

Abstract
The design of an optimal process is particularly crucial when the reactants deactivate the biocatalyst. The reaction cascades of the chemo‐enzymatic epoxidation where the intermediate peroxy acid is produced by an enzyme are still limited by enzyme inhibition and deactivation by hydrogen peroxide.
To avoid additional effects caused by interfaces (aq/org) and to reduce the process limiting deactivation by the substrate hydrogen peroxide, a single‐phase concept was applied in a fed‐batch and a continuous process (stirred tank), without the commonly applied addition of a carrier solvent. The synthesis of peroxyoctanoic acid catalyzed by Candida antarctica lipase B was chosen as the model reaction.
Here, the feasibility of this biocatalytic reaction in a single‐phase system was shown for the first time. The work shows the economic superiority of the continuous process compared to the fed‐batch process. Employing the fed‐batch process reaction rates up to 36 mmol h−1 per gramcat, and a maximum yield of 96 % was achieved, but activity dropped quickly. In contrast, continuous operation can maintain long‐term enzyme activity. For the first time, the continuous enzymatic reaction could be performed for 55 h without any loss of activity and with a space‐time yield of 154 mmol L−1 h−1, which is three times higher than in the fed‐batch process. The higher catalytic productivity compared to the fed‐batch process (34 vs. 18 gProd g−1 cat) shows the increased enzyme stability in the continuous process.
Keywords: CalB, Deactivation, Hydrogen peroxide, Octanoic acid, Organic solvent
Abbreviations
- CalB
Candida antarctida lipase B
- CSTR
continuous stirred tank reactor
- FBS
fed‐batch system
- MTSO
methyl p‐tolyl sulfoxide
- TPPO
triphenylphosphine oxide
1. Introduction
Epoxidations are industrially important chemical syntheses. The Prileshajev reaction is used to produce epoxides at industrial scale, e.g. > 200 000 tons per year of epoxidized soybean oil 1. In the traditional epoxidation process, peroxy acids produced ex situ, or strong mineral acids are used as oxidizing agents. In contrast, in a lipase‐catalyzed system, the unstable percarboxylic acid can be generated in situ from carboxylic acids and hydrogen peroxide in an enzymatic reaction, followed by spontaneous epoxidation 2, 3, 4. The application of less hazardous reactants makes the chemo‐enzymatic reaction a safe and environmentally friendly alternative to the traditional synthesis.
Candida antarctica lipase B (CalB) catalyzes the peroxidation reaction very efficiently 2, 3. Also, the immobilized form of this lipase was found to be suited for the chemo‐enzymatic epoxidation of various substrates 4, 5. The chemo‐enzymatic epoxidation with CalB has been studied intensively in the last decades, but it is still limited by enzyme deactivation. To improve the enzyme stability, the reaction was usually carried out with an additional solvent (mostly toluene) 4, 6, 7, 8. With a focus on a minimized E factor 9 and greener processes, processes where only the organic substrates and water but no additional carrier solvent are present—so‐called solvent‐free processes—were also investigated 10, 11. The main drawback is a loss of enzyme activity at conditions optimized for high initial reaction rates and yields. Particularly a high reaction temperature and a high concentration of hydrogen peroxide harm the enzyme 6, 8, 12. Also, the substrates, e.g., hydrogen peroxide and octanoic acid (>6 M) can inhibit the enzyme 7, 10, 13. From chemical reaction engineering principles it is clear that continuous operation at low substrate concentrations can circumvent this issue.
Most studies were carried out using a two‐phase system with immobilized lipase. In contrast to a two‐phase system, a single phase avoids effects caused by interfaces, e.g., high local concentrations of hydrogen peroxide or mass transfer limitations at the liquid/liquid interface, which itself can be harmful to enzymes 14. Although an organic single‐phase epoxidation system would be advantageous, the number of publications on this topic is limited. Hilker et al. used stearic acid in toluene saturated with hydrogen peroxide 15.
Typically, the enzymatic epoxidation cascade was studied and evaluated with respect to the yield of epoxide. However, the first enzymatic step was less investigated isolated from the subsequent chemical step, especially in an organic single‐phase 15, so currently the individual contributions of the enzymatic and the chemical step cannot be distinguished. Furthermore, to the best of our knowledge, carboxylic acids have not been used as single‐phase solvent‐free systems.
As a first step towards continuous chemo‐enzymatic epoxidation, a fed‐batch/enzyme recycle reactor with gradual hydrogen peroxide feeding was used by Hilker et al. for the development of a mathematical model for the epoxidation of linseed oil in a fed‐batch 15. In this enzyme recycle process 15, 16, deactivation occurred, e.g., after 300 min at 60°C (20 % aqueous hydrogen peroxide). However, Wiles et al. 17 observed no enzymatic deactivation within 24 h of continuous operation at 70°C employing a glass capillary packed‐bed reactor, filled with 0.1 g Novozym® 435. For 1‐methylcyclohexene a yield of 99.1 % and a production rate of 6.7 mg/h was reported 17, which computes to a specific production rate of . However, an acceptable economic and ecologic combination of process operation (mode) and operating parameters, which enables a long‐term productive operation has not been described yet.
As the next step towards continuous enzymatic epoxidation, this work focuses on two main topics: the development of a single‐phase process to exclude effects caused by liquid/liquid interfaces and the development of a process where deactivation by the substrate hydrogen peroxide can be avoided or minimized. Therefore, different process set‐ups were investigated. Firstly, the enzymatic reaction was carried out in batch experiments (two‐phase, Supporting information), secondly, in a single‐phase fed‐batch process, and thirdly, in a CSTR with a single‐phase influent. The biocatalysis of peroxyoctanoic acid by CalB was chosen as a model reaction for the reasons mentioned above (Fig. 1). The concentrations of hydrogen peroxide and of the enzyme were varied in the batch and fed‐batch experiments.
Figure 1.
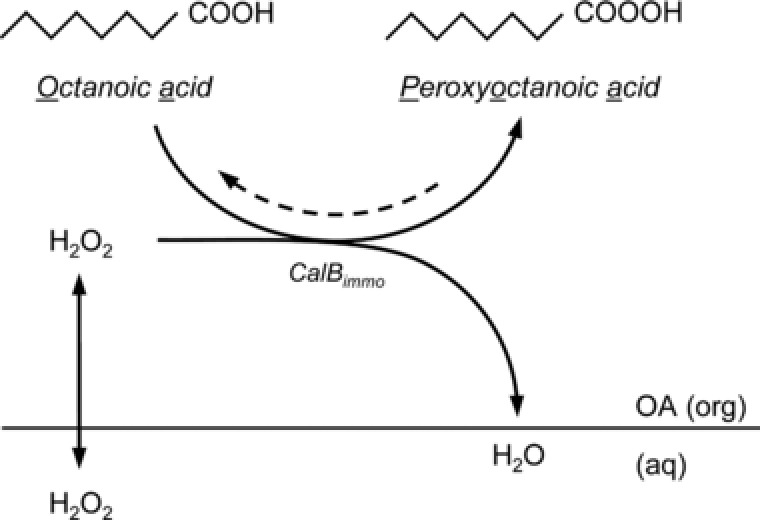
Lipase‐mediated peroxyoctanoic acid production as a first step of the chemo‐enzymatic epoxidation in the solvent‐free system.
2. Materials and methods
2.1. Enzymes and chemicals
CalB immo (immobilized lipase B from C. antarctica on methacrylate carrier) with a specific activity of 8100 LU/g was obtained from c‐LEcta GmbH (Germany).
Hydrogen peroxide (50 wt% in water) and all chemicals used for HPLC analyses and derivatization: methyl p‐tolyl sulfide (MTS 99 %), triphenylphosphine (TPP 99 %), methyl p‐tolyl sulfoxide (MTSO 97 %), triphenylphosphine oxide (TPPO 98 %), acetonitrile (ACN ≥ 99.9 % gradient grade) were purchased from Sigma‐Aldrich, Germany. Octanoic acid (≥ 99.5 % for synthesis) was obtained from Carl Roth, Germany.
2.2. Experimental setup
2.2.1. Fed‐batch operation
The single‐phase reaction was carried out in a stirred tank. The organic phase was saturated continuously with hydrogen peroxide in a liquid/liquid extraction column (reservoir tank). This saturation serves as continuous dosing of hydrogen peroxide into the stirred tank, where the reaction occurred. The fed‐batch system (FBS, Fig. 2A) consists of a glass reservoir tank (290 mL volume), where the organic phase is saturated with hydrogen peroxide, a stirred tank made of PTFE (polytetrafluoroethylene, total volume 50 mL) with a PTFE propeller stirrer (Bola, Germany) driven by an overhead digital stirrer motor (Eurostar 60 control, IKA, Germany), a pump (MCP‐Z with a Z040 micro pump head, Ismatec, Germany) and a 3‐way stopcock (Bola, Germany). To retain the enzymes, a 250 μm ETFE sieve (1) (ethylene tetrafluoroethylene, Franz Eckert GmbH, Germany) was used at the bottom of the stirred tank.
Figure 2.
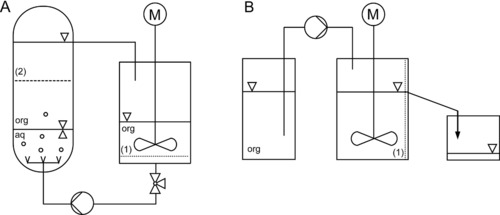
Flow sheet of the process set up used for A: fed‐batch, and B: continuous operation.
To ensure hydrogen peroxide saturation at the beginning of the experiment, the reservoir tank (aqueous hydrogen peroxide and octanoic acid) was mixed for at least 50 min before the start of the reaction. Afterward, the total system was filled with saturated octanoic acid from the reservoir tank, and the working volume (stirred tank, VR = 15 mL), the flow rate and the stirring rate (500 min−1) were set. To circumvent a concentration drop of hydrogen peroxide in the stirred tank, the system was operated at a high flow rate of 686 mL/min. A 200 μm steel sieve (2) (Spörl KG, Germany) was placed in front of the outlet of the reservoir tank to avoid water drops entrainment into the stirred tank from the aqueous phase.
The set‐up enables the saturation of octanoic acid with hydrogen peroxide and avoids the transfer of water from the reservoir into the reaction system. Therefore, it can be described as a single‐phase reaction system.
2.2.2. Continuous operation
For the design of the continuous process, the reaction rate Ṙ, determined in the fed‐batch study, was used for an estimation of the required residence time τ and enzyme mass and concentration, respectively. Figure 2B shows the flow sheet of the continuous set up (CSTR, VR = 34 mL, working volume). Through the overflow, a constant filling height was achieved, and effluent was discharged. For enzyme retention, a stainless steel sieve (200 μm, Spörl oHG, Germany) was used in front of the overflow/outlet. The experimental run was started by filling the system with the enzymes (2 g), the influent solution (see Section 2.2.2.1), setting the flow rate (1 mL/min) and the stirrer speed (500 min−1).
2.2.2.1. Preparation of the organic bi‐substrate influent solution
As an organic bi‐substrate influent solution, octanoic acid was pre‐saturated with hydrogen peroxide. The hydrogen peroxide stock solution (50 wt %) was diluted with de‐ionized water to the desired hydrogen peroxide concentration. To ensure the saturation with hydrogen peroxide, the pre‐prepared aqueous hydrogen peroxide solution and octanoic were mixed for at least 20 min (phase ratio of 1:2). For the comparison of the CSTR with the FBS, an influent concentration of 97 mM was applied. Therefore, a 12 M aqueous solution was required [Eq. (12)]. After phase separation, the aqueous phase was discharged, and only the organic phase was used as the influent solution. The influent solution was prepared and used at room temperature.
2.2.2.2. Continuous production of peroxyacetic acid
The integral mass balance for a reaction under continuous conditions [Eq. (1)], can be simplified to Eq. (2) when steady state is reached.
| (1) |
| (2) |
Using Eq. (2) for peroxyoctanoic acid, the reaction rate [Eq. (3)] and specific reaction rate [Eq. (4)] were determined.
| (3) |
| (4) |
| (5) |
The yield was calculated from Eq. (5) and the space‐time yield from Eq. (6).
| (6) |
The concentration of peroxyoctanoic acid cPOA, hydrogen peroxide in the effluent and in the influent was determined using the described HPLC method (2.3.1).
2.3. Analysis
2.3.1. Determination of the peroxyoctanoic acid and hydrogen peroxide concentration
To determine the concentration of peroxyoctanoic acid and hydrogen peroxide simultaneously, a derivatization reaction according to Pinkernell et al. was used 18. The stepwise quantitative reaction of the peroxyoctanoic acid with MTS and hydrogen peroxide with TPP produces the corresponding MTSO and TPPO. The substances were analyzed by a HPLC (module 1, Waters, USA) equipped with a LiChrosorb 100–5 RP 18, 250×4.6 mm column (Knauer GmbH, Germany) and the Millenium 2.10 chromatography software was used. The injection volume was 20 μL and the UV detection wavelength 230 nm. A constant flow of 1 mL min−1 and a gradient of acetonitrile and water (0–5 min 75 % ACN; 5–10 min ramped to 85 %, 10–20 min 75 %) were applied. The linear range (R 2>0.98) of the calibrations for MTSO and TPPO were 0.25–3.75 mM and 0.08–1.2 mM, respectively. Samples with concentrations out of the calibration range were diluted accordingly with the reaction medium octanoic acid. Typical retention times were 4.2 min for MTSO and 5.4 min for TPPO.
2.3.2. Determination of residual activities
For the determination of the enzyme activity in the fed‐batch experiments, a standard reaction procedure was defined, which employs the same substrates. At the end of the experiment, the immobilized enzymes were rinsed and filtered (250 μm ETFE sieve, Franz Eckert GmbH, Germany) twice with fresh octanoic acid to remove any residual peroxyoctanoic acid. Afterward, the washed enzymes were added to the standard reaction mix (0.57 mL of 17.5 M hydrogen peroxide and 44.43 mL of 6.3 M octanoic acid) and shaken for 1 h at 250 rpm at 30°C (incubator 3032, GLF mbH, Germany), and samples were taken. As a control reaction, 450 mg of fresh enzyme was added to the standard mix. It was confirmed that the increase of the peroxyoctanoic acid concentration was linear up to 1 h (R 2 = 0.99, data not shown), so sampling after one hour was valid. According to Eq. ((7) )the residual activity AR was determined. The reaction rate Ṙ (fed‐batch) was calculated from the measured peroxyoctanoic acid concentration cPOA in the sample (Eq. (8)).
| (7) |
| (8) |
3. Results and discussion
3.1. Distribution coefficient of hydrogen peroxide
Due to the single‐phase set up, the organic phase is the only source of hydrogen peroxide in the reaction system. Therefore, the partitioning of hydrogen peroxide between aqueous and organic phases was investigated (Fig. 3). The knowledge of hydrogen peroxide partitioning is essential to calculate the concentration of hydrogen peroxide in the organic phase in the time course of the experiment (fed‐batch) or in the influent solution (CSTR). To determine the partitioning coefficient K, a set of experiments was performed where the equilibrium hydrogen peroxide concentration in the organic phase was measured depending on the equilibrium hydrogen peroxide concentration in the aqueous phase (Fig. 3).
Figure 3.
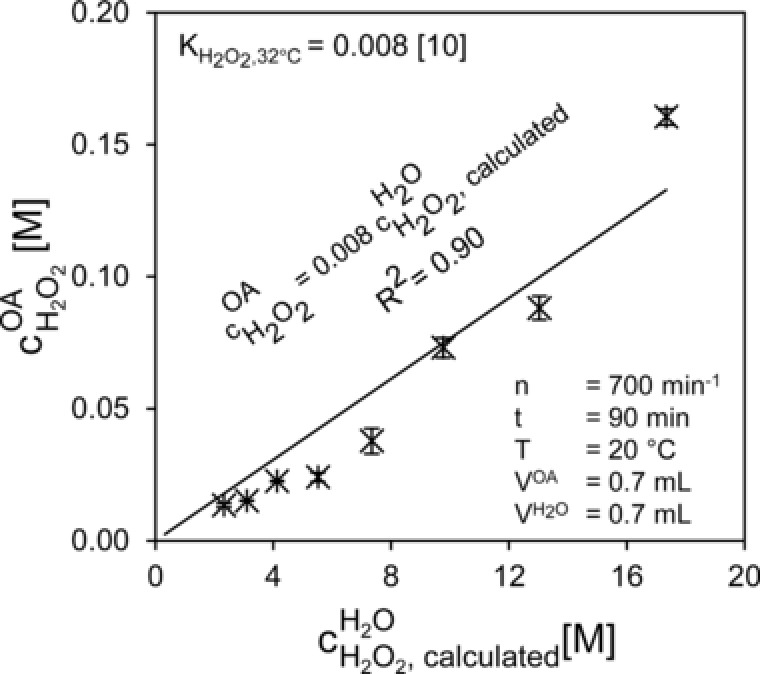
Partitioning coefficient of hydrogen peroxide between the aqueous and the organic phase, error bars represent the deviation from the mean value obtained from duplicates.
Since the volume of both phases was the same, the mass balance yields:
| (9) |
Thus, the equilibrium concentration of hydrogen peroxide in the aqueous phase can be calculated from the measured hydrogen peroxide concentration in octanoic acid [Eq. (9)]. A direct HPLC determination of hydrogen peroxide in the aqueous phase was not carried out because it requires highly diluted samples (>1:10.000) which would increase the error. All experiments were carried out at the conditions used in the actual reaction trials (T = 20°C). Despite the fact that the data suggest a non‐linear equilibrium law, a linear correlation was used in agreement with Bhattacharya 10 in order to be able to compare the obtained value with the one reported there. The distribution coefficient K was determined to be 0.008 [Eq. (10)] which is the same as the one reported by Bhattacharya 10 at 32°C.
| (10) |
Control measurements were carried out during the experiments to confirm the distribution coefficient (data not shown). Figure 3 shows the very low solubility of hydrogen peroxide in the octanoic acid phase. The low and tailored solubility of the hydrogen peroxide in octanoic acid indicates that it can be used as a model substrate because it enables a low concentration of hydrogen peroxide in the organic phase.
3.2. Fed‐batch operation
The concentration of hydrogen peroxide in octanoic acid, which is the relevant substrate concentration for the enzymatic step (octanoic acid excess), was determined from the overall hydrogen peroxide mass balance [Eq. (11)]. The total initially present amount of hydrogen peroxide consists of the hydrogen peroxide dissolved in the octanoic acid and in the aqueous phase, and of the hydrogen peroxide which is converted to peroxyoctanoic acid.
| (11) |
Substituting and leads to Eq. (12).
| (12) |
Inserting Eq. (12) into Eq. (10) and rearranging leads to Eq. (13),
| (13) |
which was used to calculate the concentration of hydrogen peroxide in the octanoic acid during the course of the reaction (FBS). Additionally, the hydrogen peroxide concentration in the octanoic acid was checked occasionally using HPLC (2.3.1).
3.2.1. Effect of enzyme concentration
In general, in a kinetically controlled enzymatic reaction, the correlation between enzyme mass and reaction rate Ṙ is linear. Figure 4a shows the amount of peroxyoctanoic acid and the hydrogen peroxide concentration during the course of the reaction using different amounts of enzyme. The hydrogen peroxide concentration was 0.6 M in the aqueous phase, and octanoic acid was used in excess.
Figure 4.
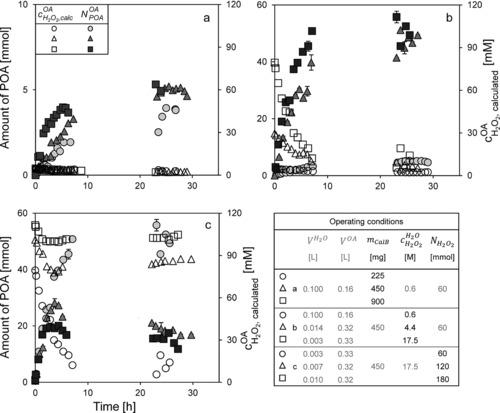
Amount of peroxyoctanoic acid and hydrogen peroxide concentration in octanoic acid (Eq. (10)) over time, FBS: (a) different amounts of enzyme (hydrogen peroxide concentration in the aqueous phase 0.6 M). The data from the experiment with 450 mg of the enzyme is repeated in b (0.6 M). (b) different hydrogen peroxide concentrations (amount of enzyme 450 mg). The error bars represent the deviation from the mean value of two HPLC injections. The data from the experiment with 17.5 M of hydrogen peroxide is repeated in c (60 mmol). (c) different amounts of hydrogen peroxide (hydrogen peroxide concentration in the aqueous phase 17.5 M).
With this experimental set‐up, it is possible to achieve a relatively constant concentration of hydrogen peroxide in octanoic acid, which was calculated by Eq. (13). In this period, less than 9 % of the substrate was converted, and a possible product inhibition is unlikely. Table 1 shows the specific reaction rate, maximum product amount and yield depending on the enzyme mass.
Table 1.
Reaction rate Ṙ, specific reaction rate ṙ, maximum product amount N and yield Y, FBS (Fig. 4). The residual activity AR was determined at the end of the experiment (Eq. (7), Section 2.3.2)
| Operating conditions | Results | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
Ṙ Initial | ṙ Initial |
|
Y | AR | ||||
| [mg] | [M] | [mmol] | [mM h−1] | [mmol h−1g−1] | [mmol] | [%] | [%] | ||||
| 225 | 2 | 1 | 5 | 7 | n.d. | ||||||
| 450 | 0.6 | 60 | 4 | 1 | 5 | 9 | n.d. | ||||
| 900 | 7 | 1 | 5 | 9 | n.d. | ||||||
| 0.6 | 4 | 1 | 5 | 9 | n.d. | ||||||
| 450 | 4.4 | 60 | 27 | 20 | 51 | 85 | 16 | ||||
| 17.5 | 50 | 36 | 58 | 96 | 7 | ||||||
| 60 | 50 | 36 | 58 | 96 | 7 | ||||||
| 450 | 17.5 | 120 | 33 | 24 | 30 | 25 | n.d. | ||||
| 180 | 37 | 27 | 21 | 11 | 3 | ||||||
Figure 4a and Table 1 show the expected correlation in the early phase of the reaction. With an increasing enzyme mass, a higher initial reaction rate was obtained. Using a low concentration of hydrogen peroxide in the aqueous phase (0.6 M), which resulted in a lower concentration in the octanoic acid phase, a low yield (7–9 %) was obtained, even though both substrates were present in sufficient amounts. In the system with 900 mg CalB, the highest yield was achieved. In this system, the product formation rate decreased after approx. 2.5 h. Nevertheless, in all trials, a maximum yield of about 9 % was detected.
To compare the single‐phase FBS with the two‐phase batch experiments (BE), the same substrate/enzyme ratio (0.13 mol/g) was applied. The productivity (yield h−1) in the initial phase of the reaction was calculated (see Supporting information Fig. SI.1a). For the FBS it is approx. 0.8 % h−1 and for the BE approx. 3.3 % h−1. The productivity using the FBS is four times lower than the productivity determined in the BE. Nevertheless, this is reached with a 130 times lower concentration of hydrogen peroxide in the single‐phase FBS (∼4.6 mM) than in the BE (0.6 M). Based on this, the single‐phase FBS has a better process potential than the BE described in the Supporting Information.
3.2.2. Effect of hydrogen peroxide concentration
As a next step, the influence of the hydrogen peroxide concentration on the peroxyoctanoic acid synthesis was investigated. The starting concentration of hydrogen peroxide in the aqueous phase in the reservoir tank was varied between 0.6 and 17.5 M.
Calculated from Eq. (13), the starting concentration of hydrogen peroxide in octanoic acid in the reactor varied in the range of 0.0046 to 0.08 M. Even bearing in mind that the linear approximation for K can lead to slight deviations in certain concentration ranges (see Fig. 3), the trends shown in Fig. 4 would remain the same and the differences between runs would still be significant. The other substrate—octanoic acid—was used in excess. An increase of the hydrogen peroxide concentration from 0.6 M to 17.5 M led to a strong increase in the initial reaction rates. As expected, also the amount of peroxyoctanoic acid increased with increasing reaction rates and a maximum yield of 96 % was achieved. Rüsch gen. Klaas and Warwel reported a similar yield of 98 % peroctanoic acid (two‐phase system, toluene as an additional solvent, determined by titration) 7.
An enzyme deactivation depending on the hydrogen peroxide concentration was observed (Table 2). Using a concentration of 17.5 M hydrogen peroxide, the enzyme was almost entirely deactivated after 27 h (residual activity 7.3 %), whereas a lower concentration (4.4 M) led to less deactivation (residual activity 16.1 %) and a similar yield, product amount, respectively. From the low residual activities but high yields (also see Table 2) we assume that deactivation rather than peroxyoctanoic acid instability) is the main reason for the plateaus in Fig. 4, which means that the impact of enzyme deactivation on the peracid concentration is significantly higher than the influence of instability.
Table 2.
Comparison between the FBS and the CSTR: 1. FBS with H2O2 concentration not constant, 2. nearly constant, 3. CSTR H2O2 concentration constant. Hydrogen peroxide influent concentration, specific reaction rate ṙ, time when deactivation was significant td, amount of product formed , catalytic productivity Pcat, yield Y. To compare the process set‐ups, the space‐time yield STY of the FBS was calculated in the initial part of the reaction
| Operation | Results | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No |
|
ṙ | td | NPOA | Pcat | Y | STY | |||
| [mM] | [mmol h−1g−1] | [h] | [mmol] | [mmol g−1] | [%] | [mmol L−1 h−1] | ||||
| start | end | |||||||||
| 1 | FBS | 80 | 5.6 | 36 | 6 | 58 | 128 | 96 | 49 | |
| 2 | 110 | 105 | 27 | 2 | 21 | 46 | 11 | 36 | ||
| 3 | CSTR | 97 | 3 | 55 | 425 | 213 | 90 | 154 | ||
3.2.3. Effect of hydrogen peroxide amount
Gradual dosing was reported to avoid or to reduce enzyme deactivation by hydrogen peroxide 13. To prevent a concentration drop of hydrogen peroxide in the octanoic acid phase during the experiments as seen in Fig. 4b and to simulate continuous dosing, the volume of the hydrogen peroxide solution in the reservoir tank was increased. However, the same concentration of hydrogen peroxide was used in all experiments, and the related amount of hydrogen peroxide within the system rose from 60 to 180 mmol.
As expected, Fig. 4c shows that higher amounts of hydrogen peroxide led to a more stable concentration over time (continuous dosing) in the organic phase over the whole experiment and to a higher starting value [Eq. (13)]. It can be seen that the higher the concentration in the octanoic acid phase, the earlier the deactivation occurs (1.5, 3, 6 h) and the lower the remaining enzymatic activity (Table 1). Providing a continuous dosing with a constant concentration of hydrogen peroxide and only ∼110 mM of hydrogen peroxide in the octanoic acid phase, which is much lower than in the aqueous phase, the deactivation occurred quickly (1.5 h, residual activity 3.2 %). In contrast, if the hydrogen peroxide in the octanoic acid phase was not constant, the deactivation occurred later and was not as strong (residual activity 7.3 %).
Using the FBS a maximum (initial) space‐time yield STY of 49 mmol L−1 h−1 was achieved (Table 2).
In comparison to the STY of 3 mol m−3 h−1 (0.5 kg m−3 h−1) reported by Bhattacharya 10 (epoxydodecene, peroxyoctanoic acid as intermediate, Lewis cell, two‐phases, 15 g of 50 % w/w silcoat‐NZ435) the maximum STY of the single‐phase FBS for the peroxyoctanoic acid production in this study is around 16 fold higher (49 mol m−3 h−1). Nevertheless, the enzyme was deactivated within the earlier mentioned 1.5 h.
3.3. Continuous operation
Since the enzyme was deactivated very quickly in the FBS, a continuous process was designed and investigated as another process alternative. From the reaction rate Ṙ obtained in the FBS, the required residence time τ and enzyme mass for the CSTR (Eq. (2)) were calculated. This design also based on the linear correlation between enzyme concentration and reaction rate (Fig. 4a, Table 1) shown in the FBS experiments. To estimate the long‐term enzyme activity or rather reusability, recycle experiments are commonly applied 8, 13, 19. These, however, include, e.g., the influence of storage and solvent drying on the enzyme support, which leads to overlapping or masking of the deactivation caused by hydrogen peroxide. Instead, actual long‐term activity in the reaction environment should be studied. Therefore, long‐term enzyme activity was investigated during continuous operation in a CSTR (τ = 34 min, T = RT). Simultaneously, the feasibility of continuous peroxyoctanoic acid synthesis was proven.
Figure 5 shows the concentration of hydrogen peroxide in the influent and the concentration of hydrogen peroxide and peroxyoctanoic acid in the effluent.
Figure 5.
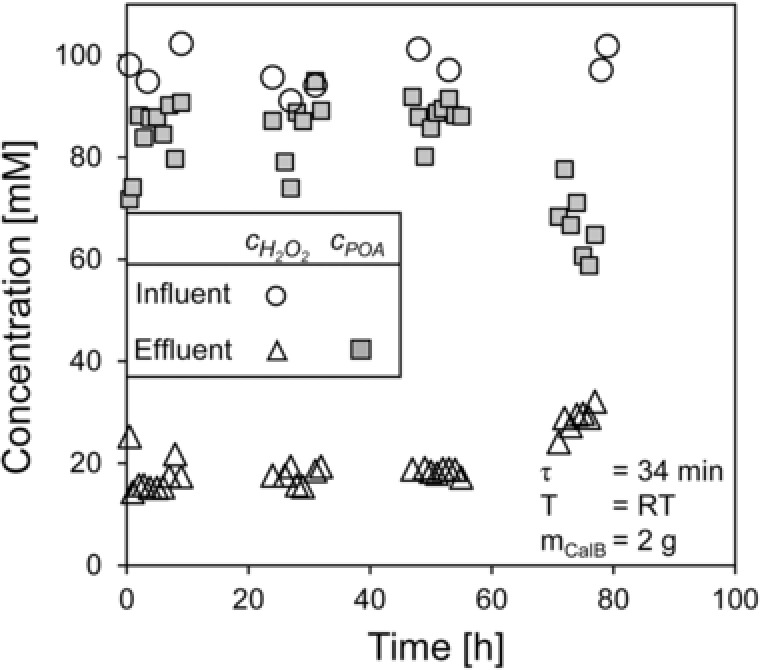
Long‐term performance of the continuous peroxyoctanoic synthesis catalyzed by CalB.
The production of peroxyoctanoic acid was stable for nearly 55 h in the CSTR, which is 37 times longer than in the FBS applying a similar hydrogen peroxide influent concentration (CSTR: 97 mM vs. FBS: 110 mM, Table 2). Generally, the enzyme stability is affected by hydrogen peroxide. With a yield of 90 % (2–55 h) the remaining concentration of hydrogen peroxide at steady state in both the effluent and stirred tank was 17 mM (Fig. 5), which is 6 times lower than in the FBS. The lower concentration leads to less deactivation in the CSTR than in the FBS. Hilker et al. also observed a deactivation free period and assumed this was the time in which the carrier material is damaged by which sensitive regions of the enzymes become exposed and therefore, deactivation becomes significant 15, 16.
From the drop in the concentration of peroxyoctanoic acid and the simultaneous increase in the concentration of hydrogen peroxide after 55 h, it can be concluded that the enzyme activity decreased.
Despite the low specific reaction rate ṙ of 3 mmol h−1g−1 a much higher STY of 154 mmol L−1 h−1 could be achieved using the CSTR in comparison to the FBS (Table 2).
Figure 5 also shows that hydrogen peroxide is stable at an average of 97.4 mM over several days in the influent tank. A comparison with the value calculated in analogy to Eq. (13) (94.5 mM) confirms that the error caused by the linear approximation of K is within an acceptable range.
The higher STY, the higher catalytic productivity and the ability to maintain enzyme activity over a longer period, demonstrate the potential of the single‐phase reaction and make the CSTR the system of choice. This work forms a sound base for an advanced coupling of the enzymatic step with the epoxidation, based on the knowledge of the individual influences of every sub‐step.
4. Concluding remarks
The feasibility of the biocatalytic production of peroxyoctanoic acid from octanoic acid and hydrogen peroxide (model reaction) was shown for the first time in a single‐phase systems, i.e. without the commonly applied addition of a carrier solvent.
With the fed‐batch system, a reaction rate of 36 mmol h−1 per gram enzymeand a maximum yield of 96 % could be achieved with an initial hydrogen peroxide concentration of 80 mM in the organic phase. Rüsch gen. Klaas and Warwel reported a similar yield of 98 % in a batch reaction 5. The highest space‐time yield applying the fed‐batch system was 49 mmol L−1 h−1 which is much higher than the space‐time yield reported by Bhattacharya using octanoic acid as substrate in a modified two‐phase (org/aq) Lewis cell 10.
In accordance with the literature 6, 8, 12 it was found that hydrogen peroxide is the driving force for reaction rates and yields during the initial phase of the reaction, but hydrogen peroxide is also the driving force for enzyme deactivation. Hydrogen peroxide was transferred into the organic phase via extraction, mimicking a gradual feeding of this component which was meant to overcome this enzyme deactivation. However, it can be stated so far that the usage of relatively low hydrogen peroxide concentrations in the octanoic acid phase (4.6–110 mM, initial) or gradual feeding (fed‐batch) is not sufficient to avoid enzyme deactivation completely.
As a process alternative to the fed‐batch, a continuous process was designed, which led to a 37 times longer stable period (1.5 vs. 55 h) using a hydrogen peroxide influent concentration in a similar range (110–105 vs. 97 mM). At a residence time of 34 minutes, a yield of 90 %, a space‐time yield of 154 mmol L−1 h−1 and a catalytic productivity of 213 mmol g−1 were observed. The economic superiority of the CSTR compared to the fed‐batch was clearly shown by the higher space‐time yield and the higher catalytic productivity.
These outcomes form a sound base for an economic and ecological coupling of the enzymatic step with the epoxidation, based on the knowledge of the individual influences of each sub‐step. A further improvement could be made by a modification of the enzyme regarding higher hydrogen peroxide stability.
Practical application
There is a keen interest of the life science industry in realizing the benefits of continuous processing like consistent product quality and high space‐time yield. The design of an optimal process is particularly crucial when the reactants or substrates deactivate the biocatalyst. The reaction cascades of the chemo‐enzymatic epoxidation, where the intermediate peroxy acid is produced by an enzyme, are still limited by enzyme inhibition and deactivation. This work deals with the identification of a suitable process to maintain enzyme activity, which is the base for an economic and ecological coupling of the multistep reaction. For the chosen model reaction, the lipase catalyzed peroxyoctanoic acid production, it was shown that (1) a single‐phase process is feasible and (2) continuous operation could be carried out for 55 h without any loss of enzyme activity and with a high catalytic productivity. The advanced process can be used for the enzymatic epoxidation of different alkenes or terpenes. From these epoxides, pharmaceutical drugs and agrochemicals can be synthesized.
The authors have declared no conflicts of interest.
Nomenclature
| A | [‐] | activity |
| c | [mM] | concentration |
| K | [‐] | distribution coefficient |
| m | [mg] | mass |
| N | [mmol] | amount |
| n | [min−1] | stirrer speed |
| P | [% h−1, mmol g−1] | productivity |
| Ṙ | [mM h−1] | reaction rate |
| ṙ | [mmol h−1 g−1] | specific reaction rate |
| STY | [mmol L−1 h−1] | space‐time yield |
| t | [h] | time |
| τ | [min] | contact time |
| V | [mL] | volume |
| Y | [%] | yield |
Subscripts and superscripts
| cat | catalytic |
| d | deactivation |
| OA | octanoic acid |
| POA | peroxyoctanoic acid |
| R | residual |
| total | hydrogen peroxide, initially present |
Supporting information
Supporting Information
Acknowledgments
The IGF‐project (17711 BG) of the research association (DECHEMA Forschungsinstitut, TU Dresden, HTW Berlin) was funded by the Federal Ministry of Economic Affairs and Energy (BMWi) as part of the program to promote joint industrial research (IGF).
5 References
- 1. Aouf, C. , Durand, E. , Lecomte, J. , Figueroa‐Espinoza, M.‐C. et al., The use of lipases as biocatalysts for the epoxidation of fatty acids and phenolic compounds. Green Chem. 2014, 16, 1740–1754. [Google Scholar]
- 2. Björkling, F. , Frykman, H. , Godtfredsen, S. E. , Kirk, O. , Lipase catalyzed synthesis of peroxycarboxylic acids and lipase mediated oxidations. Tetrahedron 1992, 48, 4587–4592. [Google Scholar]
- 3. Björkling, F. , Godtfredsen, S. E , Kirk, O. , Lipase‐mediated formation of peroxycarboxylic acids used in catalytic epoxidation of alkenes. J. Chem. Soc., Chem. Commun. 1990, 19, 1301–1303. [Google Scholar]
- 4. Warwel, S. , Rüsch gen. Klaas, M. , Chemo‐enzymatic epoxidation of unsaturated carboxylic acids. J. Mol. Catal. B: Enzym. 1995, 1, 29–35. [Google Scholar]
- 5. Rüsch gen. Klaas, M. , Warwel, S. , Lipase‐catalyzed preparation of peroxy acids and their use for epoxidation. J. Mol. Catal. A: Chem. 1997, 117, 311–319. [Google Scholar]
- 6. Orellana‐Coca, C. , Camocho, S. , Adlercreutz, D. , Mattiasson, B. et al., Chemo‐enzymatic epoxidation of linoleic acid: Parameters influencing the reaction. Eur. J. Lipid Sci. Technol. 2005, 107, 864–870. [Google Scholar]
- 7. Skouridou, V. , Stamatis, H. , Kolisis, F. N. , Lipase‐mediated epoxidation of α‐pinene. J. Mol. Catal. B Enzym. 2003, 21, 67–69. [Google Scholar]
- 8. Yadav, G. D. , Devi K., Enzymatic synthesis of perlauric acid using Novozym 435. Biochem. Eng. J. 2002, 10, 93–101. [Google Scholar]
- 9. Ni, Y. , Holtmann, D , Hollmann, F. , How green is biocatalysis? To calculate is to know. ChemCatChem 2014, 6, 930–943. [Google Scholar]
- 10. Bhattacharya, S. , Solvent‐free chemo‐enzymatic epoxidation: Experimental and kinetic modelling studies. Ph.D. thesis, Technische Universität Berlin 2012.
- 11. Orellana‐Coca, C. , Törnvall, U. , Adlercreutz, D. , Mattiasson, B. et al., Chemo‐enzymatic epoxidation of oleic acid and methyl oleate in solvent‐free medium. Biocatal. Biotransfor. 2005, 23, 431–437. [Google Scholar]
- 12. Törnvall, U. , Orellana‐Coca, C. , Hatti‐Kaul, R. , Adlercreutz, D. , Stability of immobilized Candida antarctica lipase B during chemo‐enzymatic epoxidation of fatty acids. Enzyme Microb. Tech. 2007, 40, 447–451. [Google Scholar]
- 13. Skouridou, V. , Stamatis, H. , Kolisis, F. N. , A study on the process of lipase‐catalyzed synthesis of α‐pinene oxide in organic solvents. Biocatal. Biotransfor. 2003, 21, 285–290. [Google Scholar]
- 14. Baldascini, H. , Janssen, D. B. , Interfacial inactivation of epoxide hydrolase in a two‐liquid‐phase system. Enzyme Microb. Tech. 2005, 36, 285–293. [Google Scholar]
- 15. Hilker, I. , Bothe, D. , Prüss J., Warnecke, H.‐J. , Chemo‐enzymatic epoxidation of unsaturated plant oils. Chem. Eng. Sci. 2001, 56, 427–432. [Google Scholar]
- 16. Hilker, I. , Modelling the chemo‐enzymatic epoxidation of linseed oil. Ph.D. thesis, Universität Paderborn 2002.
- 17. Wiles, C. , Hammond, M. J. , Watts, P. , The development and evaluation of a continuous flow process for the lipase‐mediated oxidation of alkenes. Beilstein J. Org. Chem. 2009, 5, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pinkernell, U. , Effkemann, S. , Karst, U. , Simultaneous HPLC determination of peroxyacetic acid and hydrogen peroxide. Anal. Chem. 1997, 69, 3623–3627. [DOI] [PubMed] [Google Scholar]
- 19. Bhattacharya, S. , Drews, A. , Lyagin, E. , Kraume, M. et al., Chemo‐enzymatic epoxidation using silcoat‐Novozyme®435: Characterizing the multiphase system. Chem. Eng. Technol. 2012, 35, 1448–1455. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information