDNA without carriers was also employed as a control. As shown in Fig. 6, the transfection efficiency of LMW PEI (PEI800) is quite low (2.38 and 2.81%, respectively) in Hela cells (∼11 fold less than PEI‐DA and ∼14 fold less than PEI‐DA‐TAT) and HEK 293 cells (∼8 fold less than PEI‐DA and ∼11 fold less than PEI‐DA‐TAT). The poor DNA cellular uptake ability of PEI800 (as shown in Fig. 5) explains the low transfection efficiency in the gene delivery. Non‐TAT labelled PEI‐DA shows a similar transfection efficiency compared with PEI25K in both Hela (26.77% vs. 24.23%) and HEK 293 cells (23.67% vs. 25.22 %) (as shown in Supporting Information Fig. S4). However, this transfection efficiency measurement is based on the viable cells after excluding the dead cells by the dye, PI. In the cytotoxicity assay, PEI‐DA shows a much lower cytotoxicity (cell viability beyond the concentration of 10 μg/mL is around 80% in Hela and HEK 293 cells) compared with PEI25K (cell viability beyond the concentration of 10 μg/mL is lower than 70% in Hela and 30% in HEK 293 cells), and the working concentration of carriers in the evaluation of DNA transfection is around 20 μg/mL. The lower cell viability of PEI25K in Hela cells and HEK293 cells explains the close transfection efficiency with PEI‐DA measured by the flow cytometry after excluding dead cells in the test. Besides, similar results in the particle size, zeta potential and buffering capacity of PEI‐DA and PEI25K could also be the reason for their similar transfection efficiencies. On the other hand, PEI‐DA‐TAT has shown high transfection efficiency in both Hela (34.82%) and HEK 293 cells (31.26%) (as shown in Supporting Information Fig. S4) with the conjugation of TAT. The improvement of the transfection efficiency of PEI‐DA‐TAT with low feeding ratio of TAT to the amino groups on PEI‐DA is around 8% compared with PEI25K, which is comparable to another TAT‐modified gene delivery carrier prepared by Torchilin et al. 47, as the transfection efficiency was increased by 5–10% in NIH/3T3 and H9C2 cells with the introduction of TAT peptide.
Figure 5.
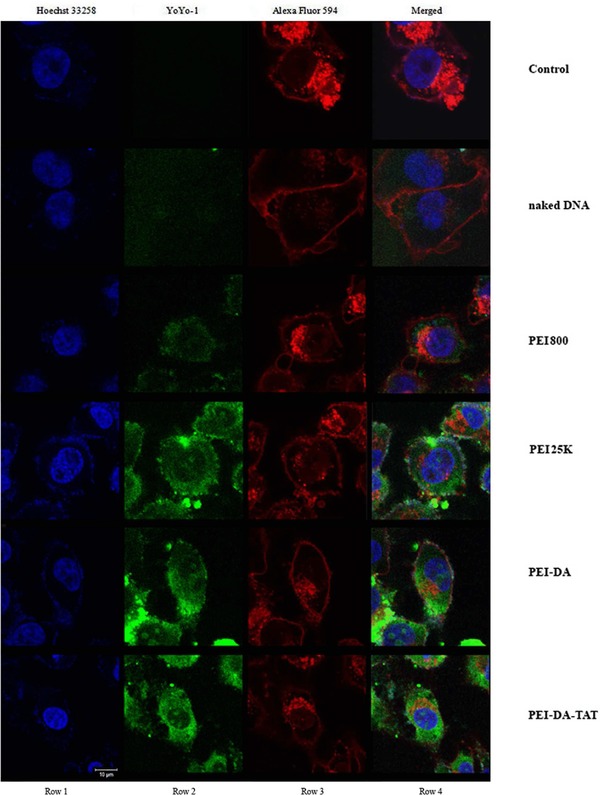
Cellular uptake of YOYO‐1 labelled pDNA complexed with different gene delivery vectors: PEI800, PEI25K, PEI‐DA and PEI‐DA‐TAT at an N/P ratio of 10 in Hela cells. Mock cells were used as a control, and naked pDNA was used as the reference.
Figure 6.
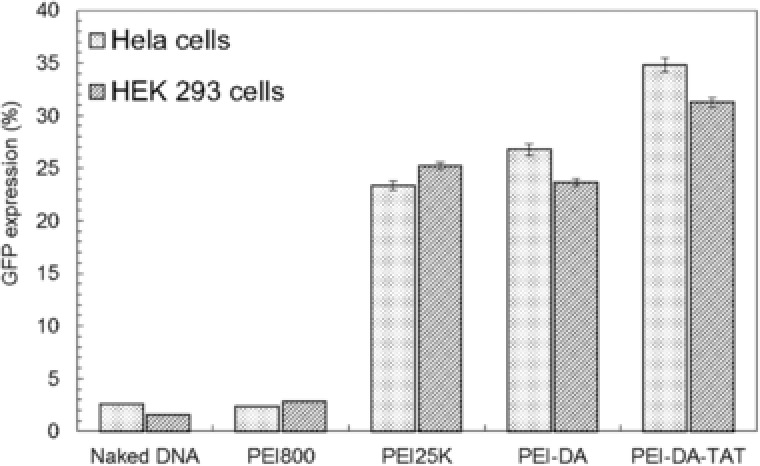
GFP expression in Hela cells and HEK 293 cells transfected by pEGFP‐N1 mediated by naked DNA, PEI800, PEI25K, PEI‐DA and PEI‐DA‐TAT at the N/P ratio of 10. The experimental data are represented as mean ± SE (standard error, n = 3).
4. Concluding remarks
In this study, we report the novel cell‐penetrating peptide labelled and degradable gene delivery carriers, PEI‐DA and PEI‐DA‐TAT, for high efficient gene delivery with low cytotoxicity. PEI‐DA and PEI‐DA‐TAT both show relatively high cell viability in Hela cells and HEK 293 cells at the working concentration at around 20 μg/mL. The particle size of PEI‐DA/pDNA complexes is lower than 200 nm beyond the N/P ratio of 10, suggesting that PEI‐DA is able to condense and retard pDNA at a low N/P ratio of carrier/pDNA, which is much better than that of LMW PEI. In the performance of gene transfection, PEI‐DA performs a comparable gene transfection to PEI25K, but the gene transfection efficiency can be further enhanced 8% after TAT conjugation. The increased transfection efficiency is attributed to the fact that TAT facilitates cell uptake. In summary, the PEI‐DA‐TAT displays satisfied nucleic acid loading ability, low toxicity, good cellular uptake ability, high gene transfection efficiency and degradability, which render it a promising carrier for high efficient and safe gene delivery systems.
Practical application
It has been demonstrated that therapeutic nucleic acids delivered into cells can treat various human diseases including cancers and genetic diseases. However, the development of satisfying gene carriers is limited by various barriers, such as cytotoxicity, transfection efficiency, and others. In this research, we synthesized a low toxic and degradable gene carrier by crosslinking low molecular weight polyethylenimine (PEI800) with 4ʹ4‐dithiodibutyric acid, and the HIV‐1 Trans‐Activator of Transcription peptide (HIV‐1 TAT) was used as a model cell‐penetrating peptide to promote cell uptake. The resulted gene carriers showed higher transfection performance and lower cytotoxicity to cells in the working concentrations compared with the commercial gene carrier, PEI25K. This gene carrier can have potential applications such as gene delivery for gene therapy and/or DNA vaccination.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting figures
5 References
- 1. Cross, D. , Burmester, J. K. , Gene therapy for cancer treatment: Past, present and future. Clin. Med. Res. 2006, 4, 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friedman, T. , Roblin, R. , Gene therapy for human genetic disease. Science 1972, 175, 949–955. [DOI] [PubMed] [Google Scholar]
- 3. Keeney, M. , Onyiah, S. , Zhang, Z. , Tong, X. et al., Modulating polymer chemistry to enhance non‐viral gene delivery inside hydrogels with tunable matrix stiffness. Biomaterials 2013, 34, 9657–9665. [DOI] [PubMed] [Google Scholar]
- 4. Kay, M. A. , Glorioso, J. C. , Naldini, L. , Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [DOI] [PubMed] [Google Scholar]
- 5. Yin, H. , Kanasty, R. L. , Eltoukhy, A. A. , Vegas, A. J. et al., Non‐viral vectors for gene‐based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [DOI] [PubMed] [Google Scholar]
- 6. Lv, H. , Zhang, S. , Wang, B. , Cui, S. et al., Toxicity of cationic lipids and cationic polymers in gene delivery. J. Controlled Release 2006, 114, 100–109. [DOI] [PubMed] [Google Scholar]
- 7. Wiseman, J. W. , Goddard, C. A. , McLelland, D. , Colledge, W. H. , A comparison of linear and branched polyethylenimine (PEI) with DCChol/DOPE liposomes for gene delivery to epithelial cells in vitro and in vivo. Gene Ther. 2003, 10, 1654–1662. [DOI] [PubMed] [Google Scholar]
- 8. Akinc, A. , Thomas, M. , Klibanov, A. M. , Langer, R. , Exploring polyethylenimine‐mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. 2005, 7, 657–663. [DOI] [PubMed] [Google Scholar]
- 9. Taranejoo, S. , Liu, J. , Verma, P. , Hourigan, K. , A review of the developments of characteristics of PEI derivatives for gene delivery applications. J. Appl. Polym. Sci. 2015, 132, 42096. [Google Scholar]
- 10. Funhoff, A. M. , van Nostrum, C. F. , Koning, G. A. , Schuurmans‐Nieuwenbroek, N. M. E. et al., Endosomal escape of polymeric gene delivery complexes is not always enhanced by polymers buffering at low pH. Biomacromolecules 2004, 5, 32–39. [DOI] [PubMed] [Google Scholar]
- 11. Kichler, A. , Gene transfer with modified polyethylenimines. J. Gene Med. 2004, 6, S3–S10. [DOI] [PubMed] [Google Scholar]
- 12. Wang, H. , Sun, Y. , Deng, J. , Yang, J. et al., Effect of peptides and their introduction methods on target gene transfer of gene vector based on disulfide‐containing polyethyleneimine. Int. J. Pharm. 2012, 438, 191–201. [DOI] [PubMed] [Google Scholar]
- 13. Kim, Y. H. , Park, J. H. , Lee, M. , Kim, Y.‐H. et al., Polyethylenimine with acid‐labile linkages as a biodegradable gene carrier. J. Control. Release 2005, 103, 209–219. [DOI] [PubMed] [Google Scholar]
- 14. Heebeom, K. , Geunwoo, J. , Hyunseo, K. , Yan, L. et al., Biodegradable branched poly(ethylenimine sulfide) for gene delivery. Biomaterials 2010, 31, 988–997. [DOI] [PubMed] [Google Scholar]
- 15. Peng, Q. , Zhong, Z. , Zhuo, R. , Disulfide cross‐linked polyethylenimines (PEI) prepared via thiolation of low molecular weight PEI as highly efficient gene vectors. Bioconj. Chem. 2008, 19, 499–506. [DOI] [PubMed] [Google Scholar]
- 16. Zha, X. , Li, Z. , Pan, H. , Liu, W. et al., Enhanced gene delivery by chitosan‐disulfide‐conjugated LMW‐PEI for facilitating osteogenic differentiation. Acta Biomater. 2013, 9, 6694–6703. [DOI] [PubMed] [Google Scholar]
- 17. Yu, H. , Russ, V. , Wagner, E. , Influence of the molecular weight of bioreducible oligoethylenimine conjugates on the polyplex transfection properties. AAPS J. 2009, 11, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mei, O. , Rongzuo, X. , Sun Hwa, K. , Bull, D. A. et al., A family of bioreducible poly(disulfide amine)s for gene delivery. Biomaterials 2009, 30, 5804–5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gosselin, M. A. , Guo, W. , Lee, R. J. , Efficient Gene Transfer Using Reversibly Cross‐Linked Low Molecular Weight Polyethylenimine. Bioconj. Chem. 2001, 12, 989–994. [DOI] [PubMed] [Google Scholar]
- 20. Christensen, L. V. , Chang, C. , Kim, W. J. , Kim, S. W. et al., Reducible poly(amido ethylenimine)s designed for triggered intracellular gene delivery. Bioconj. Chem. 2006, 17, 1233–1240. [DOI] [PubMed] [Google Scholar]
- 21. Jones, A. T. , Sayers, E. J. , Cell entry of cell penetrating peptides: tales of tails wagging dogs. J. Control. Release 2012, 161, 582–591. [DOI] [PubMed] [Google Scholar]
- 22. Verdurmen, W. P. R. , Thanos, M. , Ruttekolk, I. R. , Gulbins, E. et al., Cationic cell‐penetrating peptides induce ceramide formation via acid sphingomyelinase: Implications for uptake. J. Control. Release 2010, 147, 171–179. [DOI] [PubMed] [Google Scholar]
- 23. Kaplan, I. M. , Wadia, J. S. , Dowdy, S. F. , Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 2005, 102, 247–253. [DOI] [PubMed] [Google Scholar]
- 24. Fittipaldi, A. , Ferrari, A. , Zoppe, M. , Arcangeli, C. et al., Cell membrane lipid rafts mediate caveolar endocytosis of HIV‐1 Tat fusion proteins. J. Biol. Chem. 2003, 278, 34141–34149. [DOI] [PubMed] [Google Scholar]
- 25. Ferrari, A. , Pellegrini, V. , Arcangeli, C. , Fittipaldi, A. et al., Caveolae‐Mediated Internalization of Extracellular HIV‐1 Tat Fusion Proteins Visualized in Real Time. Mol. Ther. 2003, 8, 284–294. [DOI] [PubMed] [Google Scholar]
- 26. Truant, R. , Cullen, B. R. , The arginine‐rich domains present in human immunodeficiency virus type 1 Tat and rev function as direct importin β‐dependent nuclear localization signals. Mol. Cell. Biol. 1999, 19, 1210–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pan, L. , He, Q. , Liu, J. , Chen, Y. et al., Nuclear‐Targeted Drug Delivery of TAT Peptide‐Conjugated Monodisperse Mesoporous Silica Nanoparticles. J. Am. Chem. Soc. 2012, 134, 5722–5725. [DOI] [PubMed] [Google Scholar]
- 28. Lee, J. Y. , Choi, Y. S. , Suh, J. S. , Kwon, Y. M. et al., Cell‐penetrating chitosan/doxorubicin/TAT conjugates for efficient cancer therapy. Int. J. Cancer 2011, 128, 2470–2480. [DOI] [PubMed] [Google Scholar]
- 29. Li, Z. , Dong, K. , Huang, S. , Ju, E. et al., A smart nanoassembly for multistage targeted drug delivery and magnetic resonance imaging. Adv. Funct. Mater. 2014, 24, 3612–3620. [Google Scholar]
- 30. Torchilin, V. P. , Levchenko, T. S. , Rammohan, R. , Volodina, N. et al., Cell transfection in vitro and in vivo with nontoxic TAT peptide‐liposome–DNA complexes. Proc. Natl. Acad. Sci. 2003, 100, 1972–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen, S. , Han, K. , Yang, J. , Lei, Q. et al., Bioreducible polypeptide containing cell‐penetrating sequence for efficient gene delivery. Pharm. Res. 2013, 30, 1968–1978. [DOI] [PubMed] [Google Scholar]
- 32. Yeh, I.‐C. , Hummer, G. , System‐size dependence of diffusion coefficients and viscosities from molecular dynamics simulations with periodic boundary conditions. J. Phys. Chem. B 2004, 108, 15873–15879. [Google Scholar]
- 33. Denizot, F. , Lang, R. , Rapid colorimetric assay for cell growth and survival: Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [DOI] [PubMed] [Google Scholar]
- 34. Shi, B. , Zhang, H. , Bi, J. , Dai, S. , Endosomal pH responsive polymers for efficient cancer targeted gene therapy. Colloids Surf. B. Biointerfaces 2014, 119, 55–65. [DOI] [PubMed] [Google Scholar]
- 35. Shi, B. , Zhang, S. , Wang, Y. , Zhuang, Y. et al., Expansion of mouse sertoli cells on microcarriers. Cell Prolif. 2010, 43, 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi, B. , Deng, L. , Shi, X. , Dai, S. et al., The enhancement of neural stem cell survival and growth by coculturing with expanded sertoli cells in vitro. Biotechnol. Prog. 2012, 28, 196–205. [DOI] [PubMed] [Google Scholar]
- 37. Lin, C. , Engbersen, J. F. J. , Effect of chemical functionalities in poly(amido amine)s for non‐viral gene transfection. J. Control. Release 2008, 132, 267–272. [DOI] [PubMed] [Google Scholar]
- 38. Lin, C. , Zhong, Z. , Lok, M. C. , Jiang, X. et al., Novel bioreducible poly(amido amine)s for highly efficient gene delivery. Bioconj. Chem. 2007, 18, 138–145. [DOI] [PubMed] [Google Scholar]
- 39. Liu, Y. , Reineke, T. M. , Hydroxyl stereochemistry and amine number within poly(glycoamidoamine)s affect intracellular DNA delivery. J. Am. Chem. Soc. 2005, 127, 3004–3015. [DOI] [PubMed] [Google Scholar]
- 40. Endres, T. K. , Beck‐Broichsitter, M. , Samsonova, O. , Renette, T. et al., Self‐assembled biodegradable amphiphilic PEGePCLelPEI triblock copolymers at the borderline between micelles and nanoparticles designed for drug and gene delivery. Biomaterials 2011, 32, 7721–7731. [DOI] [PubMed] [Google Scholar]
- 41. Hess, G. T. , Humphries Iv, W. H. , Fay, N. C. , Payne, C. K. , Cellular binding, motion, and internalization of synthetic gene delivery polymers. Biochim. Biophys. Acta 2007, 1773, 1583–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baoum, A. , Xie, S. , Fakhari, A. , Berkland, C. , “Soft” Calcium crosslinks enable highly efficient gene transfection using tat peptide. Pharm. Res. 2009, 26, 2619–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thomas, M. , Klibanov, A. M. , Enhancing polyethylenimine's delivery of plasmid DNA into mammalian cells. Proc. Natl. Acad. Sci. U S A 2002, 99, 14640–14645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Teo, P. Y. , Yang, C. , Hedrick, J. L. , Engler, A. C. et al., Hydrophobic modification of low molecular weight polyethylenimine for improved gene transfection. Biomaterials 2013, 34, 7971–7979. [DOI] [PubMed] [Google Scholar]
- 45. Shi, B. , Zhang, H. , Dai, S. , Du, X. et al., Intracellular microenvironment responsive polymers: A multiple‐stage transport platform for high‐performance gene delivery. Small 2014, 10, 871–877. [DOI] [PubMed] [Google Scholar]
- 46. Peng, L. H. , Niu, J. , Zhang, C. Z. , Yu, W. et al., TAT conjugated cationic noble metal nanoparticles for gene delivery to epidermal stem cells. Biomaterials 2014, 35, 5605–5618. [DOI] [PubMed] [Google Scholar]
- 47. Torchilin, V. P. , Tat peptide‐mediated intracellular delivery of pharmaceutical nanocarriers. Adv. Drug Del. Rev. 2008, 60, 548–558. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting figures