

Abstract
Near infrared spectroscopy is a rapid and nondestructive method for compositional analysis of biological material. The technology is widely used within bioreactors and possesses potential as a standardized method for quality control in miniaturized microfluidic cell culture systems. Here, we established a method for quantification of cell density and viability of adherent HepaRG cells cultured in a translucent, miniaturized cell culture biochip. The newly developed statistical models for interpretation of near infrared spectroscopy from biochips are the basis for a novel method of fast, continuous, and contact‐free analysis of cell viability and real‐time monitoring of cell growth. The technique thus paves the way for a robust and reliable high‐throughput analysis of biochip‐embedded cell cultures.
Keywords: Biochips, Cell culture, HepaRG, Near infrared spectroscopy, Partial least squares
Abbreviations
- COC
cyclo‐olefin‐copolymere
- NIRS
near infrared spectroscopy
- PLS
partial least squares
- RMSECV
root mean square error of cross‐validation
- RMSEP
root mean square error of prediction
- RPD
residual prediction deviation
1. Introduction
Advanced cell culture methods in miniaturized biochips are a rapid growing field of interest. Cell and tissue culture in microfluidically supported biochips offer the advantage of creating in vivo like environments such as dynamic culture of cell layers under defined physiological shear stress conditions 1. Perfusion of cell cultures enables constant supply with nutrients and removal of catabolic waste products, which was shown to improve cellular functions and morphological reconstitution of endothelial cells 2, kidney cells 3, and hepatocytes 4 in vitro.
To facilitate a reliable cell culture under reproducible conditions, a nondestructive measurement of cell growth and viability is desirable. So far most cell‐based methods to determine cell viability in biochips are still endpoint analysis involving detachment or staining of cells, which potentially alters cellular metabolism 5. Optical analysis methods, such as spectroscopy, allow a rapid and contact‐free monitoring with the potential of high‐throughput measurement and continuous data acquisition 6, 7, 8 and have been applied in microfluidic cell culture systems already 9, 10. Jaccard et al. recently reported a microscopy‐based system adapted for analysis of confluency and density of various cell types in bioreactors for fast and automated cell culture monitoring 11, 12. In addition, spectroscopy‐based systems possess the advantage to further analyze multiple parameters with a single measurement 13. Those fast and easy to handle applications emerge, especially in cellular analysis, and are applicable from single cell analysis 14 up to monitoring of industrial size cell cultivation in large bioreactors 15.
It was shown that near infrared spectroscopy (NIRS) as well as Raman spectroscopy can provide data for essential components of the cell culture media, such as glucose and lactate 16, 17. Besides that, it is used as a versatile tool for quantitative and qualitative analysis of solid and liquid pharmaceutical formulations 18 and for the characterization of microorganisms such as Vibrio cholerae 19, Saccharomyces cerevisiae 20, or Escherichia coli by transmission measurements 21. As NIRS is based on molecular overtone and combination vibrations with broad and overlapping absorbance bands 22, an appropriate calibration model with defined parameters is needed for subsequent analysis of spectral data. Clavaud et al. predicted the densities of viable Chinese hamster ovary cells in bioreactors within a 2‐week period and created a predictive model in a defined range with transflective NIRS combined with chemometrics 23. This study proves the potential of NIRS for monitoring cell cultures in bioreactor devices with online readouts of cell growth. Miniaturized biochips were recently described with complex adherent cell layers, ranging from collagen embedded monolayers 24 up to the integration of four different cell types 25. Spectroscopic methods, such as NIRS, potentially represent a valuable strategy to monitor complex cell cultures. Therefore, it has to be evaluated whether in principle transmission‐based NIRS can also be used for the high‐throughput analysis of translucent cell culture biochips.
Here, we describe a transmission‐based NIRS measurement protocol in translucent custom‐built cell culture devices to predict cell densities of monolayers of the hepatic cell line HepaRG 26 as a proof of concept. Overall, 118 samples were used for the calibration data set and 19 independent samples for the validation data set. In a second approach, we calibrated a data set of 102 samples with induced apoptosis and a validation data set of 19 independent samples. To our best knowledge, this is the first report describing the application of NIRS for detection of the cell density of adherent cells in a translucent cell culture chip. The method further represents a promising tool for high‐throughput analysis of cell toxicity in pharmaceutical screening studies.
2. Materials and methods
2.1. Cell culture
HepaRG cells were obtained from Biopredic (Rennes, France). Undifferentiated cells were seeded at a density of 2.7 × 105/cm2 and cultivated in William's Medium E (Biochrom, Berlin, Germany) containing 10% v/v FBS (GIBCO, Darmstadt, Germany), 5 μg/mL insulin (Sigma‐Aldrich, Steinheim, Germany), 2 mM glutamine (GIBCO), 5 × 10−5 M hydrocortisone‐hemisuccinate (Sigma‐Aldrich), and 100 U/mL Penicillin/100 μg/mL streptomycin mixture (Pen/Strep) (GIBCO) in a humidified cell incubator at 5% CO2 and 37°C for 14 days. Subsequently cells were differentiated by the addition of DMSO (Sigma‐Aldrich) to the medium as previously described 26 and used for up to 4 weeks after differentiation. Medium was renewed twice a week.
2.2. Biochip design
The biochips of the multiorgan tissue flow chip series were produced by microfluidic ChipShop GmbH (Jena, Germany, Supporting Information Fig. S1A). The biochip devices were made from Topas®, a cyclo‐olefin‐copolymere (COC). The chip was manufactured by injection molding using a modular mold base with exchangeable metallic mold inserts. The mold inserts were structured by ultraprecision mechanical machining using a 100 μm diameter machining bit for the smallest features and finishing. First the microstructured part with microfluidic channels network, fluidic interfaces and the area for membrane integration was made by injection molding. Second, the integration of the 11 μm thick polyethylene‐terephthalate (PET) membrane with a pore diameter of 8 μm and a pore density of 2 × 105 pores/cm2 (TRAKEDGE Sabeu, Radeberg, Germany) was carried out. All four round cavities have a total cell culture area of 0.785 cm2 each. After cell seeding, the biochips were sealed at the bottom side with an extruded 140 μm thick COC film using a low‐temperature proprietary bonding method. Surface oxidation for hydrophilization was accomplished by treatment with oxygen plasma using a T200G plasma generator (PVA Tepla AG, Wettenberg, Germany). COC foils used for bottom sealing show significant oxygen permeability. Bubble formation was reduced by oxygen plasma treatment of the whole chip's surface.
2.3. Cell culture in biochips and cell density determination
Differentiated HepaRG cells were directly seeded on the porous PET membrane with indicated densities and cultured for 24 h (Supporting Information Fig. S1B). Subsequently, the top chamber of the biochips was sealed with a polystyrene cover slip ahead of NIRS spectrum acquisition. For analysis of cell number (density), as reference to NIRS measurement, the cell layer of each cavity was washed with 50 mM EDTA (Sigma‐Aldrich) and PBS (Biochrom). Cells were incubated with 0.1% collagenase solution (Biochrom) in PBS for 30 min at 37°C for maximum cell separation and stopped with 0.5 M EDTA solution. All supernatants (washing included) were pooled cavity wise and centrifuged for 7 min at 200 x g. Total cell quantities were determined using a hemocytometer (Neubeauer Feinoptik, Bad Blankenburg, Germany). All experiments were performed under static conditions.
2.4. NIRS measurement
The experimental setup for NIR measurement consisted of a plate grid spectrometer MCS611 NIR 1.7 (Carl Zeiss, Jena, Germany) with a wavelength ranging from 950 to 1700 nm and a fiber‐coupled transmission setup. The transmission setup was placed in a humidified FlowBox (Automated Lab Solutions ALS, Jena, Germany) at 5% CO2 and 37°C (Fig. 1D). The spectrometer system and transmission setup were connected via optical fibers. The measurement process was controlled by a custom‐made measurement software following the present measurement course. All measurements were done with 1000 scans to compensate pink noise. In prior measurement, the integration time was adjusted to use the dynamic range of the spectrometer system by approximately 90% at reference measurement. At least 10 min before each measurement all biochips were enclosed with a polystyrene cover slide and equilibrated. Biochip containing medium only were used as reference for NIRS analysis. For maximum coverage of cell layers and to avoid spectral outliers by inhomogeneous cell growth, three partly overlapping measurements were done at each cavity (Fig. 1B and C). These three measurements were averaged to one spectrum per sample.
Figure 1.
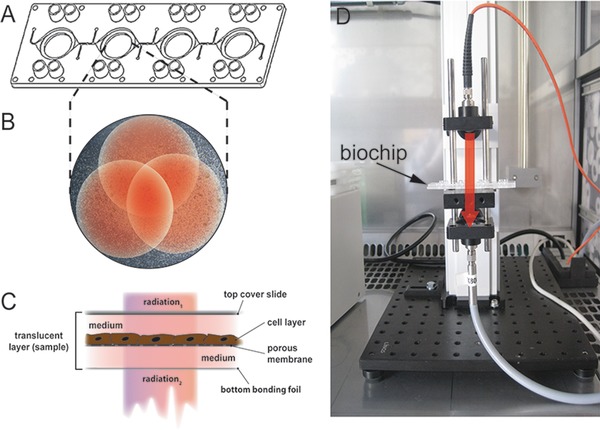
(A) Multiorgan tissue flow (MOTIF) biochip with four interconnected round cavities in microscopic slide format. Each cavity comprises a porous membrane that separates the cavity in a top and a bottom chamber. (B) Example for the three partly overlapping measurement points (red dots) in one cavity with adherent HepaRG cells. (C) Schematic cross‐section of the translucent biochip containing a HepaRG cell layer and a membrane serving as cell substrate. At top, the biochip is sealed with a polystyrene cover slide. Light beam for NIRS enters the biochip through the cover slide (radiation 1) and leaves the biochip through the bottom bonding foil (radiation 2). (D) Transmission measurement setup (NIRS). The red arrow indicates the radiation pathway through the translucent biochip.
2.5. Apoptosis assay
Cells seeded in biochips were incubated for 20 h with 2.5 μM staurosporine (Sigma‐Aldrich) at 37°C. Subsequently, biochips were sealed and NIR measurement was performed. The cell layer was washed once with medium and the supernatant containing also nonadherent cells was pooled with enzymatically detached adherent cells for subsequent flow cytometric analysis.
2.6. Flow cytometry
Nonadherent and detached cells from at least three cavities of each biochip were pooled and centrifuged for 7 min at 200 × g. The supernatant was removed and the cells were resuspended in 100 μL annexin‐V binding buffer (BD Biosciences, Heidelberg, Germany) containing 5 μL 7‐aminoactinomycin D (BD Biosciences) and 2 μL annexin‐V‐allophycocyanin (BD Biosciences) for 30 min at room temperature and immediately measured using a BD FACS Canto II (BD Biosciences). Data were analyzed with FACSDiva software (BD Bioscience) and FlowJo X software (FlowJo LLC, Ashland, OR, USA). The average (percentage) of viable, apoptotic, and dead cells of one biochip was used for the calibration of related NIRS data.
2.7. Absorbance and spectra processing
Medium filled cavities were chosen as reference and compared to cavities filled with medium and cultured cells for spectra analysis. The spectral background of the membrane was corrected by the reference spectrum for all samples. For optimal results, appropriate wavelength regions had to be identified (Table 1). The raw absorbance spectra (wavelength region of interest: 1000–1700 nm) with an average of three measurement points per sample (1000 scans each; 3000 scans/sample) was used for calibration and validation. One wavelength region offers poor signal: O‐H‐absorbance band between 1400 and 1500 nm. Reference and samples spectra (Formula 1) feature a low signal and high noise in this region. Therefore, spectra were cut at the verge of this absorbance band (between 1400 and 1500 nm) to exclude this wavelength region, similar to the reports of Mercier et al. 27 (Supporting Information Fig. S2).
| (1) |
Table 1.
Statistical parameters of the calibration models for cell density prediction
| Model | Data preprocessing (wavelength region) | No. of samples | R 2 | RMSECV (cells) | RPD | RMSEP (cells) |
|---|---|---|---|---|---|---|
| 1 | None | 118 | 93.99 | 12 700 | 4.1 | 19 000 |
| (1100–1600 nm) | ||||||
| 2 | None | 118 | 93.76 | 12 900 | 4.0 | 20 900 |
| (1000–1700 nm) | ||||||
| 3 | Kennard–Stone algorithm | 59 | 94.02 | 13 500 | 4.1 | 19 800 |
| (950–1400 nm) | ||||||
| 4 | Kennard–Stone algorithm | 59 | 94.19 | 13 300 | 4.2 | 14 900 |
| (950–1350 nm) |
Calibration/validation range (total cells): 6 × 103–2.1 × 105 for all models.
Partial least squares (PLS) regression algorithm was used to develop calibration models 28. This multivariate calibration method is used to correlate NIR spectra to the component of interest. The validation was made by cross‐validation (internal validation: leave one out). The optimal data preprocessing has to be determined iteratively. During this process, the algorithm splitted the wavelength range into 10 parts. Additionally, the following options for the iterative approach were tested to provide optimal results:
no data preprocessing,
subtraction of a constant offset,
subtraction of a line,
vector normalization (standard normal variate),
min‐max‐normalization,
multiplicative scattering correction,
first derivation,
second derivation,
first derivation and subtraction of a line,
first derivation and standard normal variate,
first derivation and multiplicative scattering correction.
During this iterative procedure, the parameters of quality coefficient of determination (R 2)(Formula 2), residual prediction deviation (RPD) (Formula 3), root mean square error of cross‐validation (RMSECV) (Formula 4), and root mean square error of prediction (RMSEP) (Formula 5) were evaluated and consequently the best calibrations chosen. The best calibration is defined by the highest RPD, lowest RMSECV, R² close to 100, and a broad range of calibration data. All iteratively tested preprocessing strategies with parameters of lower statistical quality were not considered for final calibration and validation.
| (2) |
| (3) |
| (4) |
n = number of samples for calibration
| (5) |
k = number of samples for validation
Based on the Kennard–Stone algorithm, we used a subset of 50% of all samples for analysis 29. Spectroscopic analysis and data processing were performed with Bruker OPUS 7.0 (Bruker Corporation, Billerica, MA, USA) and MatLab (The MathWorks GmbH, Ismanning, Germany).
3. Results
3.1. Cell density prediction
NIRS was performed in miniaturized cell culture biochips containing four interconnected cavities, each holding a translucent membrane serving as substrate for cultured HepaRG cells (Fig. 1A).
In a first approach, 118 NIRS samples of biochip‐embedded cell layers were measured and analyzed. The spectra of each sample was generated by averaging a total of 3000 scans at three measuring points and referred to the cell density of each cavity. Subsequently, a multivariate analysis, using Bruker OPUS software, was applied. We developed two calibration models containing the samples (Fig. 2A and B, models 1 and 2). Both calibration models have sufficient and almost similar parameters of quality and were generated without pretreatment, but cover different wavelength regions (Table 1). Models 1 and 2 were validated using 19 independent samples. The RMSEP (3) was RMSEP1 = 19 000 cells and RMSEP2 = 20 900 cells (Fig. 2C and D).
Figure 2.
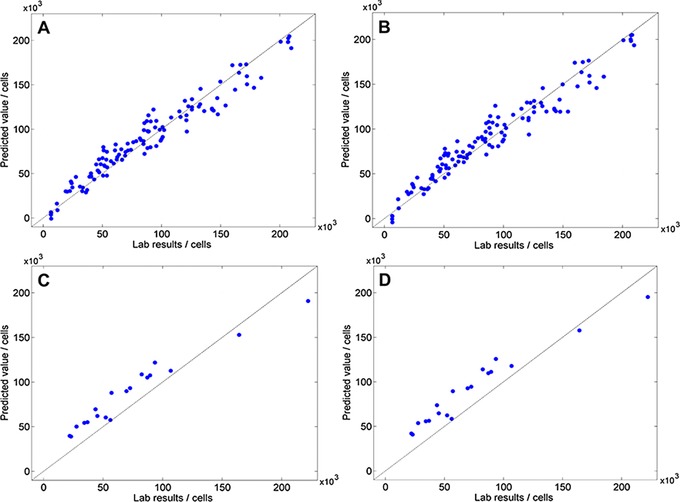
(A) Scatterplot of predicted value vs. lab results of model 1: R 2 = 93.99; RMSECV = 12 700 cells; RPD = 4.1. (B) Scatterplot of predicted value versus lab results of model 2: R 2 = 93.76; RMSECV = 12 900 cells; RPD = 4.0. (C) Validation of model 1: RMSEP = 19 000 cells. (D) Validation of model 2: RMSEP = 20 900 cells.
Due to these insufficient results, we used the Kennard–Stone algorithm to extract the samples with the highest variance in the spectra. This method efficiently selects samples with a uniform distribution throughout the whole range, starting with the two points farthest apart 29. Based on this, we developed two new calibration models containing a data set of 59 samples out of 118 samples (Fig. 3A and B), with almost similar parameters of quality as compared to Fig. 2A and B.
Figure 3.
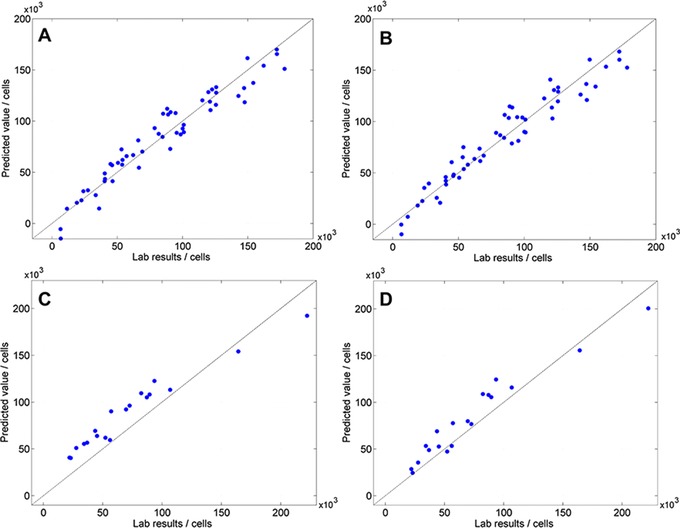
(A) Scatterplot of predicted value versus lab results of model 3: R 2 = 94.02; RMSECV = 13 500 cells; RPD = 4.1. (B) Scatterplot of predicted value versus lab results of model 4: R 2 = 94.19; RMSECV = 13,300 cells; RPD = 4.2. (C) Scatterplot of the validation of model 3: RMSEP = 19 800 cells. (D) Scatterplot of the validation of model 4: RMSEP = 14 900 cells.
The cross‐validation of these calibration models with Kennard–Stone‐selected data sets resulted in a higher RMSECV = 13 300–13 500 cells but almost similar R 2 and RPD. With a RPD ≥ 4, all models are suited for quality control 30. Additionally, the validation revealed that models 1 and 2 are less robust using independent samples. Models 1–3 have a RMSEP ranging from 19 000 to 20 900 cells. In contrast to that, model 4 proves its robustness for independent samples with the lowest RMSEP compared to the other validations (Table 1).
3.2. Spectra assembly for viability prediction
In a second approach, we developed a calibration model for the pooled samples analyzed by flow cytometry, which were treated with staurosporine, a protein‐kinase inhibitor and known inducer of apoptosis 31, 32.
The NIRS spectra of pooled samples were referred to the percentage of viable, apoptotic, and dead cells, analyzed by flow cytometric analysis. Overall, 102 samples were treated with staurosporin to induce apoptosis and correlated to NIRS spectra.
Prediction capabilities of NIRS were than extended to predict qualitative cellular parameters such as apoptosis induction. We used a flow cytometry based viability assay as reference method to evaluate the obtained NIRS data. The cytometric analysis showed 37.0 (±5.5)% viable, 46.8 (±7.9)% apoptotic, and 16.2 (±5)% dead cells (Fig. 4). All cavities of one chip were pooled for a more robust analysis. We were able to create three different plots for the prediction of viable, apoptotic, and dead cells of the single NIR spectra (Fig. 5). The highest RPD (2.7) and R 2 showed the validation of the apoptotic prediction model, whereas the statistical quality for the prediction of viable and dead cells with NIRS was lower overall (Table 2).
Figure 4.
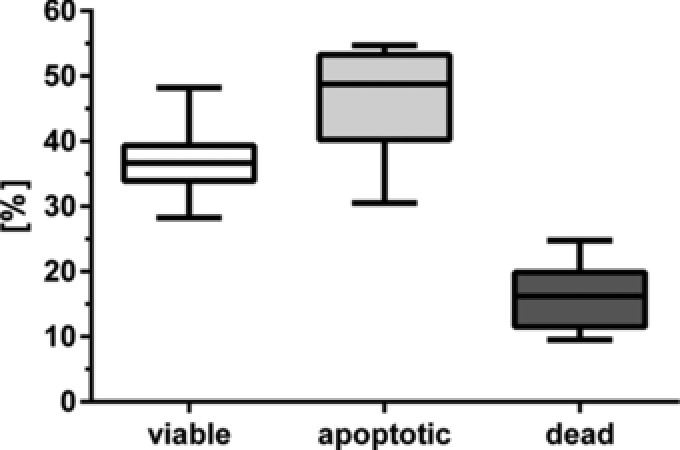
Flow cytometry data of viable, apoptotic, and dead cells after staurosporine treatment. All cavities of each chip were pooled before analysis (n = 20).
Figure 5.
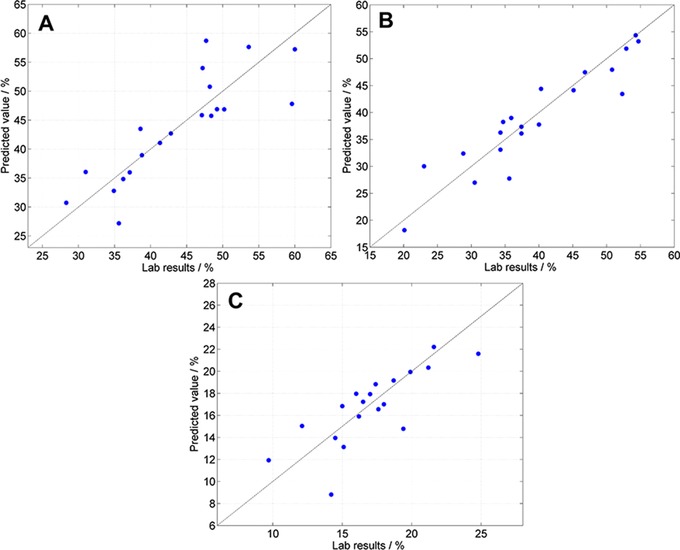
(A) Scatterplot of predicted value versus lab results of viable cells: R 2 = 66.26; RMSECV = 4.98%; RPD = 1.7. (B) Scatterplot of predicted value versus lab results of apoptotic cells: R 2 = 85.68; RMSECV = 3.75%; RPD = 2.7. (C) Scatterplot of predicted value versus lab results of dead cells: R 2 = 57.72; RMSECV = 2.21%; RPD = 1.6.
Table 2.
Validation of parameters of staurosporin‐treated HepaRG
| Model | No. of samples | Data preprocessing | R 2 | RMSECV | RPD | Range |
|---|---|---|---|---|---|---|
| (wavelength region) | (%) | (%) | ||||
| Viable | 20 | msc (1000–1600 nm) | 66.26 | 4.98 | 1.7 | 28–60 |
| Apoptotic | 20 | Subtraction of a line | 85.68 | 3.75 | 2.7 | 20–55 |
| (950–1400 nm) | ||||||
| Dead | 20 | First derivation and msc | 57.72 | 2.21 | 1.6 | 10–25 |
| (1000–1700 nm) |
msc = multiplicative scattering correction.
4. Discussion
Most NIRS‐based applications monitoring cell‐related parameters include a broad range of up to 19 different analytes 15, 16, 17, 33. Despite the variety of parameters, the applications of NIRS and Raman spectroscopy based systems available are restricted to large‐scale bioreactors 13. In order to make NIRS available as a robust technique for analysis of cell cultures in miniaturized biochip devices, we developed new measurement protocols and related statistical models to fill this need. We demonstrated that our method allows a reliable prediction of the cell density and provides an easy and rapid tool for continuing monitoring of biochip‐embedded cell cultures.
It was shown that cell confluency as well as cell density for adherent cells can be evaluated by phase contrast microscopy with precise determination for different cell types 11, 12. However, this method is not suitable for thick cell layers or multiparametric measurements due to its optical image analysis. NIRS has the potential to combine the read‐out of different analytes and cellular growth. But there are different aspects to consider before an NIRS‐based measurement can be used for biochip monitoring.
Huang et al. described that spectra preprocessing and light scattering effects are of major importance for NIR data acquisition 34. We chose a transmission measurement setup with a defined light path and standardized biochips as a basis for our work. This provides minimal variation in case of light scattering and path length variations. Furthermore, we preselected appropriate wavelength regions and tested various methods for data preprocessing as described in Section 2.1 to get optimal results. The optimal preprocessing strategy was tested iteratively and nonsuitable models with a lower quality of statistical parameters were excluded.
All four models for the prediction of cell density were calibrated based on data from cell counting with a hemocytometer. The models showed a RMSEP up to 20 000 cells, which is a mean error of approximately 10%, considering the calibration range. Cadena‐Herrera et al. showed that a trypan blue exclusion test with a hemocytometer can result in a relative standard deviation up to 8.06% depending on the cell type 35. This needs to be considered, when evaluating the mean error of the calibration/validation, because the accuracy of prediction is restricted by the reference method. Models 1 and 2 were generated without any pretreatment and showed slight differences dependent on the selection of the wavelength region (Table 1). By applying the Kennard–Stone algorithm and shrinking of the analyzed wavelength region, we were able to generate models 3 and 4. The latter showed the best validation parameters compared to the other three models. However, almost all parameter showed only slight differences, except of the RMSEP. The slight overestimation in our cell density prediction models can be explained with the PLS regression. This method is optimal for linear coherences. Due to varying and inhomogeneous cell layers, a nonlinear approximation may cause this overestimation of approximately 10 000 cells. Generally, a higher amount of samples leads to a better and more precise calibration model. However, we showed that data preprocessing with the Kennard–Stone algorithm resulted in a more robust validation by independent samples, even with a reduced amount of calibration samples. This can be explained by efficient selection of highest variance in sample spectra as described before. The validation revealed a lower RMSEP (≈15 000 cells; 7.1% mean error of the calibration range), which is close to the RMSECV with a more precise prediction capability. A broad calibration range results in a high error at low cell density values but a more robust calibration. In comparison to a recently published NIR‐model by Clavaud et al. that predicts the viable cell density, our model 4 showed a better R 2 (89 vs. 94), RPD (1.93 vs. 4.1), and a lower RMSEP (% of the calibration range, 11.6 vs. 7.1%) 23. Additionally, Mercier et al. published a PLS calibration model with no pretreatment of spectral data. This model showed a RMSEP of 2.2% of the calibration range 27. In contrast to our model, the authors used nonadherent cells with a calibration ranging from 0.76 × 106 to 91.62 × 106 cells/ mL, which is relatively high (vs. 6 × 103–2.1 × 105 total cells). Therefore, the spectroscopic setup, the calibration strategy, the statistical parameters, and the input data have to be evaluated carefully for the assessment and comparison of NIRS models.
In a second approach, we characterized apoptosis‐related cell death of staurosporin‐treated HepaRG cells. To our best knowledge, this is the first description of a novel strategy to calibrate NIRS data with cell viability measured by flow cytometry. We were able to predict the number of apoptotic cells with better statistical parameters than dead and apoptotic. Due to shrinking and compartmentalization of cell organelles during apoptosis and later on cell death, there is a variety of factors involved that have an influence on NIRS measurement. Nonattached dead cells might cause a different light scattering or absorption as cells still sticking to the cell layer 36. Furthermore, the number of samples is relatively low (n = 20) due to pooling of related spectra. Pooling is necessary to cover a broad cell density range with one measurement. We estimate to achieve improved parameters of quality (R², RMSECV, RPD) by significantly increasing the number of samples. For high‐throughput toxicity studies, a measurement of detached cells will be more sufficient for preevaluation of potential drugs rather than an NIRS viability assay. Nonetheless, a precise discrimination of viable, apoptotic, and dead cells could be more beneficial for further studies and would create new opportunities for a multiparametric measurement with NIRS.
5. Concluding remarks
In conclusion, our data prove the potential of NIRS as contact‐free and nondestructive method to analyze cell density and viability of biochip‐embedded cell cultures in real time. This rapid, cost‐efficient, and robust analysis method enables a reliable measurement within a translucent miniaturized cell culture biochip and subsequent statistical analysis of various cell densities with a RPD >4 in a defined range. The combination of these techniques with a high‐throughput analysis platform could be a valuable tool for quality control of biochip‐embedded cell cultures and their use in preclinical pharmaceutical screening studies using novel approaches such as emerging organ‐on‐a‐chip cell culture techniques.
Practical application
Near infrared spectroscopy (NIRS) is a rapid and nondestructive method for compositional analysis of biological material. Here, we describe its use as a novel, fast, and nondestructive strategy to analyze biochip‐embedded cell cultures. NIRS spectra acquisition and data analysis based on the mathematical models for prediction of cell density and viability described in the study represent a valuable tool for quality control of biochip‐embedded cell cultures. It is, thus, a suitable method to standardize biochip‐embedded cell layers and to allow reliable screening studies in the emerging field of tissue‐on‐chip cell culture techniques.
The authors have declared no conflict of interest.
Supporting information
Supplementary Information
Supplementary Information
Supplementary Figure 1. A) Drawing of the biochip design used in the study. B) Microscopic images of three cavities showing a reference sample without cells and two different typical cell numbers of membrane‐cultured HepaRG cells used to acquire NIRS spectra (cells per cavity are indicated above to image).
Supplementary Figure 2. Example of NIR spectra for staurosporine‐treated cells (red; top pair) and viable cells (blue; bottom pair). The region of high absorbance caused by water (between dashed lines) was excluded from analysis.
Acknowledgments
We are appreciating the excellent technical work of Maria Franke and Margot Voigt. The authors gratefully acknowledge support of this work by 2011 VF 0005 grant of the Thüringer Aufbaubank (Germany). M.G. and K.R. performed experiments and wrote the manuscript. M.L. developed and optimized statistical methods and wrote the manuscript. M.L., T.K., M.H., and H.P. contributed to analysis of NIRS data. A.S.M. planed the study, supervised experiments, and wrote the manuscript.
6 References
- 1. Rennert, K. , Steinborn, S. , Gröger, M. , Ungerböck, B. et al., A microfluidically perfused three dimensional human liver model. Biomaterials 2015, 71, 119–131. [DOI] [PubMed] [Google Scholar]
- 2. Raasch, M. , Rennert, K. , Jahn, T. , Peters, S. et al., Microfluidically supported biochip design for culture of endothelial cell layers with improved perfusion conditions. Biofabrication 2015, 7, 15013. [DOI] [PubMed] [Google Scholar]
- 3. Sciancalepore, A. G. , Sallustio, F. , Girardo, S. , Gioia Passione, L. et al., A bioartificial renal tubule device embedding human renal stem/progenitor cells. PLoS One 2014, 9, e87496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prot, J. M. , Aninat, C. , Griscom, L. , Razan, F. et al., Improvement of HepG2/C3a cell functions in a microfluidic biochip. Biotechnol. Bioeng. 2011, 108, 1704–1715. [DOI] [PubMed] [Google Scholar]
- 5. Press, A. T. , Ungelenk, L. , Rinkenauer, A. C. , Gröger, M. et al., A new fluorescent dye for cell tracing and mitochondrial imaging in vitro and in vivo. J. Biophotonics 2016, 9, 888–900. [DOI] [PubMed] [Google Scholar]
- 6. Qiu, J. , Arnold, M. A. , Murhammer, D. W. , On‐line near infrared bioreactor monitoring of cell density and concentrations of glucose and lactate during insect cell cultivation. J. Biotechnol. 2014, 173, 106–111. [DOI] [PubMed] [Google Scholar]
- 7. Rhiel, M. H. , Cohen, M. B. , Arnold, M. A. , Murhammer, D. W. , On‐line monitoring of human prostate cancer cells in a perfusion rotating wall vessel by near‐infrared spectroscopy. Biotechnol. Bioeng. 2004, 86, 852–861. [DOI] [PubMed] [Google Scholar]
- 8. Sampaio, P. N. , Sales, K. C. , Rosa, F. O. , Lopes, M. B. et al., In situ near infrared spectroscopy monitoring of cyprosin production by recombinant Saccharomyces cerevisiae strains. J. Biotechnol. 2014, 188, 148–157. [DOI] [PubMed] [Google Scholar]
- 9. Magnusson, E. B. , Halldorsson, S. , Fleming, R. M. T. , Leosson, K. , Real‐time optical pH measurement in a standard microfluidic cell culture system. Biomed. Opt. Express 2013, 4, 1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheri, M. S. , Shahraki, H. , Sadeghi, J. , Moghaddam, M. S. et al., Measurement and control of pressure driven flows in microfluidic devices using an optofluidic flow sensor. Biomicrofluidics 2014, 8, 54123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jaccard, N. , Griffin, L. D. , Keser, A. , Macown, R. J. et al., Automated method for the rapid and precise estimation of adherent cell culture characteristics from phase contrast microscopy images. Biotechnol. Bioeng. 2014, 111, 504–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jaccard, N. , Macown, R. J. , Super, A. , Griffin, L. D. et al., Automated and online characterization of adherent cell culture growth in a microfabricated bioreactor. J. Lab. Autom. 2014, 19, 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lourenço, N. D. , Lopes, J. A. , Almeida, C. F. , Sarraguça, M. C. et al., Bioreactor monitoring with spectroscopy and chemometrics: A review. Anal. Bioanal. Chem. 2012, 404, 1211–1237. [DOI] [PubMed] [Google Scholar]
- 14. Brundermann, E. , Bergner, A. , Petrat, F. , Schiwon, R. et al., Fast quantification of water in single living cells by near‐infrared microscopy. Analyst 2004, 129, 893–896. [DOI] [PubMed] [Google Scholar]
- 15. Arnold, S. A. , Crowley, J. , Woods, N. , Harvey, L. M. et al., In‐situ near infrared spectroscopy to monitor key analytes in mammalian cell cultivation. Biotechnol. Bioeng. 2003, 84, 13–19. [DOI] [PubMed] [Google Scholar]
- 16. Card, C. , Hunsaker, B. , Smith T., Hirsch, J. , Near‐infrared spectroscopy for rapid, simultaneous monitoring of multiple components in mammalian cell culture. Bioprocess Int. 2008, 3, 58–67. [Google Scholar]
- 17. Abu‐Absi, N. R. , Kenty, B. M. , Cuellar, M. E. , Borys, M. C. et al., Real time monitoring of multiple parameters in mammalian cell culture bioreactors using an in‐line Raman spectroscopy probe. Biotechnol. Bioeng. 2011, 108, 1215–1221. [DOI] [PubMed] [Google Scholar]
- 18. Roggo, Y. , Chalus, P. , Maurer, L. , Lema‐Martinez, C. et al., A review of near infrared spectroscopy and chemometrics in pharmaceutical technologies. J. Pharm. Biomed. Anal. 2007, 44, 683–700. [DOI] [PubMed] [Google Scholar]
- 19. Navrátil, M. , Norberg, A. , Lembrén, L. , Mandenius, C.‐F. , On‐line multi‐analyzer monitoring of biomass, glucose and acetate for growth rate control of a Vibrio cholerae fed‐batch cultivation. J. Biotechnol. 2005, 115, 67–79. [DOI] [PubMed] [Google Scholar]
- 20. Finn, B. , Harvey, L. M. , McNeil, B. , Near‐infrared spectroscopic monitoring of biomass, glucose, ethanol and protein content in a high cell density baker's yeast fed‐batch bioprocess. Yeast 2006, 23, 507–517. [DOI] [PubMed] [Google Scholar]
- 21. Arnold, S. A. , Gaensakoo, R. , Harvey, L. M. , McNeil, B. , Use of at‐line and in‐situ near‐infrared spectroscopy to monitor biomass in an industrial fed‐batch Escherichia coli process. Biotechnol. Bioeng. 2002, 80, 405–413. [DOI] [PubMed] [Google Scholar]
- 22. Reich, G. , Near‐infrared spectroscopy and imaging: Basic principles and pharmaceutical applications. Adv. Drug Deliv. Rev. 2005, 57, 1109–1143. [DOI] [PubMed] [Google Scholar]
- 23. Clavaud, M. , Roggo, Y. , von Daeniken, R. , Liebler, A. et al., Chemometrics and in‐line near infrared spectroscopic monitoring of a biopharmaceutical Chinese hamster ovary cell culture: Prediction of multiple cultivation variables. Talanta 2013, 111, 28–38. [DOI] [PubMed] [Google Scholar]
- 24. Gröger, M. , Rennert, K. , Giszas, B. , Weiss, E. et al., Monocyte‐induced recovery of inflammation‐associated hepatocellular dysfunction in a biochip‐based human liver model. Sci. Rep. 2016, 6, 21868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCarty, W. J. , Usta, O. B. , Luitje, M. , Bale, S. S. et al., A novel ultrathin collagen nanolayer assembly for 3‐D microtissue engineering: Layer‐by‐layer collagen deposition for long‐term stable microfluidic hepatocyte culture. Technology (Singap. World Sci.) 2014, 2, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gripon, P. , Rumin, S. , Urban, S. , Le Seyec, J. et al., Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mercier, S. M. , Rouel, P. M. , Lebrun, P. , Diepenbroek, B. et al., Process analytical technology tools for perfusion cell culture. Eng. Life Sci. 2016, 16, 25–35. [Google Scholar]
- 28. Martens H., Naes T. (Eds.), Multivariate Calibration, Wiley, New York: 1989. [Google Scholar]
- 29. Kennard, R. W. , Stone, L. A. , Computer aided design of experiments. Technometrics 1969, 11, 137–148. [Google Scholar]
- 30. Williams P. C., Norris K. H. (Eds.), Near‐Infrared Technology in the Agricultural and Food Industries, American Association of Cereal Chemists, St. Paul, MN: 1987. [Google Scholar]
- 31. Feng, G. , Kaplowitz, N. , Mechanism of staurosporine‐induced apoptosis in murine hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G825–G834. [DOI] [PubMed] [Google Scholar]
- 32. Mukwena, N. T. , Al‐Rubeai, M. , Apoptosis and its suppression in hepatocytes culture. Cytotechnology 2004, 46, 79–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riley, M. R. , Crider, H. M. , Nite, M. E. , Garcia, R. A. et al., Simultaneous measurement of 19 components in serum‐containing animal cell culture media by Fourier transform near‐infrared spectroscopy. Biotechnol. Prog. 2001, 17, 376–378. [DOI] [PubMed] [Google Scholar]
- 34. Huang, J. , Romero‐Torres, S. , Moshgbar, M. , Practical considerations in data Pre‐treatment for NIR and Raman spectroscopy. Am. Pharm. Rev. 2010, 13, 116–127. [Google Scholar]
- 35. Cadena‐Herrera, D. , Esparza‐De Lara, J. E. , Ramírez‐Ibañez, N. D. , López‐Morales, C. et al., Validation of three viable‐cell counting methods. Biotechnol. Rep. 2015, 7, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reed, J. C. , Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Information
Supplementary Figure 1. A) Drawing of the biochip design used in the study. B) Microscopic images of three cavities showing a reference sample without cells and two different typical cell numbers of membrane‐cultured HepaRG cells used to acquire NIRS spectra (cells per cavity are indicated above to image).
Supplementary Figure 2. Example of NIR spectra for staurosporine‐treated cells (red; top pair) and viable cells (blue; bottom pair). The region of high absorbance caused by water (between dashed lines) was excluded from analysis.