

Abstract
Directly using the promoter associated with 5′‐untranslated region of a high‐protein‐abundance gene from the genome may cause low expression activity of an expression system. A bicistronic expression part containing the short 5′ coding sequence of the source gene and an embedded Shine–Dalgarno sequence can cause higher expression levels of the recombinant gene in a bicistronic cassette. Here, we evaluated two methods to construct expression parts and exploited genomic sequence sources to provide specific functional sequences to complete the expression system. The architecture of the bicistronic part increased the expression levels of target genes and performed more reliably than conventional expression parts with the same promoter and 5′ untranslated region. For Corynebacterium glutamicum, the strongest bicistronic part, HP‐BEP4, was obtained from a heterologous sequence source, leading to a 2.24‐fold increase in the expression level of fluorescent protein over constitutively expressed pXMJ19 or the production of more than 100 mg/L single‐chain variable fragment (scFv). It could meet the needs of overexpressing key genes in C. glutamicum.
Keywords: Bicistronic design, Corynebacterium glutamicum, Genetic part, scFv, Sequence source
Abbreviations
- 5′‐UTR
5′ untranslated region
- BEP
bicistronic expression part
- MEP
monocistronic expression part
- SD sequence
Shine–Dalgarno sequence
- scFv
single‐chain variable fragment
1. Introduction
The polycistronic expression cassette is a special structure in bacteria that has been studied for many years (Fig. 1). This type of genetic structure provides the coordinated expression of proteins and the sequence economy in that several genes can be closely arranged and translated 1. The translation coupling mechanism was first found in the Escherichia coli galactose operon. Researchers have focused on the artificial development of polycistronic cassettes, especially bicistronic expression cassettes, because of their versatile applications in expressing target genes 2. The introduction of the fore‐cistron into the ribosome prevents the formation of a stable mRNA structure, facilitating the translation initiation of the second cistron 3. A hard‐to‐translate fore‐cistron with embedded Shine–Dalgarno (SD)‐like sequences can reduce the accumulation level of the second cistron, which promotes protein folding and increases production of the protein in its soluble form 4. Bicistronic architecture can also include various functional peptides such as a first‐cistronic peptide, which stabilizes the second‐cistronic protein 5. With its unique characteristics, bicistronic architecture has been investigated in terms of controlling gene expression and developing systems to synthesize biological products 6.
Figure 1.
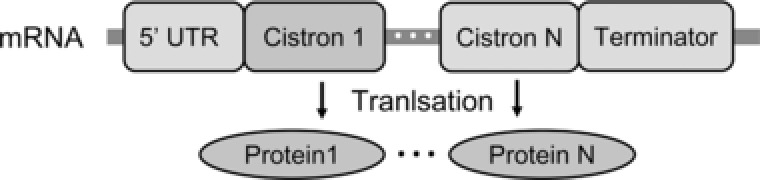
Bacterial polycistronic RNA with protein products.
The advance of omics technologies has led to an unparalleled amount of information about the genomic, genotypic, and phenotypic links within organisms, which has allowed researchers to identify particular genes and their upstream functional sequences 7. Promoters with strong activities have been identified from transcriptomic data and used to initiate the transcription of recombinant genes 8. Integrated expression constructs consisting of a promoter and the 5′ untranslated region (5′‐UTR) have been cloned and used in expression vectors 9. The direct application of these functional sequences from genomes has proven to be effective, and has avoided slow trials with randomized large libraries. However, the use of genetic parts obtained from highly expressed genes does not guarantee strong activity in a recombinant expression cassette 10. To further exploit the potential of such expression parts, we developed a bicistronic expression construct that includes not only the gene promoter and the 5′‐UTR, but also a part of the original downstream 5′ coding sequence to complete the original translation initiation region (TIR) 11. This extended functional sequence improved expression activity when the sequences were derived from several native genes with the highest expression levels in Corynebacteriunm glutamicum. For further application of this method, more sources which can provide specific sequences should be found.
C. glutamicum is an important industrial organism that has been used to produce amino acids and many other commodities 12, 13. There is a large body of information on its optimum culture conditions, as well as its metabolism, transcriptome, and proteome, which lays a stable foundation for strain engineering 14. Because C. glutamicum is non‐pathogenic strain and releases fewer endogenous proteins into the culture medium than does E. coli, C. glutamicum is considered to be a safe host with a simple downstream purification process for the production of recombinant proteins 15, 16. We have proved that placing specific functional expression sequences (promoter–5′‐UTR–short peptide–stop codon) upstream of the target gene in bicistronic expression cassette could improve the expression level of target genes, compared with the strategy of just using promoter‐5’UTR. In this study, we further exploit the bicistronic expression strategy by using functional sequences from another two different sources to construct expression parts and control the expression of our target genes. A single‐chain variable fragment (scFv) against human serum albumin (HAS), which is used to stabilize various drugs, was used to characterize the reliability of strong expression vectors 17.
2. Materials and methods
2.1. Strain and culture conditions
Table 1 lists the bacterial strains and plasmids used in this study, except for those containing expression parts. E. coli DH5α, C. glutamicum CGMCC1.15647, and Bacillus subtilis 168 were stored in our laboratory. E. coli DH5α was used to construct plasmids. C. glutamicum was used as the host for the expression of recombinant proteins and to test the bicistronic expression strategy. The genomes of C. glutamicum and B. subtilis 168 were used as templates for cloning specific functional sequences that could initiate the transcription and translation of reporter genes. E. coli strains DH5α and B. subtilis 168 were grown in Luria–Bertani (LB) broth (tryptone 10 g/L, yeast extract 5 g/L and NaCl 10 g/L, pH 7.0) or on LB plates containing 2% (wt/vol) agar at 37°C. The C. glutamicum strain CGMCC1.15647 was grown in brain heart infusion (BHI) medium (37 g/L, pH 7.0) at 30°C. Chloramphenicol was added for selection of E. coli and C. glutamicum at concentrations of 35 and 10 mg/L, respectively.
Table 1.
Bacteria and plasmids used in this study
| Strain or plasmid | Characteristics | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | F−φ80lacZ△M15△(lacZYA‐argF)U169recA1hsdR17(rk−,mk+)endA1thi‐1relA1phoAgyrA96supE44λ− | Invitrogen |
| B. subtilis 168 | Wild type | Laboratory |
| C. glutamicum | Wild type | CGMCC1.15647 |
| Plasmid | ||
| pXMJ19 | Shuttle vector(chloramphenicol resistance; lacI q Ptac pK18 oriVE. coli pBL1 oriVC. glutamicum) | 18 |
| pXMJ19‐EGFP | pXMJ19 derivative without repressor lacI q, carrying SD sequence and egfp gene | 10 |
| pXMJ19‐OR‐SCFV | pXMJ19 derivative without repressor lacI q, carrying SD sequence and origonal scFv gene (OR‐scFv). | This study |
| pXMJ19‐SCFV | pXMJ19 derivative without repressor lacI q, carrying SD sequence and codon‐optimized scFv gene | This study |
| pE‐0 | p19‐0 derivate carrying Bsa I cloning site and egfp gene | 10 |
| pOR‐SCFV‐0 | p19‐0 derivate carrying Bsa I cloning site and OR‐scFv gene | This study |
| pSCFV‐0 | p19‐0 derivate carrying Bsa I cloning site and codon‐optimized scFv gene | This study |
2.2. Construction of probe vectors for genetic expression parts
All primers used in this study are listed in Supporting Information Table 1. The probe vector pE‐0 containing enhanced green fluorescent protein (EGFP) as the reporter gene was constructed in our previous work 11. To test the reliability of expression vectors, a single‐chain variable fragment (scFv) against Homo sapiens albumin (HSA) was used as another reporter gene. Its sequence (Supplementary Sequence 1) with a downstream 6‐His tag was synthesized by Genewiz (Suzhou, China), which also performed the codon optimization for scFv gene according to the codon usage bias of C. glutamicum. The reporter gene egfp was replaced by the codon‐optimized 6‐His tagged scFv and the original 6‐His tagged scFv (OR‐scFv) using the construction method described previously to create the probe vectors pSCFV‐0 and pOR‐SCFV‐0, respectively (Fig. 2).
Figure 2.
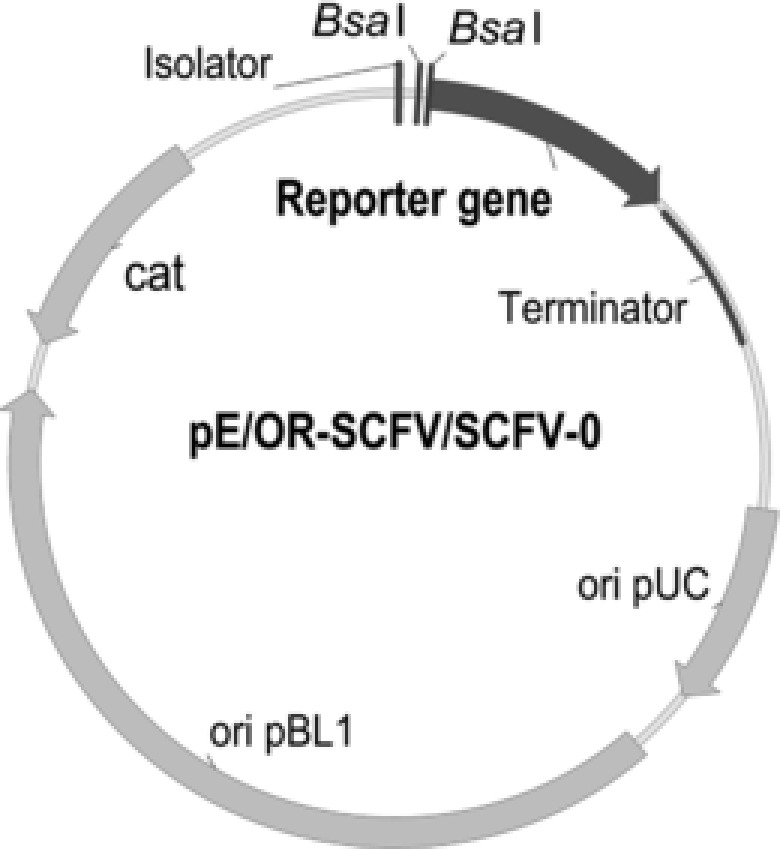
Physical map of probe vector with reporter gene egfp, OR‐scFv, or scFv.
2.3. Construction and insertion of expression parts
Two kinds of gene sources were screened to find specific sequences for genetic expression parts: C. glutamicum highly transcribed genes and B. subtilis highly expressed genes (Fig. 3). Highly transcribed genes in C. glutamicum were ranked based on transcriptomic data (GEO accession number: GSE77502), which integrated data from two cultivation conditions with different dissolved oxygen levels (Supporting Information Table 2) 18. As a well‐characterized gram‐positive bacterium, B. subtilis was used to provide heterologous functional sequences, and highly expressed genes in B. subtilis were ranked based on protein abundance according to the data in the PaxDb database (http://pax-db.org/), which is a comprehensive absolute protein abundance database 19. Based on the rank of proteins or mRNAs, the top highly transcribed or expressed genes were selected to provide sequences to construct genetic expression parts. The BDGP Neural Network Promoter Prediction tool at http://www.fruitfly.org/seq_tools/promoter.html was used to identify promoters for genomic genes according to the prediction score (Supporting Information Table 2) 20. Each promoter with its downstream functional sequence was amplified and arranged according to the architecture of a conventional monocistronic expression part (MEP); i.e. promoter–5′‐UTR, and a bicistronic expression part (BEP); i.e. promoter–5′‐UTR–short peptide–stop codon, as defined in our previous work to be two kinds of expression parts (Fig. 3) 19. In the BEP, the short peptide following the promoter–5′‐UTR was encoded by the first 38 bp of the coding sequence of the source gene, with an additional conserved SD sequence (SD2) between the 38‐bp ORF and the stop codon as a necessary element in the bicistronic expression cassette (The 5’UTR together with the 38‐bp N‐terminal coding sequence was thought sufficient to reflecting the effects of translation initiation process) 21, 22. The MEP consisted only of the promoter and the 5′‐UTR of the source gene 23.
Figure 3.
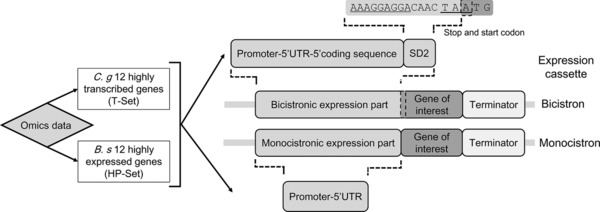
Sources of sequences and procedure used to construct parts of expression systems. Sequences of C. glutamicum highly transcribed genes (T‐Set) and B. subtilis highly expressed genes (HP‐Set) were identified from omics analysis. Then, sequences were arranged with various genetic elements in monocistronic or bicistronic expression cassettes. Bicistronic expression part contains promoter, 5′‐UTR, 38‐bp of the 5′‐coding sequence, and also a SD2 sequence overlapping 1 bp with the downstream start codon (ATG) of the gene of interest. Monocistronic expression part contains only source promoter and 5′‐UTR.
The expression parts were amplified from C. glutamicum CGMCC1.15647 and B. subtilis 168 genomic DNA, which were isolated using a genomic isolation kit (Tiangen, China). The Golden Gate method was used to seamlessly join genetic expression parts to target genes 24. The PCR products were digested by the type II restriction enzyme Bsa I and ligated into each of the probe vectors pE‐0, pOR‐SCFV‐0, and pSCFV‐0.
The two positive controls, pXMJ19‐EGFP and pXMJ19‐SCFV, expressed EGFP and scFv, respectively. Both were derived from the classic C. glutamicum expression vector pXMJ19 but lacked the lacI repressor, so that their tac promoter associated with the C. glutamicum conserved SD sequence (AAAGGAGGA) could function constitutively 25. All expression plasmids including the positive controls pXMJ19‐EGFP and pXMJ19‐SCFV and the negative controls pE‐0 and pSCFV‐0 were constructed in E. coli and transformed into C. glutamicum CGMCC1.15647.
In order to compare the total effect of BEP design with MEP design for expression initiation, the ratio of BEP to MEP was introduced and calculated as follows:
2.4. Fluorescence intensity assay
Strains were grown overnight in 24‐well deep‐well plates with 2 mL BHI medium per well, diluted 1:100 into 2 mL fresh medium, and then grown for additional 24 h. Fluorescence intensity normalized by OD600 was measured as described previously 26. The fluorescence of cells harboring EGFP plasmids was also observed using Dark Reader (Dalewe, China) after culture on solid medium and analyzed by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE).
2.5. Purification of scFv–His tag
Strains were grown overnight in BHI medium at 30°C, then diluted 1:10 into 10 mL fresh medium and grown at specific temperatures for another 24 h. The cells were harvested by centrifugation at 8000 × g for 10 min at 4°C, and then disrupted by sonication on ice. Because scFv was expressed in inclusion bodies, the insoluble fraction from the sample was collected by centrifugation, washed with isolation buffer (20 mM Tris‐HCl, 2 M urea, 0.5 M NaCl, 2% Triton‐X 100, pH 8.0) and resuspended in binding buffer (20 mM Tris‐HCl, 8 M urea, 0.5 M NaCl, 5 mM imidazole, pH 8.0). Then, scFv was purified using an AKTA purifier system (GE Healthcare, Uppsala, Sweden) with a HisTrap HP affinity column. The purity and quantity of scFv were determined by SDS‐PAGE analysis and the bicinchoninic acid (BCA) assay, respectively.
2.6. Western blot assay
After the separation of total cellular proteins by SDS‐PAGE, all protein bands were electrophoretically transferred onto a polyvinyl difluoride membrane using a Bio‐Rad transblot device (Bio‐Rad, Hercules, CA, USA). The membrane was incubated within 5% nonfat milk powder for 2 h to block nonspecific binding sites. Then, it was incubated with a monoclonal horseradish peroxidase (HRP)‐conjugated anti‐His6 antibody (1: 10,000) for the immunodetection of His‐tagged protein. The membrane was washed and the antibody was detected using an ECL kit (Amersham Biosciences, Little Chalfont, UK).
3. Results
3.1. Construction of plasmids containing genetic expression parts
Previous tests showed that if several functional expression sequences of highly expressed genes of C. glutamicum were arranged according to the architecture of a BEP rather than a MEP, there was greater expression of the target gene in C. glutamicum 11. To extensively test this method and expand the source of functional sequences, the top 12 highly transcribed genes in C. glutamicum (T‐Set) and the top 12 highly expressed genes in B. subtilis (HP‐Set) were identified as sequence provider (Supporting Information Table 2). Their sequences were used to construct expression parts in two kinds of architecture: BEP and MEP (Fig. 3). The T‐Set represented native strongly transcribed materials for T‐BEP/T‐MEP, and the HP‐Set represented heterologous strongly expressed materials for HP‐BEP/HP‐MEP. After amplifying expression parts from genomes and inserting them into probe vector pE‐0, 48‐Set ‐Set EGFP expression plasmids containing the two kinds of genetic parts derived from two source sets were respectively achieved and designated as pE‐T‐BEPn, pE‐T‐MEPn, pE‐HP‐BEPn, and pE‐HP‐MEPn. The letter ‘‘n’’ represents the rank of each candidate in terms of the mRNA or protein abundance of the corresponding source gene.
3.2. Characterization of expression parts from different kinds of sources
The growth‐normalized fluorescence intensities of C. glutamicum harboring various expression parts were measured (Fig. 4). The T and HP sets represented two kinds of sequence sources for expression parts. The results showed that there were variations in the relative performance of BEP to MEP, and in the range of expression levels. In T‐Set, eight of the twelve parts exhibited enhanced activities as BEPs (T‐BEP) compared to MEPs (T‐MEP) which have the same promoter‐5’UTR. In HP‐Set, ten of the twelve parts exhibited enhanced activities as BEPs (HP‐BEP) compared to MEPs (HP‐MEP). The ratios of BEP to MEP were 1.87 and 1.16 in T‐Set and HP‐Set, respectively. The comparison between BEPs and MEPs showed that BEPs using sequences from both the T‐Set and HP‐Set have advantage over their corresponding MEPs, and BEP form could increase the utilization of functional sequences selected from genome. Compared with the expression parts from the T‐Set, more than half of parts from the HP set showed very weak activities (Fig. 4). As noted above, especially in HP‐MEPs, the number of parts whose expression levels were lower than 2000 RFU/OD was six, which is double (or more) the number of parts in any other sets. Strong expression parts were obtained from both sources. Three expression parts (T‐BEP6, HP‐BEP4, HP‐MEP9) whose activities were higher than that of the positive control were obtained. The strongest was HP‐BEP4 (157,147 RFU/OD EGFP expression level, 2.24‐fold that of the positive control). In addition to those strong parts, the four strongest parts derived from C. glutamicum native highly expressed genes (P‐Set) in previously work were also examined 11. From each kind of source, the two strongest BEPs and MEPs were separately selected and confirmed by SDS‐PAGE and direct observation of fluorescence from C. glutamicum colonies under blue light (Fig. 5), which indicates that first the fluorescent intensity measurement matched well with SDS‐PAGE analysis and the observation under blue light. Second, the exogenous source provides us with a strong expression part HP‐BEP4 which could be used to construct overexpression vectors (Supplementary Sequence 2). Third, BEP strategy could be a more efficient way to construct strong expression vectors than that of MEP.
Figure 4.
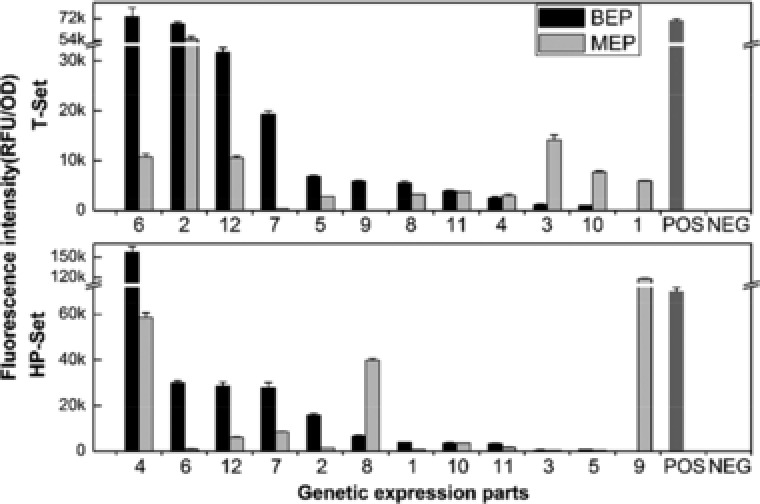
EGFP expression controlled by expression parts derived from two kinds of sequence sources. T, and HP sets were derived from C. glutamicum highly transcribed genes and B. subtilis highly expressed genes, respectively. Fluorescence intensity was normalized to OD600 of each construct. POS, positive control pXMJ19‐EGFP; NEG, negative control pE‐0; MEP and BEP, monocistronic and bicistronic expression parts, respectively. Numbers under horizontal axis indicate rank based on abundance of source genes.
Figure 5.
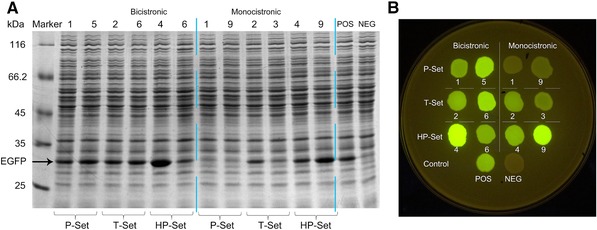
Performance of strong expression parts. Strongest two BEPs and MEPs were selected from P, T and HP‐Set. Their constructs as well as positive control (POS) and negative control (NEG) were tested. (A) SDS‐PAGE analysis of C. glutamicum cells. (B) Fluorescence image of respective cells.
3.3. Expression of scFv with different expression parts
Initially, we could not detect expression of scFv in the positive control or any of the gene expression constructs. Therefore, we adjusted three factors that could potentially affect scFv expression; codon usage bias, temperature, and expression parts. First, the sequence of the original scFv (OR‐scFv) was optimized according to the codon usage bias of C. glutamicum. Second, culture temperature was lowered. Third, we used two different constructs: pXMJ19‐SCFV and pSCFV‐HP‐BEP4 (containing the Tac‐conserved‐SD sequence and HP‐BEP4, respectively) 27, 28. After combining these factors, cells harboring recombinant plasmids pXMJ19‐OR‐SCFv, pXMJ19‐SCFV, pOR‐SCFV‐HP‐BEP4 and pSCFV‐HP‐BEP4 were cultivated and their total proteins were subjected to SDS‐PAGE and western blotting (Fig. 6A). Two factors affecting scFv expression were identified from the results. When the codons of the OR‐scFv sequence were optimized and when the HP‐BEP4 construct was used to initiate the expression of scFv, we detected this protein. The scFv was expressed in inclusion bodies (Fig. 6B).
Figure 6.
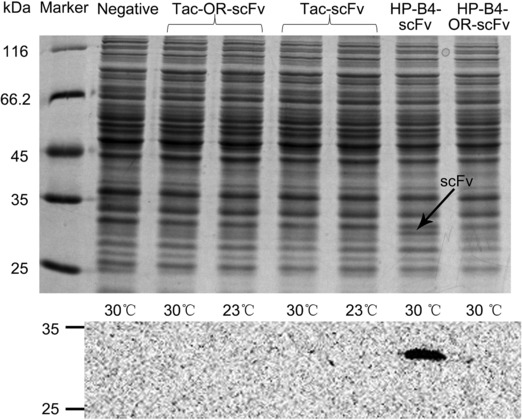
SDS‐PAGE and corresponding western blot analysis demonstrating effects of codon optimization, temperature, and expression parts. (A) Left to right: samples from cells harboring pSCFV‐0 cultured at 30°C, pXMJ19‐OR‐SCFV cultured at 30 and 23°C, pXMJ19‐SCFV cultured at 30 and 23°C, pSCFV‐HP‐BEP4 cultured at 30°C, and pOR‐SCFV‐HP‐BEP4 cultured at 30°C. In names of expression parts shown above SDS‐PAGE image, original coding sequence without optimization is indicated by OR‐scFv.
The selected 12 strong expression parts described above and the positive control pXMJ19‐SCFV and negative control pSCFV‐0 were used to express codon‐optimized scFv (Fig. 7A). Many of the expression parts exhibited strong expression of scFv. Five of the six BEPs successfully expressed scFv at a level visible after SDS‐PAGE. Of the five strong MEPs for EGFP, only two led to detectable expression of scFv, indicating that BEPs were more reliable than MEPs for gene expression. Under the control of P‐BEP5, T‐BEP6, or HP‐BEP4, scFv was expressed at >100 μg/mL, as determined by the SDS‐PAGE quantitative analysis. After cultivation in shake flasks for 24 h, cells were disrupted by sonication, centrifuged to obtain the pellet containing scFv, and then the pellet was dissolved and purified by Ni‐NTA affinity column chromatography. The SDS‐PAGE analysis showed that the purity of scFv was higher than 94% after this simple affinity purification step (Fig. 7B). About 12.5 mg scFv in the form of inclusion bodies was purified from 1 L culture. Together, the results showed that the expression part HP‐BEP4 obviously expressed a target gene in C. glutamicum.
Figure 7.
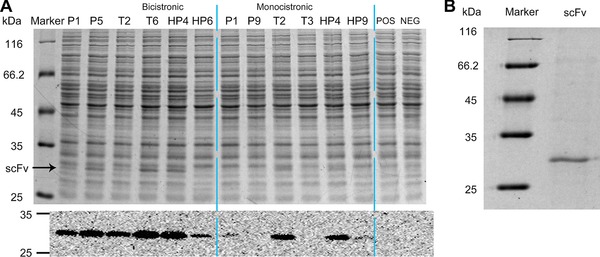
Expression and identification of scFv. (A) SDS‐PAGE and corresponding western blot analysis of recombinant strain harboring strongest expression parts used in Fig. 4. C. glutamicum (pXMJ19‐SCFV) and C. glutamicum (pSCFV‐0) were used as positive control (POS) and negative control (NEG), respectively. (B) SDS‐PAGE of purified scFv from the C. glutamicum recombinant strain harboring pSCFV‐HP‐BEP4.
4. Discussion
C. glutamicum with rich experience in industrial fermentation has considerable potential for the production of recombinant proteins 29. However, further engineering of this strain requires more advanced genetic tools and the development of a suitable expression system. In order to express recombinant genes in C. glutamicum, a synthetic approach with randomized promoter sequences was introduced 10. Although high‐throughput screening technologies are able to process a large number of samples, theoretical library size for a randomized promoter is still impossible to be achieved. Another method is directly using expression‐initiation sequences from genome. ANOVA analysis of the effect of genetic context on expressed gene showed that the promoter‐5′UTR part can explain more than 80% variance of its protein levels 30, 31. The native transcriptional or expression level can be used to evaluate the part potential, and the promoter‐5′UTR part which lead to high‐level transcription or expression of native genes have high potential of activities. Considering changing target gene could bring new influence factors including codon bias and mRNA structure on expression, it is possible that some unselected parts may lead to higher expression level of recombinant gene due to other favorable changes such as the increase of RNA stability. We assumed when functional sequences isolated from the genome are used as genetic expression parts, their performance in original genomic conditions could be better retained by preserving more sequence context, including the original TIR, as TIR are implicated in shaping expression level of target gene 21, 32.
In this study, by reusing extended functional sequences from the genome in a bicistronic expression system, high‐level expression of a target gene can be achieved, we tested the effects of the architecture of expression parts and the effect of sequence sources to provide specific functional sequences for expression parts. The functional sequences of native highly transcribed genes and heterologous highly expressed genes were used as components of monocistronic or bicistronic genetic expression systems.
The comparison between BEPs and MEPs showed that the BEP design was advantageous for exploiting the potential of functional expression sequences, leading to overall higher expression levels of recombinant genes. By contrast, those valuable materials are not expressed efficiently in the conventional promoter–5′‐UTR form. The improved expression activity resulting from the BEP design may be because it preserves the original TIR, which plays an important role in the strong translation of those original highly expressed genes 30. The BEP design provides more space to arrange the original TIR sequences in expression parts, thereby retaining the translation efficiency of the source gene.
The proportion of BEPs which exhibited higher activities than their corresponding MEPs is lower in T‐Set than in HP‐set‐Set, which may be due to the fact that those highly transcribed genes, which didn't show high protein abundance, are more likely to have strong promoters (i.e. transcriptional initiation elements) with weak TIR elements. The conventional MEP design has enough space to restore the genetic context of strong promoters, leading to good performance in EGFP expression, so there is less of a need for the BEP design to construct efficient parts from this kind of source gene.
Besides, compared with T‐Set, more functional sequences used in HP‐Set had little effect when being used to construct expression parts. This can be attributed to a heterogeneous working environment for those functional sequences from B. subtilis. In an unmatched environment with different pH, temperature, ion composition, and regulatory factors, expression parts could be inactivated or released from control from inhibitory factors. Two extremely strong expression parts, HP‐BEP4 and HP‐MEP9, were obtained from this kind of source. Their activities were greater than that of pXMJ19 and expression parts from the other two kinds of sources.
Besides strong expression levels, strong BEPs also showed reliable performance. When strong BEPs and MEPs were reused to express scFv, BEPs that performed well with EGFP also successfully expressed scFv, while two strong MEPs associated with the positive control failed to maintain their strong activities. It has been reported that the translation of an individual short peptide upstream of the gene utilizes the intrinsic helicase activities of ribosomes arriving at the second TIR, reducing the incidence of potentially inhibitory RNA structures and facilitating translation initiation at the second cistron 6, 33. Therefore, the bicistronic architecture can reduce the influence of genetic context on target gene expression, thereby improving the reliability of a strong expression vector. This is the first advantage of the bicistronic expression strategy. Second, this strategy can be used in different organisms. Although C. glutamicum was used as a typical strain to test these methods, this strategy is not limited to a specific bacterium. The third advantage is that, compared with the randomized synthetic promoter method, the bicistronic expression strategy avoids a time‐consuming step to deal with large libraries 34.
The expression‐controlling sequences for constructing a strong expression vector for C .glutamicum have been achieved. In total, three strong expression parts, T‐BEP6, HP‐BEP4 and HP‐MEP9, were obtained from 24 tested sequences. These showed better performance in expressing both EGFP and scFv than did the classic expression vector with the combination of the tac promoter and the C. glutamicum conserved SD sequence. HP‐BEP4 has the best performance, which was about 2.24 times higher than the constitutively expressed vector pXMJ19. When those expression parts were tested, the same sequence upstream those parts was T7 terminator that has no expression promoting effect, which means the function of those strong parts has less demand for a specific genetic context and a specific plasmid. Parts could also be transferred to genome. However, it has not solved the problem of the protein being localized in inclusion bodies. The production of scFv in C. glutamicum still requires favorable folding environment by a secretary system or the engineering of cytoplasmic environment. In conclusion, we developed a bicistronic expression strategy and discussed the merits of two sources of genetic sequences. With the progress of omics technologies, the highly expressed or highly transcribed genes can be identified from the genomes of a variety of bacteria. These are valuable materials to construct expression parts and be used interchangeably among bacteria. Using the bicistronic expression strategy, expression parts for efficient and reliable gene expression can be quickly constructed. We achieved an expression part, HP‐BEP4, strongly expressing the genes encoding EGFP and scFv. It is a valuable tool for the further recombinant protein production in C. glutamicum and the overexpression of key enzymes in genetically engineered organisms.
Practical application
Corynebacterium glutamiucm as an industrial host for the production of amino acids and organic acids possesses mature fermentation technologies. It is generally recognized as safe (GRAS) and able to secrete well folded protein into the culture, which catches people's attention to build a heterogonous protein expression system on this strain. In this study, we discussed the sources where the functional sequences such as promoter can be selected and how those functional sequences could be efficiently utilized to construct expression vectors for those important or potential bacteria like C. glutamicum. A strong expression vector for C. glutamicum has been constructed.
The authors have declared no conflict of interest.
Supporting information
Supplementary Table 1 Primers used in this study
Acknowledgments
This study was funded by the National Basic Research Program of China (973 Program) (grant number: 2013CB733602) and the Natural Science Foundation of Jiangsu Province (grant number: BK20150148).
5 References
- 1. Schumperli, D. , McKenney, K. , Sobieski, D. A. , Rosenberg, M. , Translational coupling at an intercistronic boundary of the Escherichia coli galactose operon. Cell 1982, 30, 865–871. [DOI] [PubMed] [Google Scholar]
- 2. Levin‐Karp, A. , Barenholz, U. , Bareia, T. , Dayagi, M. et al., Quantifying translational coupling in E. coli synthetic operons using rbs modulation and fluorescent reporters. ACS Synth. Biol. 2013, 2, 327–336. [DOI] [PubMed] [Google Scholar]
- 3. Kimura, S. , Umemura, T. , Iyanagi, T. , Two‐cistronic expression plasmids for high‐level gene expression in Escherichia coli preventing translational initiation inhibition caused by the intramolecular local secondary structure of mRNA. J. Biochem. 2005, 137, 523–533. [DOI] [PubMed] [Google Scholar]
- 4. Nishizawa, A. , Nakayama, M. , Uemura, T. , Fukuda, Y. et al., Ribosome‐binding site interference caused by Shine‐Dalgarno‐like nucleotide sequences in Escherichia coli cells. J. Biochem. 2010, 147, 433–443. [DOI] [PubMed] [Google Scholar]
- 5. Saito, Y. , Ishii, Y. , Niwa, M. , Ueda, I. , Direct expression of a synthetic somatomedin C gene in Escherichia coli by use of a two‐cistron system. J. Biochem. 1987, 101, 1281–1288. [DOI] [PubMed] [Google Scholar]
- 6. Mutalik, V. K. , Guimaraes, J. C. , Cambray, G. , Lam, C. et al., Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods 2013, 10, 354–360. [DOI] [PubMed] [Google Scholar]
- 7. D'Agostino, P. M. , Woodhouse, J. N. , Makower, A. K. , Yeung, A. C. Y. et al., Advances in genomics, transcriptomics and proteomics of toxin‐producing cyanobacteria. Environ. Microbiol. Rep. 2016, 8, 3–13. [DOI] [PubMed] [Google Scholar]
- 8. Wang, J. , Ai, X. , Mei, H. , Fu, Y. et al., High‐throughput identification of promoters and screening of highly active promoter‐5 '‐UTR DNA region with different characteristics from Bacillus thuringiensis . PLoS One. 2013, 8, e62960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu, Y. D. , Wan, H. , Li, J. G. , Zhou, J. W. , Enhanced production of L‐sorbose in an industrial Gluconobacter oxydans strain by identification of a strong promoter based on proteomics analysis. J. Ind. Microbiol. Biotechnol. 2015, 42, 1039–1047. [DOI] [PubMed] [Google Scholar]
- 10. Yim, S. S. , An, S. J. , Kang, M. , Lee, J. et al., Isolation of fully synthetic promoters for high‐level gene expression in Corynebacterium glutamicum . Biotechnol. Bioeng. 2013, 110, 2959–2969. [DOI] [PubMed] [Google Scholar]
- 11. Zhao, Z. , Liu, X. , Zhang, W. , Yang, Y. et al., Construction of genetic parts from the Corynebacterium glutamicum genome with high expression activities. Biotechnol. Lett. 2016, 38, 2119–2126. [DOI] [PubMed] [Google Scholar]
- 12. Heider, S. A. E. , Wendisch, V. F. , Engineering microbial cell factories: Metabolic engineering of Corynebacterium glutamicum with a focus on non‐natural products. Biotechnol. J. 2015, 10, 1170–1184. [DOI] [PubMed] [Google Scholar]
- 13. Dong, X. , Zhao, Y. , Zhao, J. , Wang, X. , Characterization of aspartate kinase and homoserine dehydrogenase from Corynebacterium glutamicum IWJ001 and systematic investigation of L‐isoleucine biosynthesis. J. Ind. Microbiol. Biotechnol. 2016, 43, 873–885. [DOI] [PubMed] [Google Scholar]
- 14. Wang, C. , Cai, H. , Zhou, Z. , Zhang, K. et al., Investigation of ptsG gene in response to xylose utilization in Corynebacterium glutamicum . J. Ind. Microbiol. Biotechnol. 2014, 41, 1249–1258. [DOI] [PubMed] [Google Scholar]
- 15. Liu, X. , Yang, Y. , Zhang, W. , Sun, Y. et al., Expression of recombinant protein using Corynebacterium Glutamicum: progress, challenges and applications. Crit. Rev. Biotechnol. 2016, 36, 652–664. [DOI] [PubMed] [Google Scholar]
- 16. Teramoto, H. , Watanabe, K. , Suzuki, N. , Inui, M. et al., High yield secretion of heterologous proteins in Corynebacterium glutamicum using its own Tat‐type signal sequence. Appl. Microbiol. Biotechnol. 2011, 91, 677–687. [DOI] [PubMed] [Google Scholar]
- 17. Jeong, K. J. , Jang, S. H. , Velmurugan, N. , Recombinant antibodies: Engineering and production in yeast and bacterial hosts. Biotechnol. J. 2011, 6, 16–27. [DOI] [PubMed] [Google Scholar]
- 18. Liu, X. , Yang, S. , Wang, F. , Dai, X. et al., Comparative analysis of the Corynebacterium glutamicum transcriptome in response to changes in dissolved oxygen levels. J. Ind. Microbiol. Biotechnol. 2017, 44, 181–195. [DOI] [PubMed] [Google Scholar]
- 19. Wang, M. , Weiss, M. , Simonovic, M. , Haertinger, G. et al., PaxDb, a database of protein abundance averages across all three domains of life. Mol. Cell. Proteomics 2012, 11, 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reese, M. G. , Application of a time‐delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 2001, 26, 51–56. [DOI] [PubMed] [Google Scholar]
- 21. Kudla, G. , Murray, A. W. , Tollervey, D. , Plotkin, J. B. , Coding‐sequence determinants of gene expression in Escherichia coli . Science 2009, 324, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yusupova, G. Z. , Yusupov, M. M. , Cate, J. H. D. , Noller, H. F. , The path of messenger RNA through the ribosome. Cell 2001, 106, 233–241. [DOI] [PubMed] [Google Scholar]
- 23. Huang, Y. , Zhao, K. X. , Shen, X. H. , Chaudhry, M. T. et al., genetic characterization of the resorcinol catabolic pathway in Corynebacterium glutamicum . Appl. Environ. Microbiol. 2006, 72, 7238–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Engler, C. , Kandzia, R. , Marillonnet, S. , A one pot, one step, precision cloning method with high throughput capability. PLoS One 2008, 3, e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jakoby, M. , Ngouoto‐Nkili, C. E. , Burkovski, A. , Construction and application of new Corynebacterium glutamicum vectors. Biotechnol. Technol. 1999, 13, 437–441. [Google Scholar]
- 26. Qin, X. L. , Qian, J. C. , Yao, G. F. , Zhuang, Y. P. et al., GAP promoter library for fine‐tuning of gene expression in Pichia pastoris . Appl. Environ. Microbiol. 2011, 77, 3600–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baneyx, F. , Mujacic, M. , Recombinant protein folding and misfolding in Escherichia coli . Nat. Biotechnol. 2004, 22, 1399–1408. [DOI] [PubMed] [Google Scholar]
- 28. Carton, J. M. , Sauerwald, T. , Hawley‐Nelson, P. , Morse, B. et al., Codon engineering for improved antibody expression in mammalian cells. Protein Expr. Purif. 2007, 55, 279–286. [DOI] [PubMed] [Google Scholar]
- 29. Wang, J. , Wen, B. , Wang, J. , Xu, Q. Y. et al., Enhancing l‐isoleucine production by thrABC overexpression combined with alaT deletion in Corynebacterium glutamicum . Appl. Biochem. Biotechnol. 2013, 171, 20–30. [DOI] [PubMed] [Google Scholar]
- 30. Mutalik, V. K. , Guimaraes, J. C. , Cambray, G. , Mai, Q. A. et al., Quantitative estimation of activity and quality for collections of functional genetic elements. Nat. Methods 2013, 10, 347–353. [DOI] [PubMed] [Google Scholar]
- 31. Kosuri, S. , Goodman, D. B. , Cambray, G. , Mutalik, V. K. et al., Composability of regulatory sequences controlling transcription and translation in Escherichia coli. PNAS 2013, 110, 14024–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goodman, D. B. , Church, G. M. , Kosuri, S. , Causes, effects of N‐terminal codon bias in bacterial genes. Science 2013, 342, 475–479. [DOI] [PubMed] [Google Scholar]
- 33. Jang, S. A. , Sung, B. H. , Cho, J. H. , Kim, S. C. , Direct expression of antimicrobial peptides in an intact form by a translationally coupled two‐cistron expression system. Appl. Environ. Microbiol. 2009, 75, 3980–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rytter, J. V. , Helmark, S. , Chen, J. , Lezyk, M. J. et al., Synthetic promoter libraries for Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2014, 98, 2617–2623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Primers used in this study