

Abstract
This study reports the application of peptide linker in the construction of bi‐functional formate dehydrogenase (FDH) and leucine dehydrogenase (LeuDH) enzymatic complex for efficient cofactor regeneration and L‐tert leucine (L‐tle) biotransformation. Seven FDH‐LeuDH fusion enzymes with different peptide linker were successfully developed and displayed both parental enzyme activities. The incorporation order of FDH and LeuDH was investigated by predicting three‐dimensional structures of LeuDH‐FDH and FDH‐LeuDH models using the I‐TASSER server. The enzymatic characterization showed that insertion of rigid peptide linker obtained better activity and thermal stability in comparison with flexible peptide linker. The production rate of fusion enzymatic complex with suitable flexible peptide linker was increased by 1.2 times compared with free enzyme mixture. Moreover, structural analysis of FDH and LeuDH suggested the secondary structure of the N‐, C‐terminal domain and their relative positions to functional domains was also greatly relevant to the catalytic properties of the fusion enzymatic complex. The results show that rigid peptide linker could ensure the independent folding of moieties and stabilized enzyme structure, while the flexible peptide linker was likely to bring enzyme moieties in close proximity for superior cofactor channeling.
Keywords: Bi‐functional enzyme, Formate dehydrogenase, L‐tert leucine, Leucine dehydrogenase, Peptide linker
Abbreviations
- AF
Acceleration factor
- DL
Direct link of two enzymes
- FDH
Formate dehydrogenase
- IPTG
Isopropyl‐β‐D‐thiogalactopyranoside
- KR
Ketoreductase
- L‐tle
L‐tert leucine
- LeuDH
Leucine dehydrogenase
- OE‐PCR
Overlap extension by polymerase chain reaction
- RN
N unit of rigid peptide linker
- SN
N unit of flexible peptide linker
1. Introduction
The synthesis of optically pure intermediate has become increasingly important to meet the demand of pharmaceutical industrials. Biocatalysts, in the form of whole cell or purified enzyme, have been widely applied in the production of chiral building blocks 1. The advantages of biocatalysis over chemical reaction were the higher chemo‐, region‐, enantioselectivities and ambient reaction conditions. Moreover, the development of bioinformatics and protein engineering like direct evolution and rational design has improved the efficiency of biocatalyst modification 2. The advance in bioprocess engineering has offered various hosts and strategies for heterologous protein product with high quality 3, 4.
L‐tert leucine (L‐tle) has been widely used as a chiral building block in many asymmetric reactions for the synthesis of anti‐tumor and anti‐HIV drugs 5. The enzymatic catalysis of L‐tle have been developed in past decades and several biocatalysts were found to be capable of producing L‐tle including leucine dehydrogenase 6, branched chain aminotransferase 7, penicillin acylases 8, lipase 9, amidase 10, protease 11. Among the above‐listed reports, leucine dehydrogenase (LeuDH, EC 1.4.1.9) and formate dehydrogenase (FDH, EC 1.2.1.2) based cofactor regeneration system was regarded as a desirable process with high catalytic and economic efficiency 12. Degussa AG (now Evonik AG) performed LeuDH and FDH coupling system for repetitive batch reaction with an enzyme membrane reactor at ton scale and obtained 638 g/L/day space‐time‐yield of L‐tle 13.However, to our best knowledge, there was no report on the artificial assembly of LeuDH and FDH multi‐enzyme complex using peptides linkers to accelerate the reaction rate.
Inspired by nature sophisticated protein complex mostly found in metabolic pathways like tryptophan synthase 14 and cellulosome systems 15, artificial multi‐enzymatic systems have been reported to enhance catalytic efficiency and reduce overall cost 16. Iturrate et al. 17 constructed an engineered bifunctional aldolase/kinase enzyme with five amino acid length linker (QGQGQ) which exhibited a 20‐fold increase in the reaction rate of aldol synthesis. Gao et al. 18 fused LeuDH and FDH with PDZ (PSD95/Dlg1/zo‐1) domain and corresponding ligand (PDZlig) from metazoan cells respectively for in vitro and vivo self‐assembly of multienzyme supramolecular. Both in vitro and vivo assemblies exhibited higher cofactor regeneration efficiency and structural stability than free enzyme mixture. Fu et al. 19 constructed a swinging‐arm nanostructure complex consisting of glucose‐6‐phosphate dehydrogenase, malic dehydrogenase and NAD+ attached oligonucleotide and all these components can be programmed assembly on a DNA scaffold. The cofactor attached with oligonucleotide was located between two enzymes with great proximity effect, facilitating intramolecular cofactor transfer. The close proximity of the catalytic entities in the complex gives rise to the substrate channeling phenomenon which eliminated the diffusion limitation of substrate or intermediate such as cofactor 20. Nevertheless the enhanced catalytic efficiency of some fusion enzymes were suggested to be caused by altered kinetic properties instead of channeling effect 21, 22.
In this paper, a series of bi‐functional enzyme complexes were produced by fusing LeuDH and FDH with different peptide linkers, which were expressed in E. coli, purified, and exhibited varied parental enzyme activities and varied L‐tle catalytic efficiency. The enzymatic reaction of FDH and LeuDH system for L‐tle production and cofactor regeneration was shown in Supporting Information Fig. 1. The impact of structural feature of peptide linkers and fused moieties over the properties of fusion enzymatic complex. The obtained results indicated that the fusion enzyme with suitable peptide linker is potentially more excellent than free enzymes with lower labor‐cost on purification, better thermal stability and higher catalytic efficiency compared with the free coupling of parental enzymes.
2. Materials and methods
2.1. Materials
E. coli BL21(DE3) competent cell, Isopropyl‐β‐D‐thiogalactopyranoside (IPTG), kanamycin and SDS‐PAGE kit were provided by Transgen (Beijing, China). PrimerSTAR Max DNA Polymerase, restriction enzymes, ligation enzyme were purchased from Takara (Shiga, Japan). pET‐28a(+) expression vector was from Novagen. Plasmid, gel extraction and PCR purification kits were purchased from Omega bio‐tek(USA). Modified Bradford Protein Assay Kit, LB broth, NADH and NAD+ were provided by Sangon biotech (Shanghai, China). L‐tert leucine, D‐tert leucine, Trimethylpyruvic acid, ammonium formate and sodium formate were purchased from Sigma‐Aldrich (USA).
2.2. Gene synthesis
pUC18‐LeuDH and pUC18‐FDH contained LeuDH gene from Bacillus cereus ATCC 10987 (3950125‐3951225 nucleotide, GeneBank Accession number: NC_003909.8) and FDH gene from Candida boidinii (190…1284 nucleotide, GeneBank Accession number: AF004096.1). NdeI and XhoI restriction sites were introduced in the 5’ end and 3’ end of both genes. These genes were then synthesized by Sangon biotech (Shanghai, China).
2.3. Structure analysis
Homology modeling of LeuDH and FDH was carried out utilizing the Swiss Model automated comparative protein modeling server (http://swissmodel.expasy.org) 23. According to the amino acid sequence homology, LeuDH from Bacillus sphaericus (PDB ID: 1LEH, sequence identity: 76.92) and FDH from Candida boidinii (PDB ID.: 2J6I, sequence identity: 96.69) were selected as potential structural templates to obtain the structural model of homology proteins. The three‐dimensional (3D) structural models of fusion enzyme complex in different incorporation orders were built up using the web‐based I‐TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER) 24. The molecular graphics software program PyMOL (http://www.pymol.org/) was used for illustration.
2.4. Gene construction
For the construction of parental enzyme gene, pUC‐LeuDH and pUC‐FDH plasmid was digested by NdeI and XhoI, followed by ligation with NdeI/XhoI‐linearized pET‐28a(+). The constructed plasmids were then transformed into E.coli BL21(DE3). OE‐PCR was applied for the construction of the chimeric gene encoding LeuDH and FDH with different peptide linkers. The oligonucleotides sequences encoding these peptide linkers and the corresponding amino acid sequences were shown in Supporting Information Table 1. The construction of F‐R1‐L (F indicated FDH, L indicated LeuDH, R1 indicated one unit of rigid linker) chimeric gene was taken as an example to demonstrate the cloning process. First, the FDH gene from pUC‐FDH was amplified using primer 1 (5’‐GGAATTCCATATGAAAATTGTCCTGGTCCTGT‐3’) with NdeI restriction site (underlined) and primer 2‐R1 (5’‐GCCTATGGCAAACACGATAAAAAGGAAGCTGCTGCTAAAATGACATTGGAAATCTTCGA‐3’) with sequences that encoding one repeat of rigid peptide linker (R1, underlined). The other primers for constructing other six fusion enzyme were listed in Supporting Information Table 1. Second, the LeuDH gene from pUC‐LeuDH was amplified using primer 3 (5’‐ATGACATTGGAAATCTTCGAATAT‐3’) and primer 4 (CCGCTCGAGTTACCGGCGACTAATGATGT) with XhoI restriction site (underlined). Finally, PCR products from the first two amplifications were extracted and used as templates for OE‐PCR to obtained F‐R1‐L chimeric gene using primer 1 and primer 4. Then the obtained chimeric gene was digested with NdeI and XhoI, followed by ligation with NdeI/XhoI‐linearized pET‐28a(+). The constructed plasmids were named pET‐28a‐F‐R1‐L and then transformed into E. coli BL21(DE3).
2.5. Enzyme preparation and purification
The transformed E. coli BL21(DE3) harboring fusion enzyme plasmids were cultured at 37°C in 1 L shaking flask containing 200 mL of LB media with 40 mg/L kanamycin. The expression of recombinant fusion enzyme was induced by the addition of 1 mM isopropyl‐β‐D‐thiogalactopyranoside (IPTG) at an optical density (OD600) of approximately 0.5 and the cell was grown at 16°C overnight. Cell was harvested by centrifugation and disrupted by ultrasonication. Cell debris was removed by 20‐min centrifugation at 12 000 × g at 4°C. Obtained crude cell extract was then applied to Histrap column (HisTrap HP 5 mL, GE Healthcare Corp., Piscataway, NJ, USA) pre‐equilibrated with binding buffer (20 mM sodium phosphate, 0.5 M NaCl, 20 mM imidazole, pH 7.4).The column was equilibrated with binding buffer and eluted by eluting buffer (20 mM sodium phosphate, 0.5 M NaCl, 0.5 M imidazole, pH 7.4) at a gradient concentration. Fractions with biology activity were desalted and concentrated by Macrosep Advance Centrifugal Devices (cut‐off 10 kDa, Pall, East Hills, NY, USA). The purity of the obtained enzyme was tested by 12% (w/v) SDS‐PAGE.
2.6. Enzyme characterization
Activity of LeuDH and FDH was assayed by monitoring the NADH (ε = 6.22/mM/cm) concentrations at 340 nm at 30°C using Tecan M200 Pro plate reader (Tecan Group Ltd., Männedorf, Switzerland). For LeuDH, the assay mixture contained 4.5 mM trimethylpyruvic acid, 0.2 mM NADH, 0.8 M NH4Cl‐NH3·H2O buffer (pH 10.0) and a certain amount of enzyme solution. For FDH, the assay mixture contained 162 mM sodium formate, 1.62 mM NAD+, 100 mM potassium phosphate buffer (pH 7.5) and a certain amount of enzyme solution. The volume of the reaction mixture was 200 μL in all cases. Reactions were initiated by the addition of the enzyme solutions. Enzyme activities were determined in triplicates. One unit of LeuDH and FDH activity was respectively defined as the amount of enzyme catalyzing the consumption or generation of 1 μmol NADH per minute under standard measurement conditions. Protein concentration was assayed using Modified Bradford Protein Assay Kit.
2.7. Determination of thermal stability
To determine the effect of linker property on the thermal stability of the fusion enzymes, the purified fusion enzymes (0.15 mg/mL) were incubated at 30°C and 40°C water bath for 1 h and assayed for the residual FDH and LeuDH activity. The measured activity was calculated as a percentage of the original activities assayed before incubating. All measurements were performed in triplicate.
2.8. Biocatalysis of L‐tle and product analysis
The standard reaction mixture contained 50 mM trimethylpyruvic acid, 50 mM ammonium formate, 0.04 mM NAD+, pH = 8.5 (adjusted by adding ammonia), followed by adding free enzyme mixture or constructed fusion enzymatic complex in a total reaction volume of 2 mL. The reaction was performed at 30°C with 200 rpm of horizontal shaking. Aliquots of the reaction mixture were taken and stored in ‐80°C for further analysis.
For product analysis, the reaction samples were heat denatured at 80°C for 30 min. After centrifugation at 13 000 × g for 10 min to remove precipitated protein, the samples were filtered using 0.22 μm filter and then analyzed with HPLC. The L‐tle concentration and the optical purity (ee %) were determined at 35°C with HPLC column (Phenomenex Chirex 3126D‐penicillamine). The mobile phase was 2 mM Copper (II) sulfate in water/isopropanol (95:5) at a flow rate of 1 mL/min.
3. Results and discussion
3.1. The incorporation order
For industrial process, an efficient cofactor regeneration system is indispensable to ensure the synthesis process by oxidoreductases is economically viable because the cofactor such as NAD(H) and NADP(H) is usually expensive. One of the advantages of integrating two catalytic entities is the possibility to achieve proximity effect in the multi‐enzyme complex, which could accelerate cofactor regeneration and catalytic efficiency 25. The order of the fused moieties was reported to be critical for catalytic performance. Moreover, it was not uncommon that one order resulted in completely nonactive for all the fused moieties 26, 27. To ensure favorable orientation of active sites in the fusion enzyme complex, the incorporation order of LeuDH and FDH was studied by structural modeling approach with web‐based I‐TASSER server to predict the orientation of cofactor binding domains of LeuDH‐FDH and FDH‐LeuDH. Fusing C terminal of LeuDH with N terminal of FDH (Fig. 1B) exhibited a back to back orientation of active clefts without a viable tunnel for cofactor channeling and the cofactor released from one enzyme have to diffuse in bulk solution and accomplish the regeneration process by random, unguided Brownian motion. Fusing C terminal of FDH with N terminal of LeuDH (Fig. 1A) created a favorable face to face orientation of active clefts which would promote the formation of intramolecular tunnel and accelerate the cofactor channeling between FDH and LeuDH. In addition, recent studies have shown that fusion of FDH to its C terminal would cause severe loss of enzymatic activity of FDH 28, 29. Since FDH is the rate limiting enzyme in bi‐enzymatic system, the incorporation order FDH‐LeuDH was predicted to be more suitable for efficient L‐tle production and used in further study.
Figure 1.
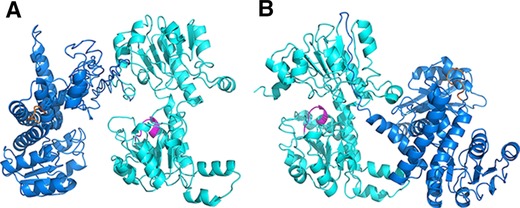
Representation of fusion enzyme structures of different incorporation orders, FDH‐LeuDH (A) and LeuDH‐FDH (B). LeuDH was displayed in blue and its cofactor binding domain was depicted in orange. FDH was displayed in cyan and its cofactor binding domain was marked in magenta.
3.2. Construction of fusion genes and activities comparison of fusion enzymes
A peptide linker was usually introduced to connect protein moieties for independent folding and preserving biological functions 30. Since linker type and length were the most concerned points that affect the properties of fused subunits 31, we proceeded to investigate the effect of linker configuration on fusion enzyme structure and L‐tle priduction efficiency. Different copy numbers of rigid linkers (EAAAK) and flexible linkers (GSSSS) were inserted into FDH and LeuDH using OE‐PCR (overlap extension by polymerase chain reaction). The primers for introducing peptide linker were listed in Method section and Supporting Information Table 1. As shown in Supporting Information Fig. 2, the fusion genes obtained by OE‐PCR were about 2200 bp which were identical to their theory sizes. The genes were then ligated with pET28 (a) plasmid and transformed into BL21 (DE3). After induced by IPTG, the cells were harvested and the whole cell proteins were analyzed by SDS ‐PAGE (Supporting Information Fig. 3). The sizes of the recombinant protein on SDS‐PAGE matched with their theoretical sizes. The fusion enzymes F‐DL‐L (DL indicated direct link), F‐R1‐L (R1 indicated one unit of rigid linker), F‐R2‐L, F‐R3‐L, F‐S1‐L (S1 indicated one unit of flexible linker), F‐S2‐L, F‐S3‐L were purified by nickel affinity column (Supporting Information Fig. 4). FDH and LeuDH activity of the parental enzyme and fusion enzyme with different peptide linkers were assayed and shown in Fig. 2. All fusion enzymes successfully retained varied degree of catalytic abilities of FDH and LeuDH. As expected, F‐DL‐L constructed without peptide only retained 24.7 and 65.2% of the parental FDH and LeuDH activities probably because direct assembly of two enzymes may interfere with structure folding. The impaired catalytic activity cause by direct fusion was consistent with previous reports 32, 33. Among the six peptide linkers, F‐R3‐L was the most active fusion which attained 145.1 and 103.6% of the parental FDH and LeuDH activities. Due to the effective separation of protein moieties, the fusion enzymes with rigid peptide linkers yielded better activities than the fusion enzymes with flexible peptide linkers.
Figure 2.
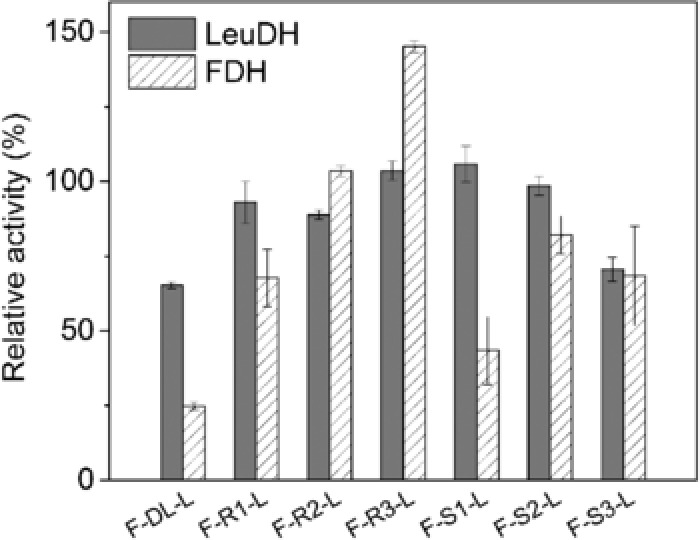
Enzyme activity of the FDH‐LeuDH fusion enzyme mediated by different peptide linker. The specific activities (in the unit of U/nmol) were assayed as described in Method section. The relative activity was expressed as a percentage of the activity of parental LeuDH and FDH. Values shown are means ± standard deviation of triplicate determinations.
The activity measurement of fusion enzymes showed that LeuDH moiety could retain more parental activity than FDH moiety (Fig. 2). Figure 3 represented the secondary structure of N, C terminal and their relative position to functionally relevant domains of FDH and LeuDH. Active site of FDH and LeuDH were located according to previous reports 34, 35. The C terminal of FDH was flexible structure containing β‐sheet and random coil which located near the cofactor binding domain and the active site, while N terminal of LeuDH formed rigid α‐helix and was located at the backside of active pocket. The N terminal of LeuDH (MTLEIFEYLEKY) contained abundant glutamic acids and lysines that could stabilize protein conformation by Glu−‐Lys+ salt bridges, which shared the same sequence pattern and stabilizing mechanism with rigid peptide linker (EAAAK) 36. We assumed that the merits of FDH C terminal rendered the fusion to take great risks of steric hindrance, while the rigid N terminal of LeuDH could behave as an intrinsic rigid peptide linker and ensure independent folding of LeuDH moietie. Thus, we suggested that the comprehensive investigation of moiety structures and linker features was necessary for efficient multi enzymatic complex construction.
Figure 3.
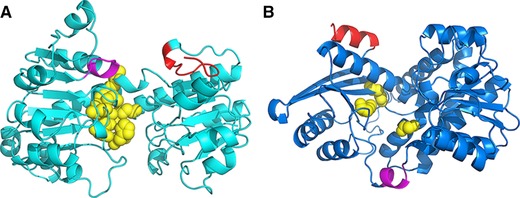
The positioning of N, C terminal and functionally relevant domains of FDH (A) and LeuDH (B). C terminal of FDH and N terminal of LeuDH was displayed in red. Cofactor binding domain was depicted in magenta. Substrate binding sites were depicted in yellow balls.
3.3. Kinetic parameters for the free and fusion enzymes
Table 1 showed a list of kinetic parameters about NADH/NAD+ of the fusion enzymes with different peptide linker and free enzymes. The kinetic parameters of LeuDH moiety was slightly changed after fusion with FDH moiety while the kinetic parameter of FDH moiety was significantly improved by the fusion. The K m value of all seven fusion enzymes was decreased by a magnitude compared with that of the free FDH. The catalytic efficiency (k cat/K m) of fusion enzymes was enhanced by 3–12 fold compared with that of the free FDH. F‐R3‐L exhibited best kinetic property among seven fusion enzyme which was in accordance with the result from enzyme activities determination in Fig. 2. F‐S2‐L showed a very high K m for NADH (151.22 μM) compared with free LeuDH and other fusion enzymes, which indicated fusion enzyme mediated by (GGGGS)2 may lead to steric hindrance near its cofactor binding domain. We speculated that altered kinetic parameters of cofactor were caused by moiety–moiety interactions in fusion enzyme or the proximity of two cofactor binding domains from LeuDH and FDH. The existence of two cofactor binding domain may explain the increased enzyme‐cofactor affinity of the bi‐functional enzymes.
Table 1.
The kinetic parameters of free enzyme and fusion enzyme
| NADH oxidation by LeuDH part | NAD+ reduction for FDH part | |||||
|---|---|---|---|---|---|---|
| Enzymes | K m (μM) | V m (U/μmol) | k cat/K m (×106M−1S−1) | K m (μM) | V m (U/μmol) | k cat/K m (×106M−1S−1) |
| LeuDH | 60.52 ± 4.32 | 615.37 ± 32.06 | 10.17 ± 0.87 | ‐ | ‐ | ‐ |
| FDH | ‐ | ‐ | ‐ | 623.22 ± 42.44 | 41.57 ± 3.61 | 0.07 ± 0.01 |
| F‐DL‐L | 41.49 ± 3.29 | 411.4 ± 20.63 | 9.91 ± 0.70 | 39.12 ± 2.40 | 10.93 ± 0.68 | 0.28 ± 0.02 |
| F‐R1‐L | 50.88 ± 4.43 | 483.61 ± 34.24 | 9.51 ± 0.82 | 37.46 ± 1.58 | 32.74 ± 1.70 | 0.87 ± 0.06 |
| F‐R2‐L | 43.57 ± 2.51 | 486.84 ± 25.30 | 11.17 ± 0.53 | 51.21 ± 3.31 | 42.32 ± 2.65 | 0.83 ± 0.06 |
| F‐R3‐L | 27.86 ± 2.11 | 628.28 ± 49.52 | 22.55 ± 0.55 | 45.16 ± 1.23 | 61.41 ± 5.91 | 1.36 ± 0.08 |
| F‐S1‐L | 22.26 ± 1.40 | 595.97 ± 47.61 | 26.78 ± 1.58 | 66.27 ± 4.76 | 22.16 ± 1.64 | 0.33 ± 0.02 |
| F‐S2‐L | 151.22 ± 10.99 | 500.37 ± 48.10 | 3.31 ± 0.20 | 52.13 ± 2.59 | 31.98 ± 3.09 | 0.61 ± 0.01 |
| F‐S3‐L | 34.91 ± 1.14 | 429.41 ± 33.79 | 12.3 ± 0.52 | 26.37 ± 2.73 | 24.94 ± 2.61 | 0.95 ± 0.04 |
Reaction conditions: 5 μM of free enzyme and fusion enzyme were used for the measurement of kinetic parameters. The reaction was performed as described in Method section using 0.0625‐0.5 mM NADH and 0.025‐1 mM NAD+ for LeuDH part and FDH part, respectively.
There is an ongoing debate whether the artificially induced proximity of enzymes by synthetic scaffolds or gene fusion is sufficient to prevent the diffusion of reaction intermediates into the bulk phase and to cause effective intermediate channeling 37. Some reports have claimed the enhanced production efficiency of fusion enzyme was entirely due to altered kinetic properties 21, 38. The analysis toward kinetic parameters of cofactor (Table 1) revealed a great difference between fusion enzymes and parental enzymes. The dramatically decrease of K m NAD+ of FDH moiety by fusion FDH with an oxidoreductases was also observed by Sührer et al. 22 in their research about an artificial fusion protein of a ketoreductase (KR) and FDH.
3.4. The influence of peptide linker on the thermal stability of fusion enzymes
The structural feature of peptide linker also affected the performance of fusion enzymes in thermal stability 39. The residual activities of the seven fusion enzymes and the parental enzymes were determined after incubating at 30 and 40°C for 1 h (Fig. 4). Most fusion enzyme with peptide linkers showed greater thermal stability than F‐DL‐L, indicating that the introduction of peptide linker was beneficial for stabilizing fused moieties. LeuDH moieties of all seven fusion enzyme complexes were more stable than free LeuDH. Fusion enzymes with a rigid linker were more stable than those with a flexible linker. F‐R2‐L even displayed higher thermal stability both in LeuDH and FDH moieties compared with the parental enzyme. This was not in accordance with the report by Lu and Feng 40 which suggested the thermal stability of β‐glucanase‐xylanase fusion enzyme increased with the copy number of inserted rigid peptide linker (EAAAK).
Figure 4.
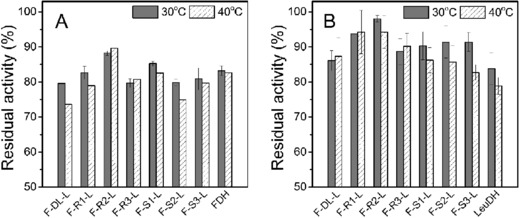
Thermal stability (A: LeuDH; B: FDH) of the parental enzyme and FDH‐LeuDH fusion enzyme mediated by different peptide linkers. Activities were assayed after incubating fusion enzymes in 30 and 40°C for 1 h. The residual activity was expressed as a percentage of the original activity measured before incubating. Values shown are means ± standard deviation of triplicate determinations.
It had been reported that the fusion of two multimeric enzyme could lead to the formation of multimeric protein network 41 which may cause steric hindrance of active domain. Soluble bi‐functional enzyme compose of two multimeric enzyme have reported to form oligomers 29, 42. LeuDH and FDH in our study usually formed octamer and dimer, respectively. The implementation of different peptide linker in FDH‐LeuDH fusion enzyme could cause differnent multimeric state, which explained the varities of catalytic activity and thermal stability of the constructued fusion enzyme 43.
3.5. Biotransformation of L‐tle
To study the cofactor channeling effect of the fusion enzyme complex, L‐tle biosynthesis by FDH‐LeuDH fusion enzyme system was implemented with two sets of biocatalysts, fusion enzyme with different peptide linker and parental enzyme mixtures whose activities were adjusted to match the fusion enzyme. We introduced acceleration factor (AF) to evaluate the cofactor channeling effect 44. AF was defined in Eq. (1), where VF and VP are the initial production rate of L‐tle catalyzed by the fusion enzyme complex and the parental enzyme mixture with same activities, respectively.
| (1) |
As shown in Fig. 5, except for F‐S2‐L, the AF of other 6 fusion enzymes were all positive. Although F‐S1‐L displayed highest AF among these fusions, which presented 1.2 times higher initial reaction rate than that of parental enzyme mixture, the FDH activity of F‐S1‐L was greatly impaired (Fig. 2). Similar phenomenon was observed in the F‐DL‐L and F‐S3‐L sets. Nevertheless, the acceleration factor of F‐R1‐L, F‐R2‐L and F‐R3‐L were 0.40, 0.18 and 0.57, respectively, while preserving relatively high catalytic ability. In addition, the fusion with FDH did not alter the enantio‐selectivity of LeuDH moiety. The product of all seven fusion enzymes exhibited enantiomeric purity of > 99% ee just like the parental LeuDH.
Figure 5.
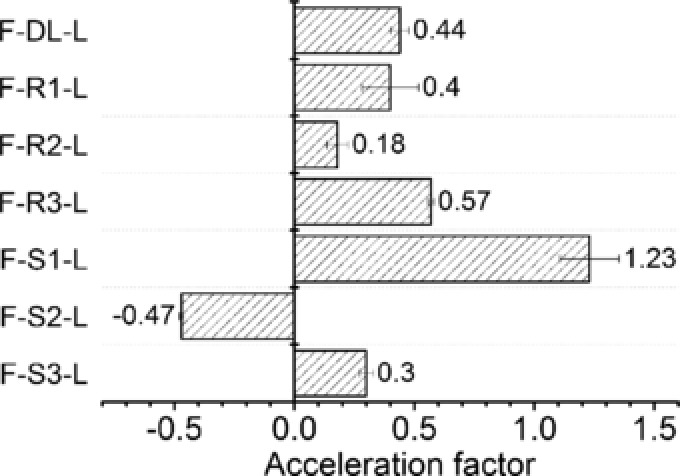
The acceleration factor of cofactor channeling in L‐tle catalysis by FDH‐LeuDH multi‐enzyme complex mediated by different peptide linker. 250 nM fusion enzymes were used for the production of L‐tle. Free LeuDH and FDH mixtures with equal activities to each fusion enzyme were used as control to calculate the acceleration factors.
The goal of fusing enzyme moieties together is to eliminate diffusion limitation of cofactor or intermediate. A few studies have been reported on fusion enzyme complex to enhance catalytic efficiency 17, 26, 32, 45. To confirm the effect of peptide linker composition on the production efficiency, the acceleration factor of each fusion enzyme complex was studied. According to our results, F‐S1‐L presented higher AF than other fusions with rigid linkers which indicating that the implement of flexible peptide may result in optimal positioning of cofactor binding domain (Fig. 5), probably because flexible peptide could retain certain domain movement 46. However, the catalytic activity and kinetic property of FDH moiety of F‐S1‐L was severely impaired (Fig. 2, Table 1) which indicated that the proximity of two catalytic moieties may cause steric influence or blockage of active sites if the peptide linker was inappropriate for the specific fusion enzyme system. In the FDH‐LeuDH enzymatic complex mediated by rigid peptide linker, the cofactor could transfer from active site to active site with increased efficiency, at the meantime, well separated fused entities could avoid structural influence caused by each other. Negative acceleration factor exhibited by F‐S2‐L indicated that fusing two enzymes together was not always beneficial for enhancing production efficiency. The application of inappropriate peptide linker could form an unfavorable orientation of cofactor binding domain and result in worse cofactor regeneration rate than free enzyme coupling system.
4. Concluding remarks
For the biotransformation of trimethylpyruvic acid into L‐tle, previous researchers have demonstrated several approaches using LeuDH and FDH in the form of free enzyme mixture, whole‐cell biocatalyst (LeuDH and FDH were expressed separately) and co‐expressed whole‐cell biocatalyst 5, 14, 47. Nevertheless, the L‐tle production by FDH‐LeuDH fusion complex has been reported for the first time in this study. In the present study, we constructed seven bi‐functional fusion enzymatic complexes comprising FDH and LeuDH moieties which inserted with different peptide linkers and compared their properties including structural property, bioactivity, thermal stability, and the ability to produce L‐tle. Although the long distance between fused moieties leads to ordinary production efficiency, the rigid peptide linker was preferable for independent folding and bioactivity retainment of fused moieties. The fusion enzymatic complex with proper flexible peptide linker was able to keep fused moieties close enough to obtain superior proximity effect, but liable to decrease bioactivity due to steric hindrance of functional domains. We suggested that the construction of bi‐functional enzymatic complex with peptide linker would be a powerful tool for constructing efficient cofactor regeneration system with satisfying cofactor regeneration rate.
In our future research, we will focus on the rational design of more delicate peptide linker in order to improve our fusion enzyme systems. In addition to protein modeling and molecular dynamic, linker selection based on natural linker is also a quite effective method for linker design. For example, Liu et al. 48 developed SynLinker, a web‐based bioinformatics tools for linkers selection and structure prediction, which contained 2150 natural linkers and 110 artificial linkers. The design of changing enzyme moiety number for activity rebalance and enhancing soluble expression of fusion enzymatic complex would be another interesting subjects for further investigation to increase L‐tle production efficiency.
Practical application
The information contained in this paper exhibits major relevance in applications like the multi‐enzyme system for fine chemical production, or biosensor based on multi‐enzyme reaction. The construction of bi‐functional enzyme can reduce the cost for recombinant protein purification and enhance the cofactor regeneration rate. Our work investigated the effect of different peptide linker configuration on the enzyme activities and productivity of obtained bi‐functional enzymes. The pros and cons of rigid and flexible peptide linker for FDH‐LeuDH was studied by both experiments and enzyme structural analysis. The study shall be useful to those who are interested in optimizing the biotransformation process of L‐ter leucine. Also, this work may be extended to other bi‐enzyme system for selecting an optimal peptide linker.
The authors have declared no conflict of interest.
Supporting information
Supporting Information
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 41176111, No. 41306124), the State Key Program of National Natural Science Foundation of China (No. 21336009), the Foundation of South Oceanographic Research Center of China in Xiamen (No. 14GYY011NF11) and the Fundamental Research Funds for the Central Universities (No. 20720170033), and the Public science and technology research funds projects of ocean (No. 201505032‐6).
Contributor Information
Shizhen Wang, Email: szwang@xmu.edu.cn.
Baishan Fang, Email: fbs@xmu.edu.cn.
5 References
- 1. Patel, R. N. , Biocatalysis: synthesis of key intermediates for development of pharmaceuticals. Acs Catal. 2011, 1, 1056–1074. [Google Scholar]
- 2. Bornscheuer, U. T. , Huisman, G. W. , Kazlauskas, R. J. , Lutz, S. et al., Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- 3. Xu, Y. , Tao, F. , Ma, C. , Xu, P. , New constitutive vectors: Useful genetic engineering tools for biocatalysis. Appl. Environ. Microbiol. 2013, 79, 2836–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Palomares, L. A. , Estrada‐Mondaca, S. , Ramírez, O. T. , Production of recombinant proteins: challenges and solutions. Methods Mol. Biol. 2004, 267, 15–52. [DOI] [PubMed] [Google Scholar]
- 5. Bommarius, A. S. , Schwarm, M. , Stingl, K. , Kottenhahn, M. et al., Synthesis and use of enantiomerically pure tert‐leucine. Tetrahedron: Asymmetry 1995, 6, 2851–2888. [Google Scholar]
- 6. Kragl, U. , Vasic‐Racki, D. , Wandrey, C. , Continuous production of L‐tert‐leucine in series of two enzyme membrane reactors. Bioprocess Eng. 1996, 14, 291–297. [Google Scholar]
- 7. Hong, E. Y. , Cha, M. , Yun, H. , Kim, B.‐G. , Asymmetric synthesis of l‐tert‐leucine and l‐3‐hydroxyadamantylglycine using branched chain aminotransferase. J. Mol. Catal. B Enzym. 2010, 66, 228–233. [Google Scholar]
- 8. Liu, W. , Luo, J. , Zhuang, X. , Shen, W. et al., Efficient preparation of enantiopure l‐tert‐leucine through immobilized penicillin G acylase catalyzed kinetic resolution in aqueous medium. Biochem. Eng. J. 2014, 83, 116–120. [Google Scholar]
- 9. Turner, N. J. , Winterman, J. R. , McCague, R. , Parratt, J. S. et al., Synthesis of homochiral L‐(S)‐tert‐leucine via a lipase catalysed dynamic resolution process. Tetrahedron Lett. 1995, 36, 1113–1116. [Google Scholar]
- 10. Stelkes‐Ritter, U. , Beckers, G. , Bommarius, A. , Drauz, K. et al., Kinetics of peptide amidase and its application for the resolution of racemates. Biocatal. Biotransform. 1997, 15, 205–219. [Google Scholar]
- 11. Agosta, E. , Caligiuri, A. , D'Arrigo, P. , Servi, S. et al., Enzymatic approach to both enantiomers of N‐Boc hydrophobic amino acids. Tetrahedron: Asymmetry 2006, 17, 1995–1999. [Google Scholar]
- 12. Bommarius, A. S. , Schwarm, M. , Drauz, K. , Biocatalysis to amino acid‐based chiral pharmaceuticals—examples and perspectives. J. Mol. Catal. B Enzym. 1998, 5, 1–11. [Google Scholar]
- 13. Liese A., Seelbach K., Wandrey C. (Eds.), Industrial Biotransformations. Wiley‐VCH, Weinheim: 2006. [Google Scholar]
- 14. Dunn, M. F. , Niks, D. , Ngo, H. , Barends, T. R. et al., Tryptophan synthase: the workings of a channeling nanomachine. Trends Biochem. Sci. 2008, 33, 254–264. [DOI] [PubMed] [Google Scholar]
- 15. Hyeon, J. E. , Jeon, S. D. , Han, S. O. , Cellulosome‐based, Clostridium‐derived multi‐functional enzyme complexes for advanced biotechnology tool development: Advances and applications. Biotechnol. Adv. 2013, 31, 936–944. [DOI] [PubMed] [Google Scholar]
- 16. Hirakawa, H. , Haga, T. , Nagamune, T. , Artificial protein complexes for biocatalysis. Top. Catal. 2012, 55, 1124–1137. [Google Scholar]
- 17. Iturrate, L. , Sánchez‐Moreno, I. , Doyagüez, E. G. , García‐Junceda, E. , Substrate channelling in an engineered bifunctional aldolase/kinase enzyme confers catalytic advantage for C–C bond formation. Chem. Commun. 2009, 13, 1721–1723. [DOI] [PubMed] [Google Scholar]
- 18. Gao, X. , Yang, S. , Zhao, C. , Ren, Y. et al., Artificial Multienzyme supramolecular device: highly ordered self‐assembly of oligomeric enzymes in vitro and in vivo. Angew. Chemie Int. Ed. 2014, 53, 14027–14030. [DOI] [PubMed] [Google Scholar]
- 19. Fu, J. , Yang, Y. R. , Johnson‐Buck, A. , Liu, M. et al., Multi‐enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nanotechnol. 2014, 9, 531–536. [DOI] [PubMed] [Google Scholar]
- 20. Zhang, Y. H. P. , Substrate channeling and enzyme complexes for biotechnological applications. Biotechnol. Adv. 2011, 29, 715–725. [DOI] [PubMed] [Google Scholar]
- 21. Pettersson, H. , Pettersson, G. , Kinetics of the coupled reaction catalysed by a fusion protein of β‐galactosidase and galactose dehydrogenase. Biochim. Biophys. Acta 2001, 1549, 155–160. [DOI] [PubMed] [Google Scholar]
- 22. Sührer, I. , Haslbeck, M. , Castiglione, K. , Asymmetric synthesis of a fluoxetine precursor with an artificial fusion protein of a ketoreductase and a formate dehydrogenase. Process Biochem. 2014, 49, 1527–1532. [Google Scholar]
- 23. Arnold, K. , Bordoli, L. , Kopp, J. , Schwede, T. , The SWISS‐MODEL workspace: a web‐based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [DOI] [PubMed] [Google Scholar]
- 24. Roy, A. , Kucukural, A. , Zhang, Y. , I‐TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Conrado, R. J. , Varner, J. D. , DeLisa, M. P. , Engineering the spatial organization of metabolic enzymes: mimicking nature's synergy. Curr. Opin. Biotechnol. 2008, 19, 492–499. [DOI] [PubMed] [Google Scholar]
- 26. Kim, Y. M. , Ko, E. A. , Kang, H. K. , Kim, D. , Construction, expression and characterization of fusion enzyme from Arthrobacter oxydans dextranase and Klebsiella pneumoniae amylase. Biotechnol. Lett. 2009, 31, 1019–1024. [DOI] [PubMed] [Google Scholar]
- 27. Kufner, K. , Lipps, G. , Construction of a chimeric thermoacidophilic beta‐endoglucanase. BMC Biochem. 2013, 14, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li, B. , Li, Y. , Bai, D. , Zhang, X. et al., Whole‐cell biotransformation systems for reduction of prochiral carbonyl compounds to chiral alcohol in Escherichia coli . Sci. Rep. 2014, 4, 10.1038/srep06750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hölsch, K. , Weuster‐Botz, D. , Enantioselective reduction of prochiral ketones by engineered bifunctional fusion proteins. Biotechnol. Appl. Biochem. 2010, 56, 131–140. [DOI] [PubMed] [Google Scholar]
- 30. Yu, K. , Liu, C. , Kim, B. G. , Lee, D. Y. , Synthetic fusion protein design and applications. Biotechnol. Adv. 2015, 33, 155–164. [DOI] [PubMed] [Google Scholar]
- 31. Elleuche, S. , Bringing functions together with fusion enzymes—from nature's inventions to biotechnological applications. Appl. Microbiol. Biotechnol. 2015, 99, 1545–1556. [DOI] [PubMed] [Google Scholar]
- 32. Bai, Y. , Shen, W. C. , Improving the oral efficacy of recombinant granulocyte colony‐stimulating factor and transferrin fusion protein by spacer optimization. Pharm. Res. 2006, 23, 2116–2121. [DOI] [PubMed] [Google Scholar]
- 33. Bergeron, L. M. , Gomez, L. , Whitehead, T. A. , Clark, D. S. , Self‐renaturing enzymes: design of an enzyme‐chaperone chimera as a new approach to enzyme stabilization. Biotechnol. Bioeng. 2009, 102, 1316–1322. [DOI] [PubMed] [Google Scholar]
- 34. Menzel, A. , Werner, H. , Altenbuchner, J. , Gröger, H. , From enzymes to ‘designer bugs’ in reductive amination: a new process for the synthesis of L‐tert‐leucine using a whole cell‐catalyst. Eng. Life Sci. 2004, 4, 573–576. [Google Scholar]
- 35. Schirwitz, K. , Schmidt, A. , Lamzin, V. S. , High‐resolution structures of formate dehydrogenase from Candida boidinii . Protein Sci. 2007, 16, 1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lazo, N. D. , Downing, D. T. , Effects of Na2SO4 on hydrophobic and electrostatic interactions between amphipathic α‐helices. J. Pept. Res. 2001, 58, 457–463. [DOI] [PubMed] [Google Scholar]
- 37. Castellana, M. , Wilson, M. Z. , Xu, Y. , Joshi, P. et al., Enzyme clustering accelerates processing of intermediates through metabolic channeling. Nat. Biotechnol. 2014, 32, 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pettersson, H. , Olsson, P. , Bülow, L. , Pettersson, G. , Kinetics of the coupled reaction catalysed by a fusion protein of yeast mitochondrial malate dehydrogenase and citrate synthase. Eur. J. Biochem. 2000, 267, 5041–5046. [DOI] [PubMed] [Google Scholar]
- 39. Arai, R. , Ueda, H. , Kitayama, A. , Kamiya, N. et al., Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001, 14, 529–532. [DOI] [PubMed] [Google Scholar]
- 40. Lu, P. , Feng, M. G. , Bifunctional enhancement of a β‐glucanase‐xylanase fusion enzyme by optimization of peptide linkers. Appl. Microbiol. Biotechnol. 2008, 79, 579–587. [DOI] [PubMed] [Google Scholar]
- 41. Bülow, L. , Mosbach, K. , Multienzyme systems obtained by gene fusion. Trends Biotechnol. 1991, 9, 226–231. [DOI] [PubMed] [Google Scholar]
- 42. Prachayasittikul, V. , Ljung, S. , Isarankura‐Na‐Ayudhya, C. , Bülow, L. , NAD (H) recycling activity of an engineered bifunctional enzyme galactose dehydrogenase/lactate dehydrogenase. Int. J. Biol. Sci 2006, 2, 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fernandez‐Lafuente, R. , Stabilization of multimeric enzymes: strategies to prevent subunit dissociation. Enzyme Microb. Tech. 2009, 45, 405–418. [Google Scholar]
- 44. You, C. , Myung, S. , Zhang, Y. H. P. , Facilitated substrate channeling in a self‐assembled trifunctional enzyme complex. Angew. Chemie Int. Ed. 2012, 124, 8917–8920. [DOI] [PubMed] [Google Scholar]
- 45. Orita, I. , Sakamoto, N. , Kato, N. , Yurimoto, H. et al., Bifunctional enzyme fusion of 3‐hexulose‐6‐phosphate synthase and 6‐phospho‐3‐hexuloisomerase. Appl. Microbiol. Biotechnol. 2007, 76, 439–445. [DOI] [PubMed] [Google Scholar]
- 46. Chen, X. , Zaro, J. L. , Shen, W. C. , Fusion protein linkers: property, design and functionality. Adv. Drug Deliver. Rev. 2013, 65, 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li, J. , Pan, J. , Zhang, J. , Xu, J. H. , Stereoselective synthesis of L‐tert‐leucine by a newly cloned leucine dehydrogenase from Exiguobacterium sibiricum . J. Mol. Catal. B Enzym. 2014, 105, 11–17. [Google Scholar]
- 48. Liu, C. , Chin, J. X. , Lee, D. Y. , SynLinker: an integrated system for designing linkers and synthetic fusion proteins. Bioinformatics 2015, 31, 3700–3702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information