

Abstract
SilCoat‐biocatalysts are immobilized enzyme preparations with an outstanding robustness against leaching and mechanical stress and therefore promising tools for technical synthesis. They consist of a composite material made from a solid enzyme carrier and silicone. In this study, a method has been found to enable provision of these catalysts in large scale. It makes use of easily scalable fluidized‐bed technology and, in contrast to the original method, works in almost complete absence of organic solvent. Thus, it is both a fast and safe method. When the Pt‐catalyst required for silicone formation is cast on the solid enzyme carrier before coating, resulting composites resemble the original preparations in morphology, catalytic activity, and stability against leaching and mechanical forces. Only the maximum total content of silicone in the composites lies about 10% w/w lower resulting in an overall leaching stability below the theoretical maximum. When the Pt‐catalyst is mixed with cooled siloxane solution before coating, surficial coating of the enzyme carriers is achieved, which provides maximum leaching stability at very low silicone consumption. Thus, the technology offers the possibility to produce both composite and for the first time also core‐shell silCoat‐particles, and optimize leaching stability over mechanical strength according to process requirements.
Keywords: Biocatalyst, Coating, Entrapment, Enzyme, Silicone
Abbreviations
- EDX
energy dispersive X‐ray spectroscopy
- MCC
microcrystalline cellulose
- NZ435
Novozym 435®
- PDMS
poly(dimethyl siloxane)
- PLU
propyl laurate units
- SEM
scanning electron microscopy
1. Introduction
Immobilization of isolated enzymes is an accepted way to simplify their storing and handling and enable their continuous or repetitive use. Thus, it has become almost a prerequisite for the technical application of enzymes in biocatalysis. Among the multitude of methods described to date 1, 2, physical adsorption onto solid carriers has gained particular importance for industrial use. This is due to the simplicity and cost‐effectiveness of enzyme adsorption as well as the exertion of an overall low impact on catalytic activity by this method. A major drawback, however, lies in the frequently observed continuous leaching of enzyme from the carrier under technical process conditions 3. A way to overcome this problem has recently been introduced with the so‐called silCoat‐technology 4, 5, 6. This deposits silicone on carriers with adsorbed enzyme and forms a composite material with significantly improved stability against enzyme leaching. Additionally, these silCoat‐composites are less susceptible to disintegration by mechanical forces 7, 8. It was demonstrated for Novozym 435® (NZ435; Novozymes, Denmark) and related carrier‐bound enzymes that their technical applicability can thus be considerably extended 9.
Preparation of silCoat‐material as described to date involves mixing of silicone precursors, enzyme carrier, and cross‐linking catalyst (Pt‐derivative) in an organic solvent followed by polymer curing and final drying. Depending on the temperature, the overall preparation time from start to finish ranges between two and four hours at a maximum scale of 10 g per batch. This is fine for lab‐scale experiments, but has considerable drawbacks for preparative scale (>100 kg) provision of biocatalysts: Consumption of large amounts of expensive and hazardous organic solvents is neither economical nor environmentally friendly. In addition, severe safety risks (explosion hazard) might arise from the combination of easily flammable solvents and potentially electrostatic material such as enzyme carriers.
Here, we report on the development of a solvent‐free, fast, and scalable method for the preparation of silCoat‐biocatalysts. The method is based on a “Wurster”‐design‐fluidized‐bed reactor as a well‐understood and particularly suitable device for the coating of solid particles 10, which was adapted to the specific needs of silicone formation via hydrosilylation. Resulting preparations were evaluated with regard to morphology and operational robustness using NZ435 and a silicone made from α,ω‐divinyl‐terminated PDMS (poly(dimethyl siloxane)) with an average chain length of 100 (100 SiO‐units per molecule) and a comb‐like SiH‐activated PDMS with five SiH‐groups on an average chain length of 50 7 as model system. Transferability to alternative carrier systems was demonstrated for selected examples.
2. Materials and methods
2.1. Material
Coating experiments were performed in the commercial fluidized‐bed reactors “Mini‐Glatt” and “Uni‐Glatt Pilot” (Glatt Ingenieurtechnik GmbH, Weimar, Germany). Novozym 435® was obtained from Novozymes A/S (Bagsvaerd, Denmark). α,ω‐Divinyl‐terminated PDMS A100: CH2 = CH‐(SiMe2O)98‐SiMe2O‐CH = CH2, SiH‐activated PDMS B5: Me3SiO‐(SiMe2O)43‐(SiHMeO)5‐SiMe3, and Karstedt‐catalyst were provided by Evonik Industries AG (Essen, Germany). All other chemicals were purchased from Sigma‐Aldrich (Germany) and were used as obtained.
2.2. Activity assay
Catalytic activity of NZ435‐preparations was determined at the esterification of lauric acid and propanol, and was assessed as propyl laurate units (PLU). One PLU corresponds to the amount of NZ435‐preparation (g) that catalyzes the production of 1 μmol propyl laurate per minute. Reactions were performed at 60°C in closed glass vessels containing 10–20 mg of particles and an equimolar, solvent‐free solution of lauric acid and 1‐propanol (5 mL). Samples (50 μL) were taken from the solution every 5 min over a total period of 25 min and were diluted with decane (950 μL) containing 4 mM dodecane as internal standard. Product formation was analyzed via gas chromatography on a BTX column as described previously 4.
2.3. Determination of enzyme leaching
Hundred milligrams of NZ435‐preparations were placed in sealable glass vessels, covered with 5 mL MeCN/H2O (1:1) and stirred at 45°C for 30 min. Particles were recovered through filtration, rinsed with 100 mL pure water, dried for 3h at 50°C and investigated for residual activity.
2.4. Determination of mechanical stability
One gram of NZ435‐preparations and 4 g of glass beads (4 mm ∅) were placed in an electric swing mill (MM301, Retsch, Haan, Germany) and pounded for 5 min at a frequency of 30 s−1. The particles were then rinsed with an aqueous detergent solution (1% w/w) through a series of sieves (mesh sizes 50–800 μm), washed with 100 mL of pure water, and dried on the sieves at 50°C for 1 h. The sieves were gravimetrically analyzed and the relative amount of different sized particles was determined.
2.5. Characterization of particles
Scanning electron microscopy (SEM) was conducted on a Hitachi S‐2700 instrument at an acceleration voltage of 20 kV. Element distribution mappings were performed via energy dispersive X‐ray spectroscopy (EDX) using a Röntec‐XFlash detector attached to the scanning electron microscope at beam currents of 20 nA. Cross‐sections of particles were prepared by quick freezing under liquid nitrogen and successive grinding in a mortar. Samples were fixed on double‐sided adhesive foils, water was evaporated, and samples were then spluttered with gold films for SEM and with carbon films for EDX, respectively.
3. Results and discussion
3.1. Fluidized‐bed procedure for NZ435‐coating with silicone
Coating in a “Wurster”‐design fluidized‐bed reactor in principal involves mobilization of solid particles or powders via an air stream passing the material in a way that a state of “solid fluidization” is achieved. The particle stream is channeled upwards through a central cylinder (the “Wurster”‐tube), and breaks up at its top letting the particles drop slowly back in the periphery of the reactor. The coating material is unidirectionally sprayed into the particle stream to connect with the solids (Fig. 1). This system was particularly developed for coating of small particles to prevent premature agglomeration as observed in standard bottom‐spray systems since the particle circulation increases the drying rate and reduces the potential for particle agglomeration through rapid relative movement of particles 10. Considering the edge conditions connected with preparation of silCoat‐biocatalysts (i.e. coating of small‐sized particles with highly viscous and “sticky” silicone) this seemed the appropriate technology to adapt. Application to silCoat‐formation required identification of measures to (1) appropriately fluidize the specific particles, (2) introduce siloxanes into the fluidized‐bed, and (3) enable silicone curing on the solid supports.
Figure 1.
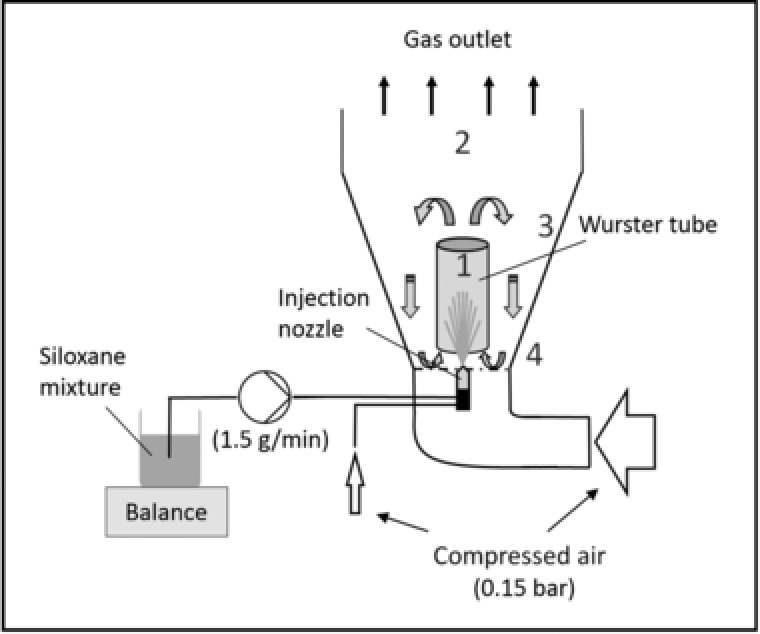
Scheme of the “Wurster”‐design‐fluidized‐bed reactor with (1): up‐bed region, (2): highest fluidization point, (3): deceleration region, (4): down‐bed region.
3.1.1. Fluidization of NZ435
Fluidization of particles in a (“Wurster”‐design) fluidized‐bed reactor is principally determined by the amount of particles filling the reactor and by the intensity of the air stream entering the reactor. Here, 40 and 370 g of NZ435 were introduced into the Mini‐Glatt laboratory reactor and the Uni‐Glatt pilot scale reactor, respectively. According to El Mafadi et al. 11 this filling corresponds to 30 and 50% of the respective maximum capacity for particle uptake of the two reactors (see Eq. (1)) and should ensure both quick uptake of coating material and a coating efficiency of up to 98%.
| (1) |
with M: total reactor's capacity for particle uptake in kg; r1: radius of the reactor chamber; r2: radius of the “Wurster”‐tube; L: length of the “Wurster”‐tube; ρP: particle density.
For fluidization of this amount of particles, compressed air was injected into the reactor with an initial pressure of 0.1–0.12 bar. Due to the increase in specific mass of the particles during the coating process, successive increase of the pressure to a final value of 0.2 bar was necessary to keep the fluidized‐bed stable. This adaption was performed manually and required a good deal of experience in order to avoid overdue collapse of the fluidized‐bed on the one hand and drag‐out of particles in the air stream on the other hand.
3.1.2. Introduction of silicone‐precursors into the fluidized‐bed
Injection of siloxanes into the fluidized‐bed was accomplished “bottom‐up” by use of a two‐component jet nozzle with external mixing creating fluid drops with a size of 30–50 μm in diameter. This small drop size enabled homogeneous distribution of the highly viscous coating material (50–150 Pas−1) in the fluidized‐bed without addition of solubilizing organic solvents such as cyclohexane. For safety reasons and under consideration of eco‐friendliness, solvent‐free operation is an important requirement for application of this technology in large scale. In addition, the small droplet size ensures fast drying during the coating process 12, and favors unhindered permeation of the material into the pores of NZ435 for composite formation. The fluid was produced with a constant pressure of 0.15 bar at the jet nozzle. A mixture of PDMS A100 and PDMS B5 was transferred with a peristaltic pump from a 100‐ to 500‐mL reservoir into the jet nozzle at a speed of 1.5 g/min until the fluidized‐bed collapsed. Depending on the final silicone loading (see the following section) this involved up to 31.4 g in the Mini‐Glatt reactor and 290.7 g in the Uni‐Glatt reactor and was completed within 21 and 194 min, respectively. For tubing, Marpren tubes were chosen to avoid swelling and leaching in the presence of siloxane. Transportation of the viscous coating material within these tubes was facilitated by a large outer diameter (4.8 mm) and small inner diameter (2.4 mm).
3.1.3. Silicone curing on fluidized particles
Curing of divinylated PDMS and SiH‐activated PDMS to form a continuous three‐dimensional silicone elastomer can be achieved through Pt‐catalyzed hydrosilylation. SilCoat‐formation, i.e. formation of interpenetrating network composites, occurs when the reaction is run in presence of particulate enzyme carriers such as NZ435 7. A major problem in the transfer of this process to a fluidized‐bed reactor lies in the obligation to ensure that curing of silicone proceeds only after collision of siloxanes with particles in order to avoid abundant production of carrier‐less silicone matrices and “coating” of reactor internals. Two approaches were developed to address this issue: Provision of the Pt‐catalyst on the particle surface for locally defined hydrosilylation, and temperature‐controlled retardation of hydrosilylation in a siloxane‐catalyst‐mixture, respectively.
3.1.3.1. Composite formation through provision of Pt‐catalyst on the particle surface
The Pt‐catalyst (Karstedt‐catalyst) was deposited onto the surface (and into the pores) of NZ435 by incubating NZ435 in a solution of the catalyst in cyclohexane until the solvent was completely evaporated. The resulting amount of Pt on the carrier was below the detection limit of EDX applied to examine cross‐sections of the treated carriers for Pt‐content. The carriers were fluidized and contacted with siloxane spray (see previous section) at a process temperature of 60°C. Formation of carrier‐less silicone particles or silicone layers on reactor internals did not occur under these conditions. Siloxane uptake by the NZ435‐particles to a maximum loading of 44% (w/w, weight silicone per total weight of particles) was accomplished, as was calculated from the balanced mass of siloxane solution introduced into the reactor. This maximum loading, however, was only achieved when the coating process (i.e. siloxane injection) was repeatedly interrupted for a short time to allow full silicone uptake and curing (visible as drying) and thus unstick aggregated particles. Coating without intermediate interruptions yielded a silicone loading of only up to 30–35% w/w. When the silicone loading exceeded 44% w/w a wet siloxane film formed on the particle surfaces, which enhanced the undesired aggregation of particles and led to the irreversible collapse of the fluidized‐bed. It was not possible to “dry” this film through an increased curing time indicating that no Pt‐catalyst was accessible on the outer carrier surface. Accordingly, coverage of the outer carrier surface as described for composite silCoat‐NZ435 obtained from preparation in a vessel with a silicone loading of 52–54% w/w was not possible with this method. Treatment of loaded carriers (44% w/w) with toluene or cyclohexane did not extract significant amounts of PDMS indicating that silicone formation within the carrier was successful and a composite must consequently have formed. Judging from the different color of the obtained particles (white as of uncoated particles to slightly yellow as of fully coated particles) silicone loading was rather heterogeneous (see Fig. 2). This is probably due to different fluidization behavior, loading capacity, and curing time of different sized particles and therefore corresponds with the enormous particle size distribution of commercial NZ435 (150–900 μm in diameter) (see Fig. 4). Additionally, temporary adhesion of particles to the reactor walls resulting from electrostatic charging might also play a role. The overall lower loading of silCoat‐composites with silicone by preparation in the fluidized‐bed reactor might be due to a slower penetration of PDMS into the carrier in the absence of organic solvent, or to an enhanced curing of PDMS in the periphery of the particles upon first contact with the immobilized Pt‐catalyst. A definite explanation cannot be offered at this point.
Figure 2.

Photographical picture of a typical coating batch from composite formation through provision of Pt‐catalysts on the particle surface at an overall silicone loading of 44% w/w.
Figure 4.
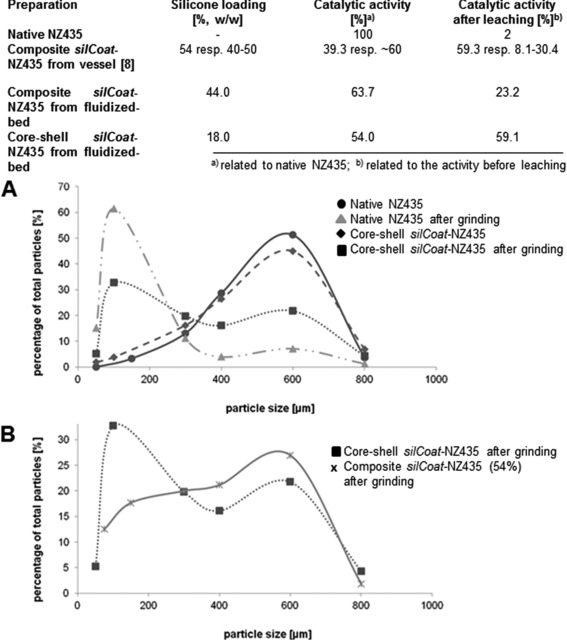
Catalytic activity and robustness of native NZ435 and silCoat‐NZ435 derived from different preparation methods. (A) Particle size distribution (diameters) of native NZ435 and core‐shell silCoat‐NZ435 before and after treatment in a bead mill. (B) Particle size distribution (diameters) of core‐shell silCoat‐NZ435 and composite silCoat‐NZ435 (54% silicone w/w) before and after treatment in a bead mill.
3.1.3.2. Composite formation through temperature‐controlled retardation of hydrosilylation in a siloxane‐catalyst‐mixture
In general, hydrosilylation of divinylated and SiH‐activated PDMS proceeds upon first contact with the Pt‐catalyst. Accordingly, mixing of PDMS and catalyst in the coating solution before injection into the fluidized‐bed severely increased viscosity and caused untimely solidification in the tubing, the injection nozzle, and the reaction chamber. Additionally, the reaction mixture hardly entered the particles any more. However, the speed of hydrosilylation is strongly dependent on catalyst concentration and temperature 13. Accordingly, we observed that the Pt‐catalyst used in this study (Karstedt‐catalyst) did hardly initiate hydrosilylation at a temperature of 0°C. Based on this finding, a coating process relying on a temperature‐controlled retardation of silicone curing in a reservoir solution that already contained the Pt‐catalyst was developed. The Pt‐catalyst was dissolved in a small amount of cooled toluene for homogeneous distribution and mixed with the cooled siloxane solution (0°C) directly before the coating process was started. The solvent content in the mixture was only 1% v/v and therefore exhibited no risks for exposure in the fluidized‐bed reactor. The mixture was pumped into the injection nozzle at a rate of 3–5 g/min and injected into the fluidized‐bed with a pressure of 1.5 bar. Fast curing after entering the fluidized‐bed was ensured by setting the temperature in the reactor to 70–80°C. Curing was complete within time ranges of three minutes (Mini‐Glatt reactor) and 27 min (Uni‐Glatt reactor) at maximum, during which a negative effect on the catalytic activity of the under nonaqueous conditions highly stable NZ435‐catalyst must not be expected 9.
With this procedure, an enhanced tendency to particle aggregation was observed right from the start. Resolution of the aggregates via manual interventions such as knocking on the reactor with a rubber mallet (as is common practice in industry) or increasing the flow rate was successful only for a short time. Accordingly, irreversible collapse of the fluidized‐bed occurred at a silicone loading of only 18% w/w. SEM on the resulting particles revealed a very smooth surface with small circular patches (see Fig. 2, left side), which can usually be observed on particles with a silicone layer on the outer surface 8. With the original method for silCoat‐preparation such particles were only obtained at a silicone loading of 52–54% w/w 8, which indicated that the structure of particles produced in the here described set‐up of the fluidized‐bed significantly differed from the original silCoat. In fact, EDX for silicon distribution (i.e. the element marking PDMS) on cross‐sections of the treated carriers revealed that silicone is strongly concentrated in the carrier pores near the surface forming a shell‐like structure (see Fig. 3) instead of being evenly distributed over the carrier as in the original silCoat‐preparations 8. Obviously, curing of the siloxane‐Pt‐catalyst mixture on the particles was so fast that a penetration to inner pores did not occur. Hence, this method did not produce typical silCoat‐composites, but was able to produce a silicone shell (i.e. a “real” silicone coating) for the first time.
Figure 3.
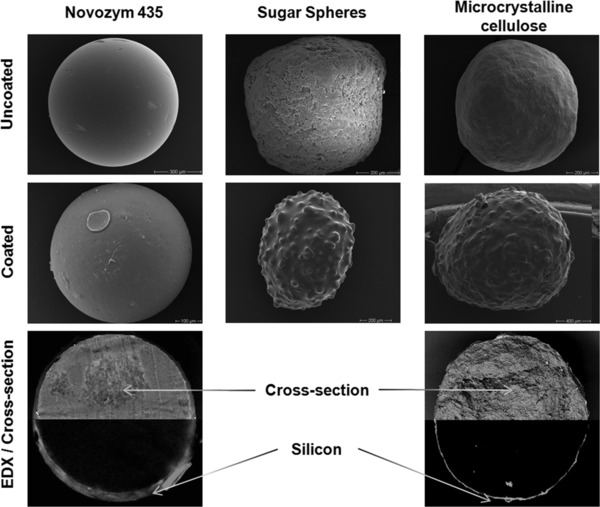
SEM pictures of native and core‐shell silCoat‐preparations of NZ435, sugar spheres, and microcrystalline cellulose (MCC), and EDX‐scans of cross‐sections of core‐shell silCoat‐NZ435 and –MCC showing the elemental distribution of silicon.
3.2. Catalytic activity and robustness of silCoat‐NZ435 from fluidized‐bed production
Both particle types obtained from fluidized‐bed production, composite silCoat‐NZ435 (44%, w/w) and core‐shell silCoat‐NZ435 (18%, w/w), were investigated for catalytic esterification activity, stability against enzyme leaching, and stability against mechanical stress according to literature proceedings 7, 8. No differences between preparations from different scale fluidized‐bed reactors (Mini‐Glatt and Uni‐Glatt) were observed.
Composite silCoat‐NZ435 showed a residual esterification activity of 63.7% (related to the activity of native NZ435). After leaching with a mixture of acetonitrile and water, 23% of that activity remained. Both values are in very good agreement with the reported activities of composite silCoat‐NZ435 from stirred vessels at a comparable silicone loading (40–50%, w/w) 8 (see Fig. 4). The mechanical strength was high; treatment in a bead mill had only a minor effect on the composite particles. The overall bead size decreased from initially about 500 μm to 430 μm, while under the same conditions native NZ435 retained an overall bead size of only 312 μm. These results are comparable to silCoat‐composites obtained with the original preparation method and demonstrate that composite formation in a fluidized‐bed reactor using Pt‐doped carriers yields similar particles. This makes the fluidized‐bed reactor a suitable measure to scale‐up silCoat production. Due to the limitation of silicone loading on fluidized‐bed derived particles to 44% (w/w; see above) and prevention of formation of a closed silicone layer on the particle surface, the highest possible resistance against leaching that the silCoat‐technology can in principle provide 8, cannot be achieved with fluidized‐bed derived composite particles. However, studies on the performance of silCoat‐NZ435 in technically relevant reactions demonstrated that under realistic leaching conditions (not the very harsh treatment performed for evaluation) silCoat‐composites with a silicone loading of 45% w/w can reveal sufficient stability 9, while benefitting from the higher specific activity of these particles compared to particles with higher silicone loading.
Core‐shell silCoat‐particles had a residual esterification activity of 54%. This is about 5% more than for composite silCoat‐particles with comparable silicone coverage on the surface (at a total silicone loading of 54% w/w; see Fig. 3). After leaching, 59.1% of the residual activity of the core‐shell silCoat‐particles remained. This is in the same range as for the above‐mentioned composite silCoat‐particles with a silicone loading of 54% w/w and thus equals the highest leaching stability observed for silCoat‐NZ435 so far (see Fig. 3) 8. In case of core‐shell silCoat‐particles, however, this leaching stability only requires a silicone content of 18% w/w and thus considerably lowers silicone consumption. The stability of native NZ435 against mechanical forces also improved through formation of core‐shell silCoat‐NZ435, i.e. fewer particles disrupted to smaller pieces upon grinding (see Fig. 3A). However, the mechanical stability of core‐shell silCoat‐NZ435 was considerably lower than of composite silCoat‐NZ435 with a silicone content of 54% w/w (see Fig. 3B). The results support the hypothesis that surficial coverage of particles with silicone particularly favors stability against enzyme leaching, while composite formation maintains integrity of the carrier material in the first place 8.
3.3. Core‐shell silCoat‐particles from alternative carrier systems
The method for production of core‐shell silCoat‐particles was further investigated by checking performance with microcrystalline cellulose (MCC) and sugar spheres (SS) as alternative carriers. Both materials are hardly porous and accordingly reveal only small specific surface areas (MCC: 1–3 m2/g; SS: 0.–1 m2/g). They are typically used in coating processes for pellet formulation, but resemble nonporous materials with attractiveness for biocatalytic processes 14. By fluidized‐bed coating with a cooled siloxane‐Pt‐mixture core‐shell silCoat‐particles were formed with both MCC and SS (see Fig. 2). The maximum silicone content before irreversible agglomeration was 2–4% w/w and 6–7% w/w, respectively. A closed layer of silicone was formed on the particle surface. In case of MCC this was considerably thinner than on porous NZ435 (SS could not be investigated by EDX since grinding of the particles in a mortar was not possible), which corresponds with the very low silicone content after coating.
4. Concluding remarks
In this study, fluidized‐bed reactor technology was successfully adapted to the production of silCoat‐enzyme preparations. Depending on the way for provision of the hydrosilylation catalyst (Pt), this technology provides two different types of particles. Composite silCoat‐particles with catalytic properties and robustness fully comparable to silCoat described in literature 8 are obtained when the Pt‐catalyst is cast on the carrier surface before coating. Core‐shell silCoat‐particles with only a layer of silicone on the outer surface derive from using a cold mixture of Pt and siloxane as the coating solution. The two preparations emphasize either mechanical strength or leaching stability. All preparations run without excessive use of organic solvents. Thus, a solvent‐free, fast, and scalable method for the preparation of silCoat‐biocatalysts for technical synthesis is now available. It for the first time offers the possibility to generate also core‐shell instead of composite silCoat‐particles and thus optimize leaching stability over mechanical strength according to process requirements.
Practical application
SilCoat‐biocatalysts, i.e. enzymes or whole cells bound to a carrier and covered with silicone, are extremely useful material for development of biocatalysed processes. They improve both leaching stability and mechanical strength and thus enable reuse or continuous application of the cost‐intensive biocatalysts. With the presented technology, silCoat‐biocatalysts can be prepared in large scale. In addition, it enables pronunciation of leaching stability or mechanical strength during production to match specific requirements of individual processes.
The authors have declared no conflicts of interest.
Acknowledgments
Financial support of this work by the FNR e.V. (Gülzow, Germany, FKZ: 22005507) and COST865 is gratefully acknowledged. The work was scientifically supported by the Cluster of Excellence “Unifying Concepts in Catalysis” coordinated by TU Berlin. We thank J. Nissen from ZELMI (TU Berlin, Germany) for performing SEM and EDX experiments and A. Maudhuit from ENITIAA for expert help with the fluidized‐bed reactor.
5 References
- 1. Cao, L. L. , Carrier‐Bound Immobilized Enzymes, Wiley‐VCH, Weinheim: 2005. [Google Scholar]
- 2. Ansorge‐Schumacher, M. B. , Immobilization of biological catalysts, in: Ertl G., Knözinger H., Schüth F., Weitkamp J. (Eds.), Handbook of Heterogeneous Catalysis, Wiley‐VCH, Weinheim: 2008, pp. 644–655. [Google Scholar]
- 3. Mazeaud, I. , Poulsen, P. B. R. , Christensen, M. W. , Brask, J. , Immobilization of enzymes. EP1934342 2006.
- 4. Thum, O. , Ansorge‐Schumacher, M. B. , Wiemann, L. , Enzyme preparations for use as biocatalysts. US2009/0017519(A1) 2009.
- 5. Thum, O. , Ansorge‐Schumacher, M. B. , Wiemann, L. O. , Buthe, A. , Enzyme preparations. EP2011865B1 2009.
- 6. Thum, O. , Ansorge‐Schumacher, M. B. , Wiemann, L. O. , Ferenz, M. , Naumann, M. , Enzyme preparations. EP2163617B1 2012.
- 7. Wiemann, L. O. , Weißhaupt, P. , Nieguth, R. , Thum, O. , Ansorge‐Schumacher, M. B. , Enzyme stabilisation by deposition of silicone coatings. Org. Proc. Res. Dev. 2009, 13, 617–620. [Google Scholar]
- 8. Wiemann, L. O. , Nieguth, R. , Eckstein, M. , Naumann, M. et al., Novel composite particles of Novozyme 435 and silicone: Advancing technical applicability of macroporous enzyme carriers. ChemCatChem 2009, 1, 455–462. [Google Scholar]
- 9. Nieguth, R. , Eckstein, M. , Wiemann, L. O. , Thum, O. , Ansorge‐Schumacher, M. B. , Enabling industrial biocatalytic processes by application of silicone‐coated enzyme preparations. Adv. Synth. Catal. 2011, 353, 2522–2528. [Google Scholar]
- 10. Tenoue, E. , Poncelet, D. , Batch and continuous fluid bed coating—review and state of the art. J. Food Eng. 2002, 53, 325–340. [Google Scholar]
- 11. El‐Mafadi, S. , Hayert, M. , Poncelet, D. , Fluidization control in the Wurster coating process. Chem. Ind. 2003, 57, 641–644. [Google Scholar]
- 12. Dybdahl‐Hede, P. , Fluid Bed Particle Processing, Bookboon.com, London, Copenhagen: 2006. [Google Scholar]
- 13. Tracton, A. A. , Coatings Technology Handbook, Taylor & Francis Group, Oxford: 2006. [Google Scholar]
- 14. Wu, C.‐W. , Lee, J.‐G. , Lee, W.‐C. , Protein and enzyme immobilization on non‐porous microspheres of polystyrene. Biotechnol. Appl. Biochem. 1998, 27, 225–230. [Google Scholar]