

Abstract
Bacillus subtilis is a model organism for Gram‐positive bacteria and widely used in the study of cellular functions and processes including protein secretion, sporulation, and signal transduction. It is also an important industrial host for the production of proteins and chemicals. Generally, genome editing of B. subtilis often needs the construction of integration vectors in Escherichia coli, linearizing the constructed plasmids, and subsequent transformation of the linear deoxyribonucleic acid via natural competence or electroporation. In this work, we examined the feasibility to directly transform and integrate B. subtilis using linear deoxyribonucleic acid from Gibson assembly without the need for cloning in E. coli. Linear deoxyribonucleic acid of 8–10 kb showed the highest transformation efficiency which was similar to that of using linearized plasmids constructed in E. coli. This method shortens the overall process from 1 week to 1 day and allows the integration of multiple genes in one step, providing a simple and fast method for genome editing in B. subtilis.
Keywords: DNA assembly, gene knockout/knockin, genome editing mutant libraries, transformation efficiency
Abbreviations
- amyE
gene for α‐amylase
- cotC‐petase
gene for a fusion protein CotC‐Petase
- cotG‐mhetase
gene for a fusion protein CotG‐Mhetase
- DNA
deoxyribonucleic acid
- LB
Luria‐Bertani
1. INTRODUCTION
The Gram‐positive model organism Bacillus subtilis has been extensively studied to understand bacterial cell biology, cell cycle, and differentiation, and widely used as industrial hosts for the production of proteins, antibiotics, and food additives 1, 2, 3. However, genetic engineering tools available for B. subtilis are far less efficient than those developed for the Gram‐negative model bacterium Escherichia coli, mainly due to the low genetic transformation efficiency. Many B. subtilis strains are found to possess natural competence, the ability to uptake single‐stranded deoxyribonucleic acid (DNA). Ever since Spizizen developed a two‐step minimal media procedure to activate the natural competence in B. subtilis, tremendous work has been done to improve the transformation efficiency and natural competence based protocols remain the most commonly used methods for genetic transformation in B. subtilis 4, 5, 6. With the elucidation of the regulatory network of natural competence 7, 8, the gene encoding the master regulator ComK was placed under the control of inducible promoters to transiently switch on the natural competence and significantly increased the transformation efficiency 9, 10. DNA uptake specificity was investigated to uncover the relationship among DNA topology, sequence and transformation efficiency 4, 11, 12. Electroporation and protoplast fusion were also used for genetic transformation in B. subtilis, particularly for strains without natural competence 13, 14. Both natural competence based transformation using multimeric plasmids and electroporation of high osmolarity have achieved 105–106 transformants/μg DNA 15, 16.
Although a few self‐replicating plasmids have been developed for B. subtilis 17, most practice is to integrate the target DNA into the chromosome to avoid genetic instability due to the homologous recombination system in B. subtilis. The general procedure for inserting/deleting a gene into the chromosome is to use an E. coli–B. subtilis shuttle vector, which is first constructed to carry the desired genes in E. coli through routine cloning procedures, followed by the transformation of linearized or circular plasmids to B. subtilis, depending on whether it is double‐crossover or single‐crossover homologous recombination 18, 19, 20, 21. As shown in Figure 1, a circular plasmid is typically used to add an epitope tag or fluorescent protein tag to a gene on the chromosome through single‐crossover homologous recombination; linearized DNA is for gene knockout or knockin via double‐crossover homologous recombination. The major steps of this method for genetic engineering in B. subtilis are listed in Figure 1C. It takes about a week assuming every step goes smoothly.
Figure 1.
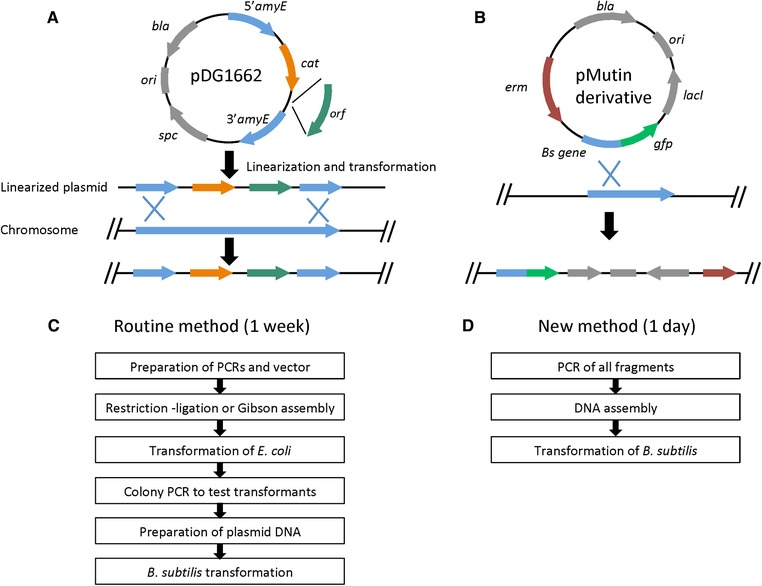
Schematic representation of homologous recombination in B. subtilis and comparison of the flow schemes of the routine method based on cloning in E. coli and the method of this work. (A) an example vector for double‐crossover recombination for gene knockout or knockin; (B) an example vector for single‐crossover recombination for tagging a protein; (C) routine method based on cloning in E. coli; (D) new method using assembled linear DNA
Along with the fast progress of DNA synthesis, various DNA assembly methods have been developed, such as Biobricks, Golden Gate, and Gibson assembly 22, 23, 24. Toolkits like EcoFlex have been designed to construct large integrative plasmids from multiple pieces of DNA using Golden Gate assembly 25, which significantly reduce the workload to insert heterologous or synthetic pathways into B. subtilis. Although these DNA assembly methods are designed to obtain circular form of DNA via ligation in vitro, effort has been made to adapt them to assemble linear DNA that can be directly used to transform B. subtilis. A method named Ordered Gene Assembly in B. subtilis (OGAB) was developed to assemble multimeric linear DNA from a dozen of fragments using the restriction enzyme SfiI. Taking advantage of the homologous recombination system in B. subtilis, the multimeric linear DNA could circularize into plasmids inside the cell and be purified for subsequent applications. Recently this method was improved to assemble over 50 DNA fragments using Type IIS restriction enzymes from the Golden Gate assembly method 26, 27.
Gibson assembly is another popular method for DNA assembly in vitro. Compared with Golden Gate assembly, it can only accommodate up to four to six fragments and the efficiency is low when assembling more than four fragments in one step. However, it is not limited by the presence of Type IIS restriction enzyme cutting sites in the DNA to be assembled and can be used to assemble much longer fragments. Repeated sequences could be problematic for Gibson assembly but could be overcome with proper design. So far Gibson assembly is mainly used for the construction of circular plasmids and has not been reported for the assembly of large linear DNA. To reduce the time needed for gene knockout or knockin in B. subtilis, we attempted to assemble linear DNA fragments from PCR products using Gibson assembly and directly use the assembled DNA to transform B. subtilis, which could bypass the cloning and screening in E. coli and decrease the whole process from 1 week to 1 day (Figure 1D). In this work, the regular Spizizen transformation protocol was used to transform B. subtilis 168 with DNA prepared either from linearizing an integrative plasmid (the routine method) or directly from Gibson assembly. It was that the assembled linear DNA can directly transform B. subtilis with efficiency comparable to the routine method. This new simple protocol will significantly improve the workflow of genome editing in B. subtilis.
2. MATERIALS AND METHODS
2.1. Bacterial strains and growth conditions
E. coli DH5α and B. subtilis 168 used in this study were grown at 37ºC. E. coli strains were grown in Luria–Bertani (LB) broth (10 g/L peptone, 5 g/L NaCl, 5 g/L yeast extract) and LB solid plates with 15 g/L agar. B. subtilis strains were grown in LB broth and TBAB solid plates (10 g/L Tryptose, 3 g/L beef extract, 5 g/L NaCl, 15 g/L agar). Ampicillin was added to a final concentration of 100 μg/mL for E. coli strains and chloramphenicol was added to a final concentration of 5 μg/mL for B. subtilis strains when required.
PRACTICAL APPLICATION
Bacillus subtilis is an important bacterium for both academic study and industrial processing. Extensive effort has been made to improve the efficiency of genetic engineering in B. subtilis. Many integration vectors have been developed. Genome editing generally starts with inserting the target genes into these integration vectors in Escherichia coli and then linearizing the resulted circular plasmids for B. subtilis transformation. The whole process usually takes at least a week. The method we developed in this work directly used assembled linear DNA for transformation and reduced the workflow from one week to one day. It will significantly improve the efficiency of genome editing in B. subtilis. When combined with error‐prone PCR and more efficient transformation protocols, this could also be used for the fast generation of mutant libraries and directed evolution, making B. subtilis a better host for synthetic biology and metabolic engineering.
2.2. Construction of plasmids and assembly of linear DNA
The primers, plasmids, and assembled linear DNA fragments are listed in Table 1. Individual DNA fragments were amplified using Q5 polymerase via PCR. They were designed to have overlapping homologous sequences so that they can be assembled into circular plasmids or linear DNA by Gibson assembly (NEB). Our design was based on the shuttle vector pDG1662, which can be linearized and integrated into the gene for α‐amylase (amyE) locus on the chromosome of B. subtilis. PCR3, which is the backbone of pDG1662, is used to construct circular plasmids: PCR3 and PCR6 were assembled into the plasmid pBS1; PCR3 and PCR7 into pBS2; PCR3, PCR8, and PCR9 into pBS3. PCR1 or PCR4 carries the 5′ amyE and chloramphenicol resistance gene camR and PCR2 or PCR5 carries the 3′ amyE. They were assembled with DNA fragment(s) to be integrated into linear DNA ranging from 4 to 18 kb. The detailed information about all PCRs and assembled DNA fragments are listed in Table 1. For the assembly of circular plasmids, the amount of insert to vector was 3:1. For the assembly of linear DNA, all fragments were mixed in an equimolar way. The mixtures were incubated at 50ºC for 2 h to assemble the fragments together. The assembly of the linear DNA fragments was confirmed by gel electrophoresis.
Table 1.
Plasmids, oligonucleotides, and DNA fragments used in this study
| Plasmids, primers, or DNA fragments | Description, sequence, or primers/template | Note |
|---|---|---|
| pDG1662 | B. subtilis integration vector, amyE locus, ampR and specR in E. coli, cmR in B. subtilis | Guerout‐Fleury, AM et.al. 8973347 |
| pBS1 | pDG1662 derivative, cotC‐petase | This work |
| pBS2 | pDG1662 derivative, cotG‐mhetase | This work |
| pBS3 | pDG1662 derivative, cotC‐petase, cotG‐mhestase | This work |
| 1178R | Ggttagtgacattagaaaaccgactg | This work |
| 1219F | Cagtcggttttctaatgtcactaacctgtaggataaatcgtttgggcc | This work |
| 1221F | Cagtcggttttctaatgtcactaaccagtgtccctagctccgagaaaaaatcc | This work |
| 1224F | Tcgacatggatgagcgatgatgatatccg | This work |
| 1230F | Aattctccagtcttcacatcggtttg | This work |
| 1233R | Gcaaatgcagacaatatcagcatcc | This work |
| 1342F | Tcttgagtccaacccggtaagacac | This work |
| 1343R | Cgatagttaccggataaggcgcag | This work |
| 1476R | cggatatcatcatcgctcatccatgtcgattaatgatggtggtggtggtgagagcag | This work |
| 1478R | Cggatatcatcatcgctcatccatgtcgattaaggaggagcagcgcaagcg | This work |
| 1479R | Ctttgagtgatctgataccgctcg | This work |
| 1480F | Cgagcggtatcagctcactcaaagtgtaggataaatcgtttgggcc | This work |
| 1629F | Cagtcggttttctaatgtcactaaccgactgcaacgggcaatatgtctctg | This work |
| 1630R | Ggcccaaacgatttatcctacagcatattgatccgccactgcctgg | This work |
| 1631F | Tgtaggataaatcgtttgggcc | This work |
| 1632R | Ggcccaaacgatttatcctacaaaccgacatcgctttcaacattg | This work |
| 1633R | Ggcccaaacgatttatcctacagagatatcatcaccaacaagctg | This work |
| 1634R | Ggcccaaacgatttatcctacatcatcgcgaccggcaataagagg | This work |
| 1635R | Ggcccaaacgatttatcctacactgcggtgccagcgcaatctatctg | This work |
| PCR1 | 1342F, 1178R, pDG1662, 5′ amyE + camR | This work |
| PCR2 | 1224F, 1343R, pDG1662, 3′ amyE | This work |
| PCR3 | 1224F, 1178R, pDG1662 backbone | This work |
| PCR4 | 1230F, 1178R, pDG1662, 5′ amyE + camR | This work |
| PCR5 | 1224F, 1233R, pDG1662, 3′ amyE | This work |
| PCR6 | 1219F, 1476R, synthesized DNA, cotC‐petase | This work |
| PCR7 | 1221F, 1478R, synthesized DNA, cotG‐mhetase | This work |
| PCR8 | 1221F, 1479R, synthesized DNA, cotC‐petase | This work |
| PCR9 | 1480F, 1476R, synthesized DNA, cotG‐mhetase | This work |
| PCR10 | 1631F, 1343R, assembled linear DNA 3, cotC‐petase + 3′ amyE | This work |
| PCR11 | 1629F, 1630R, E. coli chromosome | This work |
| PCR12 | 1629F, 1632R, E. coli chromosome | This work |
| PCR13 | 1629F, 1633R, E. coli chromosome | This work |
| PCR14 | 1629F, 1634R, E. coli chromosome | This work |
| PCR15 | 1629F, 1635R, E. coli chromosome | This work |
| DNA 1 | PCR4 + PCR6 + PCR5, 4 kb | This work |
| DNA 2 | PCR4 + PCR7 + PCR5, 6 kb | This work |
| DNA 3 | PCR1 + PCR6 + PCR2, 8 kb | This work |
| DNA 4 | PCR1 + PCR8 + PCR9 + PCR2, 10 kb | This work |
| DNA 5 | PCR1 + PCR12 + PCR10, 12 kb | This work |
| DNA 6 | PCR1 + PCR13 + PCR10, 14 kb | This work |
| DNA 7 | PCR1 + PCR14 + PCR10, 16 kb | This work |
| DNA 8 | PCR1 + PCR15 + PCR10, 18 kb | This work |
2.3. B. subtilis transformation
Standard Spizizen transformation protocol was used to transform B. subtilis 168 28. Assembled linear DNA was directly used for B. subtilis transformation. 0.15 pmol of each fragment was used for the assembly and the mixture was used for transformation. Plasmids were first constructed in E. coli and verified by colony PCR. Then they were prepared using ZymoPURE Plasmid Miniprep Kit from ZYMO Research. 0.15 pmol of each plasmid was linearized by the restriction enzyme ScaI and used to transform B. subtilis. Eight transformants from each sample were tested by colony PCR to verify the integration of the target DNA fragment.
3. RESULTS AND DISCUSSION
The essential elements on an integration vector for double‐crossover homologous recombination in B. subtilis are the two homologous regions and the antibiotic resistance gene between them, while the other parts are resistance markers and origin of replication in E. coli, not needed for B. subtilis transformation. We first amplified the 5′amyE+cmR (PCR4) and the 3′amyE (PCR5) from the shuttle vector pDG1662 and assembled them with the fragments to be integrated, cotC‐petase (PCR6) and cotG‐mhetase (PCR7). The resulted linear DNA 1 was about 4 kb and linear DNA 2 was about 6 kb. Although elements essential for integration in B. subtilis were all present, the transformation of the assembled mixture either failed to work or had very low efficiency (Figure 2A). We further increased the concentration of the assembled DNA by amplifying the entire fragment via PCR and transforming the PCR products. Still, it did not work well.
Figure 2.
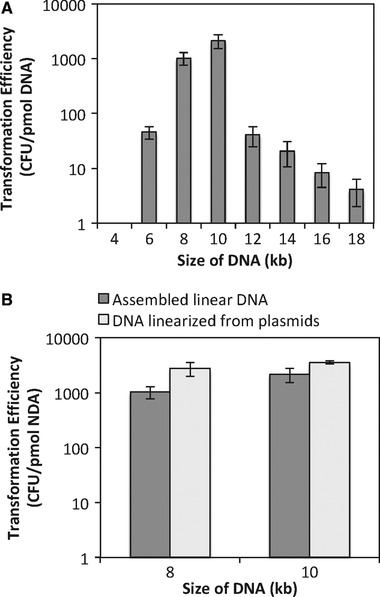
Transformation efficiency of linear DNA. (A) The effect of DNA size on transformation efficiency; (B) Comparison of linear DNA prepared via Gibson assembly and that from linearizing plasmids constructed in E. coli. Three parallel experiments were done for each transformation. The error bar represented standard deviation of the three samples
Size, topology, and sequence of DNA have been mentioned in previous work investigating properties of DNA that affect natural competence, but no quantitative results are available to correlate the size of DNA with transformation efficiency 11. Given that most commonly used double‐crossover integration vectors are about 6–10 kb, we added non‐essential sequences to our assembled linear DNA to make it longer. PCR1 and PCR2 carried 5′amyE+cmR and the 3′amyE, respectively, and also the flanking non‐essential sequences. Using these two fragments, we assembled linear DNA 3 (about 8 kb), which has cotC‐petase between them, and linear DNA 4 (about 10 kb), which has cotC‐petase and cotG‐mhetase between them. The fragments to be integrated (cotC‐petase) and (cotC‐petase and cotG‐mhetase) were also cloned into pDG1662. Then the plasmids were linearized using the restriction enzyme ScaI and used as the control for transformation. To test the effect of DNA size on transformation efficiency, we further expanded the range to 4–18 kb by incorporating DNA fragments from E. coli into DNA 3 (Table 1). As shown in Figure 2A, for DNA fragments tested from 4 to 18 kb, transformation of DNA of 8–10 kb gave the highest efficiency. Outside of this range, the efficiency dramatically decreased. The transformation efficiency with DNA of the same size was compared using DNA from linearizing circular plasmid or DNA directly assembled from PCR products. As shown in Figure 2B, the transformation efficiency using assembled linear DNA was slightly lower than that of DNA linearized from plasmids, but within the same order of magnitude, all about 1–3×103 colony forming units/μg DNA. The slightly lower efficiency is probably due to that in the assembled linear DNA samples, not all of the DNA molecules are assembled but we calculated the efficiency using the total amount of DNA in the assembly mixture. Given that the essential elements for integration including homologous sequences and the antibiotic resistance gene add together about 2 kb, this method allows one‐step integration of target DNA fragments up to 8 kb into B. subtilis chromosome. Eight transformants were randomly picked from each sample and the presence of the target DNA fragment was verified by colony PCR. Overall 75–100% of colonies showed positive results.
Gibson assembly is widely used for the assembly of self‐replicating circular plasmids 23. Compared with Golden Gate assembly, the number of DNA fragments it can assemble is limited to 4–6 fragments with efficiency much lower when it is above four. However, it does not rely on any restriction enzymes and offers flexibility in DNA assembly. While Golden Gate has been used with the OGAB method to assemble multimeric linear DNA that can be directly used to transform B. subtilis, the feasibility of Gibson assembly in constructing linear DNA fragments for direct transformation of B. subtilis has not reported before. In this work, we have demonstrated that B. subtilis can be successfully transformed and integrated with linear DNA from Gibson assembly. This offers a new method to bypass the traditional insertion of DNA into a shuttle vector in E. coli and subsequent linearization of the plasmid, which could significantly reduce the time needed for genome editing in B. subtilis.
The transformation efficiency is comparable to that of the routine method when assembling three or four pieces of DNA together, which is similar to the assembly of circular plasmids. The size of the linear DNA fragments is vital for the transformation and integration in B. subtilis. Our results showed that 6 kb (or 5 kb) is the lower limit and 12 kb is the upper limit for successfully transforming B. subtilis via natural competence. The two ends of shorter DNA may be subject to degradation during transformation, which is likely the reason why linear DNA 1 with 5′amyE and 3′amyE on both ends did not work. For linear DNA longer than 12 kb, the low efficiency may be due to the challenge of integration of long fragments through double‐crossover homologous recombination. The OGAB‐Golden Gate method has been reported to transform B. subtilis with multimeric DNA longer than 30 kb, which is circularized into a plasmid via homologous recombination once entering the cell. The purpose of OGAB‐Golden Gate is to assemble large plasmids, but not integrate them into the chromosome. However, integration of genes into the chromosome is necessary for the stability of the engineered B. subtilis strains. Further operations are needed to integrate the plasmid constructed using the OGAB method into the chromosome of B. subtilis.
In this work, we used the regular Spezizen transformation protocol, although there are more efficient methods, such as inducing and transiently activating the master regulator ComK (or ComKS) and electroporation using osmoprotectant, which are reported to have an efficiency about 106 transformants/μg DNA 15, 16. Our method is to prepare the linear DNA in a faster, more efficient way and the assembled DNA is compatible with other transformation protocols. When combined with the more efficient transformation methods, it will provide an easy way to construct mutant libraries, which is crucial to develop B. subtilis as a host for synthetic biology and systems biology.
4. CONCLUDING REMARKS
In this work, we examined the feasibility of using Gibson assembly to construct long linear DNA fragments and successfully transformed B. subtilis with the assembled linear DNA. It achieved comparable transformation efficiency when compared with linearized plasmids, which relies on the cloning in E. coli. This could significantly decrease the time needed for gene knockout or knockin in B. subtilis from 1 week to 1 day. In addition, we quantitatively examined the effect of the DNA size on the transformation efficiency and showed that linear DNA of 8–10 kb gave the highest efficiency for B. subtilis transformation and integration. Overall, this work provides a simple and fast method for genome editing in B. subtilis and will potentially aid in the study of B. subtilis as a model organism or engineering of B. subtilis strains.
CONFLICT OF INTEREST
The authors have declared no conflict of interest.
ACKNOWLEDGMENT
This work is supported by the research funds from the University of New Hampshire. The authors also wish to acknowledge the Bacillus Genetic Stock Center for providing the integration vectors.
Wu G, Drufva E, Wu K. Fast genome editing in Bacillus subtilis . Eng Life Sci. 2019;19:471–477. 10.1002/elsc.201800164
REFERENCES
- 1. Cui, W. , Han, L. , Suo, F. , Liu, Z. , Zhou, L. , Zhou, Z. , Exploitation of Bacillus subtilis as a robust workhorse for production of heterologous proteins and beyond. World. J. Microbiol. Biotechnol. 2018, 34(10), 145. [DOI] [PubMed] [Google Scholar]
- 2. Stein, T. , Bacillus subtilis antibiotics: structures, syntheses and specific functions. Mol. Microbiol. 2005, 56(4), 845–857. [DOI] [PubMed] [Google Scholar]
- 3. Wong, S. L. , Advances in the use of Bacillus subtilis for the expression and secretion of heterologous proteins. Curr. Opin. Biotechnol. 1995, 6(5), 517–522. [DOI] [PubMed] [Google Scholar]
- 4. Kumpfmuller, J. , Kabisch, J. , Schweder, T. , An optimized technique for rapid genome modifications of Bacillus subtilis. J. Microbiol. Meth. 2013, 95(3), 350–352. [DOI] [PubMed] [Google Scholar]
- 5. Spizizen, J. , Transformation of biochemically deficient strains of Bacillus Subtilis by deoxyribonucleate. Proc. Natl. Acad. Sci. U S A 1958, 44(10), 1072–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vojcic, L. , Despotovic, D. , Martinez, R. , Maurer, K. H. , Schwaneberg, U. , An efficient transformation method for Bacillus subtilis DB104. Appl. Microbiol. Biotechnol. 2012, 94(2), 487–493. [DOI] [PubMed] [Google Scholar]
- 7. Berka, R. M. , Hahn, J. , Albano, M. , Draskovic, I. , Persuh, M. , Cui, X. , Sloma, A. , Widner, W. , Dubnau, D. , Microarray analysis of the Bacillus subtilis K‐state: genome‐wide expression changes dependent on ComK. Mol. Microbiol. 2002, 43(5), 1331–1345. [DOI] [PubMed] [Google Scholar]
- 8. Gamba, P. , Jonker, M. J. , Hamoen, L. W. , A novel feedback loop that controls bimodal expression of genetic competence. PLos Genet. 2015, 11(6), e1005047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rahmer, R. , Morabbi Heravi, K. , Altenbuchner, J. , Construction of a super‐competent Bacillus subtilis 168 using the P mtlA ‐comKS Inducible Cassette. Front Microbiol. 2015, 6, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang, X. Z. , Zhang, Y. , Simple, fast and high‐efficiency transformation system for directed evolution of cellulase in Bacillus subtilis. Microb. Biotechnol. 2011, 4(1), 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohse, M. , Kawade, K. , Kusaoke, H. , Effects of DNA topology on transformation efficiency of Bacillus subtilis ISW1214 by electroporation. Biosci. Biotechnol. Biochem. 1997, 61 (6), 1019–1021. [DOI] [PubMed] [Google Scholar]
- 12. Mell, J. C. R. , Redfield, R. J. , Natural competence and the evolution of DNA uptake specificity. J. Bacteriol. 2014, 196(8), 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang, S. , Cohen, S. N. , High frequency transformation of Bacillus subtilis protoplasts by plasmid DNA. Mol. Gen. Genet. 1979, 168(1), 111–115. [DOI] [PubMed] [Google Scholar]
- 14. Kusaoke, H. , Hayashi, Y. , Kadowaki, Y. , Kimoto, H. , Optimal conditions for electric pulse‐mediated gene transfer to Bacillus subtilis cells. Agric. Biol. Chem. 1989, 53, 6. [Google Scholar]
- 15. Meddeb‐Mouelhi, F. , Dulcey, C. , Beauregard, M. , High transformation efficiency of Bacillus subtilis with integrative DNA using glycine betaine as osmoprotectant. Anal. Biochem. 2012, 424(2), 127–129. [DOI] [PubMed] [Google Scholar]
- 16. You, C. , Zhang, X. Z. , Zhang, Y. H. , Simple cloning via direct transformation of PCR product (DNA Multimer) to Escherichia coli and Bacillus subtilis. Appl. Environ. Microbiol. 2012, 78(5), 1593–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nguyen, H. D. , Nguyen, Q. A. , Ferreira, R. C. , Ferreira, L. C. , Tran, L. T. , Schumann, W. , Construction of plasmid‐based expression vectors for Bacillus subtilis exhibiting full structural stability. Plasmid. 2005, 54(3), 241–248. [DOI] [PubMed] [Google Scholar]
- 18. Feucht, A. , Lewis, P. J. , Improved plasmid vectors for the production of multiple fluorescent protein fusions in Bacillus subtilis. Gene 2001, 264(2), 289–297. [DOI] [PubMed] [Google Scholar]
- 19. Guerout‐Fleury, A. M. , Frandsen, N. , Stragier, P. , Plasmids for ectopic integration in Bacillus subtilis. Gene 1996, 180(1‐2), 57–61. [DOI] [PubMed] [Google Scholar]
- 20. Hartl, B. , Wehrl, W. , Wiegert, T. , Homuth, G. , Schumann, W. , Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J. Bacteriol. 2001, 183(8), 2696–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaltwasser, M. , Wiegert, T. , Schumann, W. , Construction and application of epitope‐ and green fluorescent protein‐tagging integration vectors for Bacillus subtilis. Appl. Environ. Microbiol. 2002, 68(5), 2624–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Engler, C. , Kandzia, R. , Marillonnet, S. , A one pot, one step, precision cloning method with high throughput capability. PLoS One, 20083(11), e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gibson, D. G. , Young, L. , Chuang, R. Y. , Venter, J. C. , Hutchison, C. A., 3rd , Smith, H. O. , Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Meth. 20096(5), 343–5. [DOI] [PubMed] [Google Scholar]
- 24. Ho‐Shing, O. , Lau, K. H. , Vernon, W. , Eckdahl, T. T. , Campbell, A. M. , Assembly of standardized DNA parts using BioBrick ends in E. coli. Methods Mol. Biol. 2012, 852, 61–76. [DOI] [PubMed] [Google Scholar]
- 25. Moore, S. J. , Lai, H. E. , Kelwick, R. J. , Chee, S. M. , Bell, D. J. , Polizzi, K. M. , Freemont, P. S. , EcoFlex: a multifunctional MoClo Kit for E. coli synthetic biology. ACS Synth. Biol. 2016, 5(10), 1059–1069. [DOI] [PubMed] [Google Scholar]
- 26. Tsuge, K. , Matsui, K. , Itaya, M. , One step assembly of multiple DNA fragments with a designed order and orientation in Bacillus subtilis plasmid. Nucleic Acids Res. 2003, 31(21), e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsuge, K. , Sato, Y. , Kobayashi, Y. , Gondo, M. , Hasebe, M. , Togashi, T. , Tomita, M. , Itaya, M. , Method of preparing an equimolar DNA mixture for one‐step DNA assembly of over 50 fragments. Sci. Rep. 2015, 5, 10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anagnostopoulos, C. , Spizizen, J. , Requirements for transformation in Bacillus Subtilis. J Bacteriol. 1961, 81(5), 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]