

Abstract
Old yellow enzymes are able to catalyze asymmetric C=C reductions. A mediated electroenzymatic process to regenerate the NADPH in combination with an old yellow enzyme was investigated. Due to the fact that the overall process was affected by a broad set of parameters, a design of experiments (DoE) approach was chosen to identify suitable process conditions. Process conditions with high productivities of up to 2.27 mM/h in combination with approximately 90% electron transfer efficiency were identified.
Keywords: C=C reduction, Design of experiments, Electroenzymatic synthesis, Mediated electron transfer, Old yellow enzyme
Abbreviations
- CE
current efficiency
- DoE
design of experiments
- OYE
old yellow enzyme
- TF
turn over frequency
- TsOYE
old yellow enzyme from Thermus scotoductus
1. Introduction
First found in yeast in 1932 by Warburg and Christian 1 the family of so called old yellow enzymes (OYEs) is now receiving an increased interest by the academic and industrial community because of their potential application in the production of fine chemicals, pharmaceuticals, and agrochemical products as well as in the decontamination of areas polluted with the trinitrotoluene 2, 3, 4. Their substrate scope spans a broad range of compounds containing conjugated C=C double bonds 2. The catalytic mechanism of OYE entails a Michael‐type hydride transfer to the β‐C atom of the conjugated C=C double bond. The hydride is transferred from a reduced, enzyme‐bound flavin (FMN) cofactor that had previously been generated from NAD(P)H (Fig. 1.) 5. The prohibitive high cost of the latter necessitates their use in catalytic amounts together with a suitable in situ regeneration approach. In the last decades several co‐factor regeneration systems have been investigated. One method of choice is electrochemistry. In general, electrochemical methods can be used to generate hydrogen peroxide 6, 7, 8, 9, regenerate cofactors 9, 10, 11, 12, 13, or even to substitute cofactors 14, 15. One recent study has shown the interactions of electrode reactions as well as reduced electrochemical mediators with an oxygen‐dependent reduction by monooxygenases 14. From this work it can be concluded that the combination of an oxygen‐independent enzyme reaction with an electrochemical reaction is more advisable. Therefore, a mediated electroenzymatic process to regenerate the NADPH in combination with a reaction catalyzed by OYE was investigated. As model enzyme we chose the OYE homologue from Thermus scotoductus OYE (TsOYE) 4, 16 due to its high degree of stability, ability to function over a wide‐temperature range, and relative ease of purification 17, 18. The enzyme was optimally active at 65°C but was also active over a wide temperature range, retaining 70% of its activity at 80°C 19. Carvone has served as a substrate for OYEs in several publications 17, 20. When carvone is reduced by OYE, the resulting reaction product is dihydrocarvone, which serves as a precursor for insect antifeedants 21.
Figure 1.
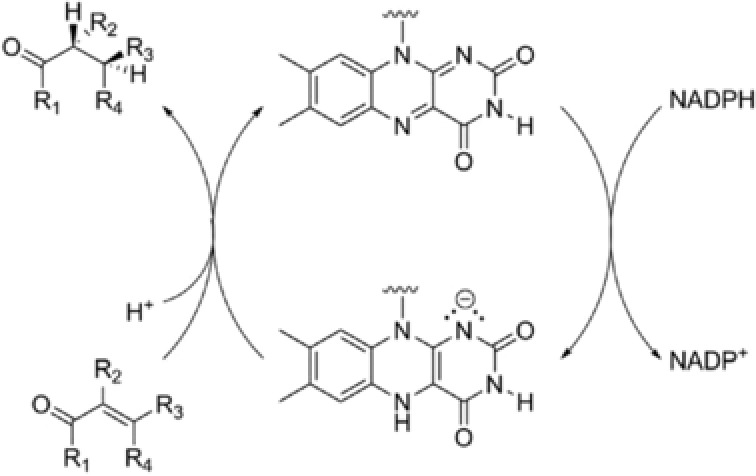
Catalytic cycle of the asymmetric reduction of activated double bonds by OYE. The active reductant within OYEs is a reduced flavin. The catalytic cycle is initiated by nicotinamide dependent reduction of the flavin.
2. Materials and methods
TsOYE (NCBI accession no. AM902709.1) was synthesized and cloned into pET28b(+) via the NdeI and EcoRI restriction sites (Genscript). Protein expression was performed in Escherichia coli BL21(DE3). Transformed strains were grown in LB medium (10 g/L peptone, 5 g/L yeast extract, and 10 g/L NaCl) containing 35 mg/L kanamycin to an OD600 of approximately 0.8–1.0 (30°C, 200 rpm). Protein expression was induced by adding IPTG to a final concentration of 1 mM. Cells were harvested by centrifugation (4000 rpm, 10 min, 4°C) after 4 h, washed with MOPS‐NaOH buffer (50 mM, 10 mM CaCl2, pH 7) and stored at –20°C. The cells were disrupted by sonication (pause on: 1 s, pause off: 2 s, overall time: 2 min, amplitude: 10%) where after E. coli proteins were denatured by a heat precipitation step (90 min, 70°C). After centrifugation (4000 rpm, 10 min, 4°C) the supernatant was used for the further experiments. Enzyme concentrations were determined using BCA assay. [Cp*Rh(bpy)Cl]Cl was synthesized following the procedure described in the literature 22. All electrochemical experiments were conducted with the potentiostat PCI4 300 (Gamry Instruments, Warminster, PA, USA). Before each experiment the mediator containing solution was purged with nitrogen in order to remove oxygen. After the addition of enzyme and substrate to the solution, only the head space of the reactor was purged with nitrogen to avoid enzyme deactivation and reduce substrate and product evaporation. Temperature was kept constant using a water recirculator. Enzyme concentration in the reaction mixture was always 1 μM.
The mediators (Fig. 2) were tested in an 18 mL reactor with a commercial 3D glassy carbon electrode in a bulk electrolysis cell (ALS Co. Ltd., Tokyo, Japan) under continuous stirring. A platinum wire counterelectrode and an Ag/AgCl (3 M KCl) reference electrode were used. A DoE tool (Design Expert 9, Stat‐Ease Inc., Minneapolis, MN, USA) was applied in order to identify the optimal NADP+ and mediator concentrations, temperature, and applied potential (Table 1). These experiments were conducted in an 85 mL reactor with a 3D glassy carbon foam (ALS Co. Ltd.) working electrode. The Ag/AgCl counterelectrode was inserted into the anodic chamber. A platinum wire as counterelectrode was separated by a glass frit, the anodic chamber was filled with buffer solution. A TRIS buffer was used (100 mM, pH 7, 10 mM CaCl2) as electrolyte/buffer in combination with Cp*Rh(bpy)(H2O)]2+ as mediator. MOPS buffer (50 mM, pH 7, 10 mM CaCl2) was used for the other mediator experiments. Five hundred microliter samples were taken after 0, 30, and 60 min. Samples were extracted with 500 μL of EtOAc containing 5 mM cyclohexanol as internal standard. (−)‐Carvone and reaction products were detected via GC using an Agilent J&W DB‐WAXetr column and GC 17A (Shimadzu Deutschland GmbH, Duisburg, Germany).
Figure 2.
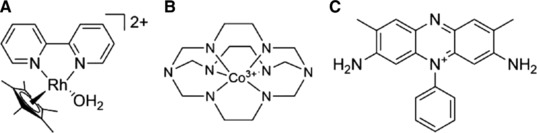
Structures of the used mediators (A) [Cp*Rh(bpy)(H2O)]2+; (B) cobalt sepulchrate, (C) safranin T.
Table 1.
Reaction conditions to optimize mediated electron transfer between electrodes and an OYE
| Entry | c (Mediator) (mM) | c (NADP+) (mM) | T (°C) | U (mV) |
|---|---|---|---|---|
| 1 | 0.01 | 0.2 | 80 | –860 |
| 2 | 0.05 | 0.2 | 60 | –860 |
| 3 | 0.05 | 0.2 | 80 | –760 |
| 4 | 0.01 | 0.1 | 80 | –860 |
| 5 | 0.05 | 0.1 | 80 | –760 |
| 6 | 0.05 | 0.1 | 60 | –860 |
| 7 | 0.05 | 0.1 | 60 | –760 |
| 8 | 0.01 | 0.2 | 60 | –860 |
| 9 | 0.01 | 0.1 | 60 | –860 |
| 10 | 0.01 | 0.2 | 60 | –760 |
| 11 | 0.01 | 0.2 | 80 | –760 |
| 12 | 0.05 | 0.2 | 60 | –760 |
| 13 | 0.01 | 0.1 | 80 | –760 |
| 14 | 0.05 | 0.1 | 80 | –860 |
| 15 | 0.05 | 0.2 | 80 | –860 |
| 16 | 0.01 | 0.1 | 60 | –760 |
3. Results and discussion
In an attempt to identify an effective mediator for the direct regeneration of the active site of TsOYE, a mediator screening in the 18 mL reactor was performed. Cobalt sepulchrate, safranin T, and [Cp*Rh(bpy)(H2O)]2+ were used as electron transfer mediators. Although small product amounts were detected in all three experiments, in no experiment was there a significant increase compared to a negative control (without enzyme and mediator, Fig. 3). Therefore, it can be concluded that the small amount of product resulted from impurities in the substrate or a nonenzymatic product formation. In order to ensure the activity of the enzyme in the reactions mixtures, NADPH was added after 18 h to the reaction with safranine T, resulting in a fast eightfold increase of product concentration (data not shown). Given that there was no significant increase in product concentration compared to the negative control, it can be ruled out that there was any electron transfer to the enzyme, neither from the electrode nor from the mediator.
Figure 3.
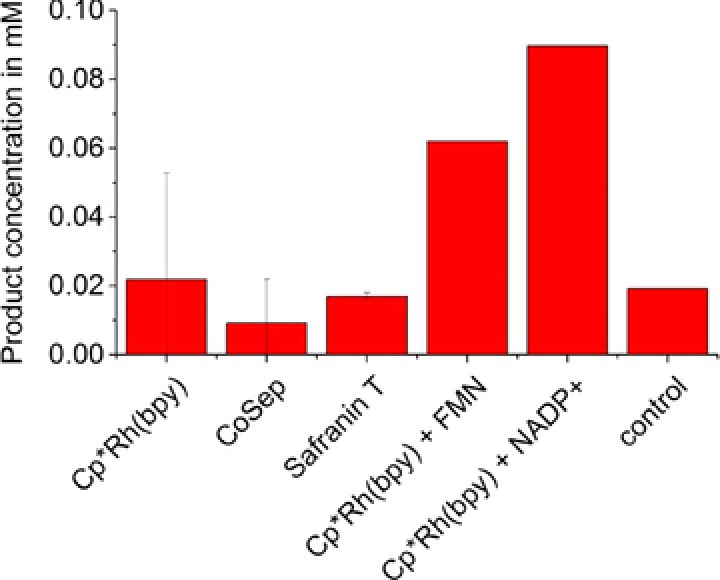
Product formation of dihydrocarvone at different reactions for a mediated cofactor substitution or regeneration (reaction conditions: 5 mM R‐(−)‐carvone, experiments with Cp*Rh(bpy) were performed in TRIS buffer (100 mM, pH 7, 10 mM CaCl2), all other experiments in MOPS buffer (50 mM, pH 7, 10 mM CaCl2), Cp*Rh(bpy), CoSep, and Safranin T: 250 μM mediator; Cp*Rh(bpy) + FMN: 100 μM mediator and 100 μM FMN; Cp*Rh(bpy) + NADP+: 50 μM mediator and 100 μM NADP+ (n = 2). An additional experiment with safranine T as mediator in the Tris/HCl buffer was performed. Again, no product concentration could be measured. Therefore it can be concluded that the buffer has no significant influence on the results.
As [Cp*Rh(bpy)(H2O)]2+ is known to regenerate NADPH and FMN 11, 12, 17, these systems were used to investigate the cofactor regeneration in combination with the OYE. The reaction system is shown in Fig. 5. A significant increase in product concentration compared to the negative control could be measured in these investigations. Due to the fact that the combination of [Cp*Rh(bpy)(H2O)]2+ and NADP+ showed the highest productivity, this system was further investigated in order to scale‐up the reaction up to 85 mL scale and to optimize it with a DOE approach. Table 1 shows the reaction conditions for 16 runs to optimize the reaction conditions and Fig. 4 the resulting product formation rates.
Figure 5.
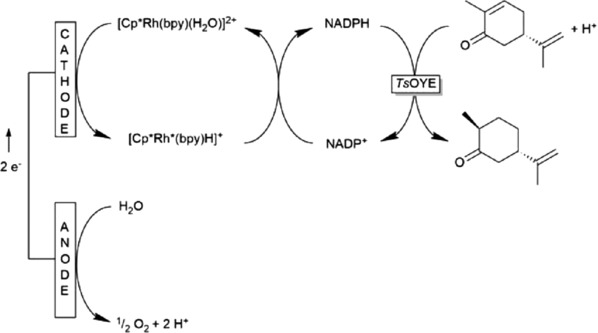
Electroenzymatic process involving OYEs and mediated cofactor regeneration, the substrate (−)‐carvone was converted into (+)‐dihydrocarvone.
Figure 4.
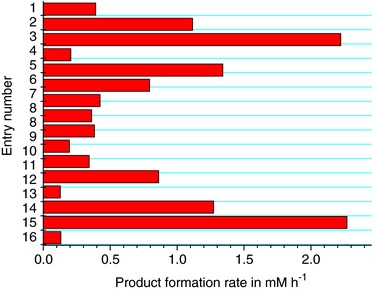
Product formation rate at different reaction conditions in order to optimize the electroenzymatic conversion of carvone by using an OYE (n = 1).
Several interactions were observed to have an effect on the product formation rate. Increasing the mediator concentration would lead to a higher product formation rate at high temperatures rather than at lower ones. Increasing the mediator concentration at higher NADP+ concentrations leads to a stronger increase of the reaction rate than at lower NADP+ levels. Obviously, there is an interaction between NADP+ concentrations and temperature. Increasing temperature has a more positive effect at high NADP+ levels. Finally, it can be concluded, that the highest product formation rate will be obtained when both concentrations are at the highest level. The highest productivity was measured with 0.05 mM mediator, 0.2 mM NADP+, 80°C, and a potential of −760 mV versus Ag/AgCl as reference electrode (entry 3). The second highest productivity was found by using similar conditions (entry 15), only the potential was changed to −860 mV versus Ag/AgCl instead of −760 mV versus Ag/AgCl. So far, one of the major issues with electroenzymatic processes comes from an insufficient energy and mediator efficiency, causing, especially under aerobic conditions, very low electron efficiencies (maximum value of 21%, 13, 14, 15) and turn over frequencies (TFs; between 0.125 and 291 h−1 11, 13, 23). Therefore, these parameters were investigated in detail. Figure 6 shows the calculated current efficiency (CE) and TF in the different runs. CE or electron transfer efficiencies were calculated according to Eq. (1), where CE is the current efficiency, z is the equivalent of electrons transferred in each reaction, F is the Faraday constant, n Product is the amount of product formed and is the overall amount of electrons transferred.
| (1) |
Figure 6.
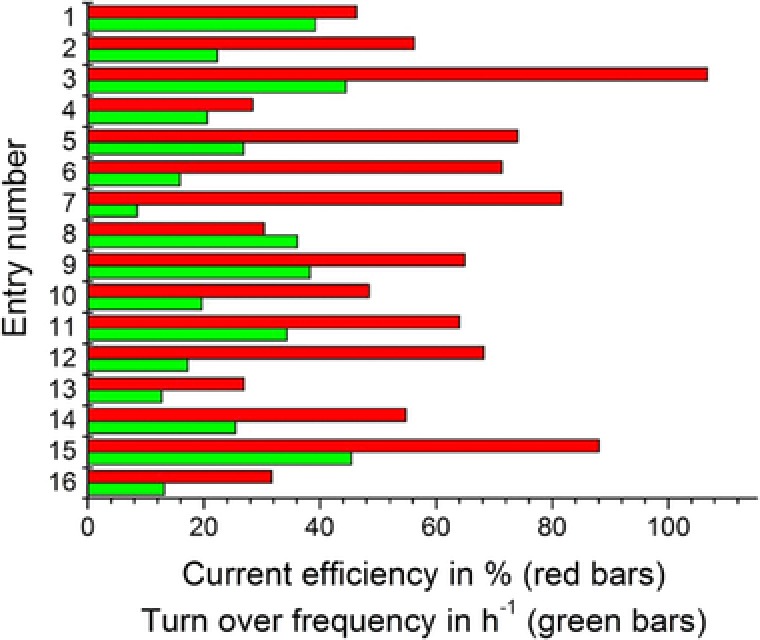
CE and TF at different reaction conditions in order to optimize the electroenzymatic conversion of carvone by using an OYE (n = 1).
The highest CE was detected under the conditions (entry 3) that lead to the above‐mentioned highest productivity. From the DoE approach only one general interaction was observed for the electron efficiency. NADP+ concentration and temperature show an antagonistic interaction. At low NADP+ levels, increasing the temperature will reduce the electron efficiency, while at high NADP+ levels it will lead to an increase of the electron efficiency. Most probably interactions between reduced cofactor and the mediator takes place and are more pronounced at low NADP+ concentrations. At higher NADP+ concentration mainly the desired reaction took place. Furthermore, it was concluded that an increased reaction rate shifts the ratio of carvone conversion and the unwanted reaction in the direction of the desired reaction. Mediator TFs varied between 10 and 45 h−1. Several interactions with effect on the TF can be observed. While at high mediator levels, an increase of the potential has almost no effect, it causes a decreased TF at low mediator concentrations. Another interaction occurs between the potential and temperature. While at 80°C, a change in potential has little effect, increasing the potential at 60°C will cause a reduced TF. An additional interaction can be observed between the temperature and mediator concentration. The TF increases with rising mediator levels only at 80°C. At 60°C, the opposite effect is observed. Independently of the mediator concentration, the TF is always higher at 80°C than at 60°C. Temperature interacts also with NADP+ concentrations. While the TF is higher at 0.2 mM NADP+ and 60°C than at 0.1 mM and 60°C, it can be further increased by raising the temperature to 80°C. At low NADP+ levels on the other hand, changing the temperature has no significant effect on the TF of the mediator. The DoE in regard to the TF indicates that the highest TF values can be achieved by working at high mediator, temperature, and NADP+ levels. Compared to values given in the literature, we have observed not only the second highest TF 13 relative to the mediator reported for an electroenzymatic processes, but also the highest electron efficiency in these processes. It should be mentioned that a CE higher than 100% is not reasonable, therefore the value of entry 3 must be regarded as approximately 100% CE. The most likely causes for the discrepancy are sampling errors or fluctuation in the calculation of the current efficiencies. By comparing the results of the above‐mentioned entries 3 and 15, the calculated current efficiencies vary between 106 and 88%. Therefore, it can be concluded that the application of 0.05 mM mediator, 0.2 mM NADP+ and a reaction temperature of 80°C resulted in current efficiencies of approximately 90%.
In general, more negative potential leads to a higher amount of side reactions and therefore to a reduced CE. Nevertheless, the observed high current efficiencies enable processes with high energy efficiency and a minor formation of side products. The values for the enantiomeric excess varied, except for one case, between 70 and 90% (data not shown). A similar enantiomeric excess of approximately 90% was also reported for the natural NADPH‐driven carvone reduction 17. An interaction between mediator concentration and temperature appeared to be relevant for the outcome of the enantiomeric excess. The two parameters show an antagonistic interaction: at high mediator concentrations, it does not seem relevant whether the temperature is at a high or low level, while at low mediator concentrations, lower temperatures favor a higher enantiomeric excess.
Finally, it must be mentioned that a long‐term experiment under the chosen electrochemical reaction conditions in combination with an increased amount of substrate did not result in the expected increase of product concentration. Most probably the reaction was limited by inhibitory high concentrations of the substrate and/or the product. Recently, it was shown that biphasic reaction can be used to overcome these limitations 4, 23, 24. Therefore, further optimization in regard to the combination of the electrochemical system and a biphasic system are currently ongoing in our laboratory.
4. Concluding remarks
The development of different systems for regeneration and substitution of NADPH as natural OYE cofactor may help granting access to technical in vitro OYE applications in the future. Clearly, enzymatic cofactor regeneration systems are already of high industrial potential and have already proven to be well suited for commercial applications, as is the case, for instance, with alcohol dehydrogenase‐catalyzed syntheses. In each case, only catalytic levels of NAD(P)+ are required but stoichiometric levels of a cosubstrate are needed to drive the recycling enzyme. When driving an enzymatically catalyzed reaction by an electrode, instead of using an enzymatic cofactor regeneration system, process costs could be reduced drastically. Electrochemical cofactor regeneration would result in a cosubstrate‐ and coproduct‐free reaction setup. As this system does not need a cosubstrate, no by‐products are produced, which facilitates the recovery of the desired product. Our results show that high productivities and high current efficiencies of approximately 90% can be achieved by using enzymes working at anaerobic conditions such as OYE in combination with electrochemical cofactor regeneration. By using oxygen‐dependent enzymes (e.g. P450s) in similar approaches, only current efficiencies up to 21% can be measured 13, 14, 15. The cathode potentials required for efficient reduction of the mediators are more negative than the O2 reduction potential. Hence, direct cathodic O2 reduction occurs during the electrolyses, reducing the current yield 25.
Fisher et al., investigated an electrochemical cofactor substitution system in combination with OYE and measured product formation rates up to 1.25 mM/h 23. Here, we investigated the cofactor regeneration and identified conditions for a productivity of up to 2.27 mM/h (corresponding to 38 μM/s). Both approaches show clearly the high potential of the electroenzymatic processes with OYE. Which cofactor regeneration or substitution system is finally “greener” must be evaluated in detail. For this evaluation the long‐term stability of all components, the cost for the production of the enzymes, and the synthesis of the different mediators as well as the impact of the mediators on the environment must be considered. Certainly, the recently investigated photochemical approaches 16, 24, 26 as well as electrochemical systems to substitute or regenerate cofactors in combination with OYE have a high potential to develop efficient processes with a low impact on the environment and small E‐factor values 27 due to the avoidance of high amounts of side products. Finally, it can be concluded that electroenzymatic processes under anaerobic conditions can lead to more sustainable processes compared to processes in the presence of oxygen and using oxygenases 13, 14, 15. The presented productivities are in the same range as in electroenzymatic processes to produce hydrogen peroxide [6, 7, 8, 28] or the regeneration of oxidized nicotinamide cofactors 10, 29. It can be concluded that anaerobic regeneration of oxidized cofactors, anaerobic regeneration, or substitution of cofactors as well as the electrochemical production of hydrogen peroxide can be successfully implemented in relevant industrial processes.
Practical application
New production routes for fine and bulk chemicals are important to establish further sustainable processes in industry. Besides the identification of new biocatalysts and substrates the optimization of existing processes in regard to an improved utilization of resources such as cofactors is needed. In this paper, we describe the successful development of a mediated electroenzymatic process to regenerate the NADPH as reducing agent for OYEs. The enzyme family of OYE is now receiving an increased interest by the academic and industrial community because of their broad applicability. The prohibitive high cost of the needed NADPH necessitates their use in catalytic amounts together with a suitable in situ regeneration approach. Due to the fact that the overall process was affected by a broad set of parameters, a design of experiments (DoE) approach was chosen to identify suitable process conditions. Our investigations resulted in a process with high productivities in combination with high electron transfer efficiencies. In comparison with other electroenzymatic processes as well as with established reaction systems with OYE, our process design allows high space‐time yields with a reduced demand on energy and chemicals.
The authors have declared no conflict of interest.
5 References
- 1. Warburg, O. , Christian, W. , Uber ein neues Oxydationsferment und sein Absorptionsspektrum. Biochem. Z 1932, 254, 438–458. [Google Scholar]
- 2. Williams, R. E. , Bruce, N. C. , “New uses for an old enzyme”—The old yellow enzyme family of flavoenzymes. Microbiology (Reading, England) 2002, 148, 1607–1614. [DOI] [PubMed] [Google Scholar]
- 3. Toogood, H. S. , Gardiner, J. M. , Scrutton, N. S. , Biocatalytic reductions and chemical versatility of the old yellow enzyme family of flavoprotein oxidoreductases. ChemCatChem 2010, 2, 892–914. [Google Scholar]
- 4. Toogood, H. S. , Knaus, T. , Scrutton, N. S. , Alternative hydride sources for ene‐reductases: Current trends. ChemCatChem. 2014, 6, 951–954. [Google Scholar]
- 5. Vaz, A. D. N. , Chakraborty, S. , Massey, V. , Old yellow enzyme: Aromatization of cyclic enones and the mechanism of a novel dismutation reaction. Biochemistry 1995, 34, 4246–4256. [DOI] [PubMed] [Google Scholar]
- 6. Holtmann, D. , Krieg, T. , Getrey, L. , Schrader, J. , Electroenzymatic process to overcome enzyme instabilities. Catalysis Commun. 2014, 51, 82–85. [Google Scholar]
- 7. Krieg, T. , Hüttmann, S. , Mangold, K.‐M. , Schrader, J. et al., Gas diffusion electrode as novel reaction system for an electro‐enzymatic process with chloroperoxidase. Green Chem. 2011, 13, 2686–2689. [Google Scholar]
- 8. Lütz, S. , Vuorilehto, K. , Liese, A. , Process development for the electroenzymatic synthesis of R‐methylphenylsulfoxide by use of a 3‐dimensional electrode. Biotechnol. Bioeng. 2007, 98, 525–534. [DOI] [PubMed] [Google Scholar]
- 9. Varničić, M. , Vidaković‐Koch, T. , Sundmacher, K. , Gluconic Acid Synthesis in an Electroenzymatic Reactor. Electrochim. Acta 2015, 174, 480–487. [Google Scholar]
- 10. Kochius, S. , Park, J. B. , Ley, C. , Könst, P. et al., Electrochemical regeneration of oxidised nicotinamide cofactors in a scalable reactor. J. Mol. Catal. B Enzym. 2014, 103, 94–99. [Google Scholar]
- 11. Hollmann, F. , Schmid, A. , Steckhan, E. , The first synthetic application of a monooxygenase employing indirect electrochemical NADH regeneration. Angew. Chem. Int. Ed. 2001, 40, 169–171. [DOI] [PubMed] [Google Scholar]
- 12. Hollmann, F. , Witholt, B. , Schmid, A. , [Cp∗ Rh (bpy)(H 2 O)]2+: A versatile tool for efficient and non‐enzymatic regeneration of nicotinamide and flavin coenzymes. J. Mol. Catal. B Enzym. 2002, 19, 167–176. [Google Scholar]
- 13. Ruinatscha, R. , Dusny, C. , Buehler, K. , Schmid, A. , Productive Asymmetric Styrene Epoxidation Based on a Next Generation Electroenzymatic Methodology. Adv. Synth. Catal. 2009, 351, 2505–2515. [Google Scholar]
- 14. Tosstorff, A. , Dennig, A. , Ruff, A. J. , Schwaneberg, U. et al., Mediated electron transfer with monooxygenases—Insight in interactions between reduced mediators and the co‐substrate oxygen. J. Mol. Catal. B Enzym. 2014, 108, 51–58. [Google Scholar]
- 15. Çekiç, S. Z. , Holtmann, D. , Güven, G. , Mangold, K.‐M. et al., Mediated electron transfer with P450cin. Electrochem. Commun. 2010, 12, 1547–1550. [Google Scholar]
- 16. Grau, M. M. , van der Toorn, J. C. , Otten, L. G. , Macheroux, P. et al., Photoenzymatic Reduction of C=C Double Bonds. Adv. Synth. Catal. 2009, 351, 3279–3286. [Google Scholar]
- 17. Bernard, J. , van Heerden, E. , Arends, I. W. C. E. , Opperman, D. J. et al., Chemoenzymatic reduction of conjugated C=C double bonds. ChemCatChem. 2012, 4, 196–199. [Google Scholar]
- 18. Opperman, D. J. , Sewell, B. T. , Litthauer, D. , Isupov, M. N. et al., Crystal structure of a thermostable old yellow enzyme from Thermus scotoductus SA‐01. Biochem. Biophys. Res. Commun. 2010, 393, 426–431. [DOI] [PubMed] [Google Scholar]
- 19. Opperman, D. J. , Piater, L. A. , van Heerden, E. , A novel chromate reductase from Thermus scotoductus SA‐01 related to old yellow enzyme. J. Bacteriol. 2008, 190, 3076–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goretti, M. , Ponzoni, C. , Caselli, E. , Marchigiani, E. et al., Biotransformation of electron‐poor alkenes by yeasts: Asymmetric reduction of (4S)‐(+)‐carvone by yeast enoate reductases. Enzyme Microb. Technol. 2009, 45, 463–468. [Google Scholar]
- 21. Jansen, B. J. M. , Kreuger, J. A. , De Groot, A. , The conversion of (−)‐ and (+)‐dihydrocarvone into chiral intermediates for the synthesis of (−)‐polygodial, (−)‐warburganal and (−)‐muzigadial. Tetrahedron 1989, 45, 1447–1452. [Google Scholar]
- 22. Kölle, U. , Grützel, M. , Organometallic Rhodium(III) Complexes as Catalysts for the Photoreduction of Protons to Hydrogen on Colloidal TiO2 . Angew. Chem. Int. Ed. Eng. 1987, 26, 567–570. [Google Scholar]
- 23. Fisher, K. , Mohr, S. , Mansell, D. , Goddard, N. J. et al., Electro‐enzymatic viologen‐mediated substrate reduction using pentaerythritol tetranitrate reductase and a parallel, segmented fluid flow system. Catal. Sci. Technol. 2013, 3, 1505–1511. [Google Scholar]
- 24. Peers, M. K. , Toogood, H. S. , Heyes, D. J. , Mansell, D. et al., Light‐driven biocatalytic reduction of α, β‐unsaturated compounds by ene reductases employing transition metal complexes as photosensitizers. Catal. Sci. Technol. 2016, 6, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Holtmann, D. , Hollmann, F. , The oxygen dilemma: A severe challenge for the application of monooxygenases? Chembiochem 2016, 17, 1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taglieber, A. , Schulz, F. , Hollmann, F. , Rusek, M. et al., Light‐driven biocatalytic oxidation and reduction reactions: Scope and limitations. Chembiochem 2008, 9, 565–572. [DOI] [PubMed] [Google Scholar]
- 27. Ni, Y. , Holtmann, D. , Hollmann, F. , How green is biocatalysis? To calculate is to know. Chem. Cat. Chem. 2014, 6, 930–943. [Google Scholar]
- 28. Getrey, L. , Krieg, T. , Hollmann, F. , Schrader, J. et al., Enzymatic halogenation of the phenolic monoterpenes thymol and carvacrol with chloroperoxidase. Green Chem. 2014, 16, 1104–1108. [Google Scholar]
- 29. Kochius, S. , Paetzold, M. , Scholz, A. , Merkens, H. et al., Enantioselective enzymatic synthesis of the α‐hydroxy ketone (R)‐acetoin from meso‐2, 3‐butanediol. J. Mol. Catal. B Enzym. 2014, 103, 61–66. [Google Scholar]