

Abstract
The oleaginous yeast Rhodosporidium toruloides has been considered as an economical lipid producer because it transforms carbohydrates from lignocellulosic hydrolyzate into triglycerides; however, R. toruloides cannot survive in hydrolyzate due to the inhibitors co‐produced by hydrolysis. We have previously reported a plasma mutagenesis‐generated mutant strain M18 that had strong tolerance for the stress environments of hydrolyzate. Here, we applied transcriptomic and proteomic approaches to analyze the global metabolic responses to the stress in hydrolyzate of R. toruloides and elucidate the tolerant mechanism of the mutant strain. The results showed that 57% genes matched and correlated well with their corresponding proteins. Five hundred and seven genes and 366 proteins had their transcription and expression levels changed, respectively, and 39 key genes with significantly changed transcription and expression levels (≥5‐fold changes) were identified. The results demonstrated that four cellular processes and their key genes are likely related to the mechanism of tolerance of M18 strain. Enhanced expression of the key genes in R. toruloides could improve the cellular stress tolerance to lignocellulosic hydrolyzate, while the altered expression of most key genes is probably not caused by mutagenesis, but induced by stressful environments of the hydrolyzate.
Keywords: Lignocellulosic hydrolyzate, Proteomics, Rhodosporidium toruloides, Transcriptomics, Tolerance
Abbreviations
- TAG
triglyceride
- ARTP
atmospheric room temperature plasma
- KEGG
Kyoto encyclopedia of genes and genomes
- GO
gene ontology
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- iTRAQ
isobaric tags for relative and absolute quantitation
- RNA‐seq
RNA sequencing
1. Introduction
Biodiesel refers to mono‐alkyl esters of long chain fatty acids produced by trans‐esterification of triglycerides (TAGs) with alcohols in the presence of different catalysts, and it is a typical form of attractive biofuel for a long time 1, 2. Conventional triglycerides resources are from animals or plants, resulting in competition with the food industry and agricultural production 3. Recently, increasing attention has been paid to microbial lipids prepared from oleaginous yeasts, bacteria, and microalgae which could be employed to produce biodiesel due to their multiple advantages and comparable properties compared with those derived from typical animal grease and plant oils 4, 5.
Some oleaginous yeast species could accumulate lipid to more than 65% of the dry cell weight using many kinds of lignocellulosic biomass such as spruce, wheat straw, or sugarcane bagasse as the carbon sources 6, 7. Under present circumstances of rapid shrink of global petroleum reserves and environmental crisis, biological conversion of renewable lignocellulosic biomass is expected to play a prominent role in reductions in dependence on fossil fuels and greenhouse gas emissions.
Rhodosporidium toruloides is one of the promising oleaginous yeasts and considered for potential applications in the lipid production 8, 9. A wide variety of substrates such as xylose, crude glycerol 9, 10, hydrolyzate of sunflower meal 10, hydrolyzate of cassava starch 11, bioethanol wastewater 12, and lignocellulosic hydrolyzate 13 can be utilized by R. toruloides as feedstock for lipid production. Rhodosporidium toruloides utilize glucose and xylose released from the lignocellulosic hydrolyzate and accumulate lipid in the nitrogen limited culture condition. In most cases, detoxification needs to be performed prior to cell culture so as to remove the toxic compounds in the hydrolyzate; R. toruloides cells may otherwise fail to grow in the hydrolyzate 13. The toxic compounds and inhibitors such as weak organic acid (formic acid, acetic acid, levulinic acid), furan derivatives (5‐hydroxymethylfurfural) and phenolic compounds (vanillin) could easily diffuse across the plasma membrane, cause harm and loss of integrity of membranes, impair ATP generation and redox balance, and eventually suppress most cellular metabolic processes 14. However, detoxification process has not been proven very effective because it is impossible to remove all the inhibitory compounds. In our previous work, several R. toruloides mutant strains obtained using atmospheric room temperature plasma (ARTP) mutagenesis showed strong tolerance to the inhibitory compounds in lignocellulosic hydrolyzate 15. Three robust mutants M11, M14, and M18 could grow and accumulate lipids in lignocellulosic hydrolyzate without detoxification. Compared with the wild‐type strain, the three R. toruloides mutants exhibited multiple resistances to the toxic or inhibitory compounds in the hydrolyzate 15. However, the resistant mechanisms remain unknown. For yeast cells, it has been concluded that there are multiple functional key genes responsible for tolerance to ethanol, salt, oxidative stress, etc. 16, 17. In those studies, high‐throughput transcriptomic and proteomic assays are often utilized for genome‐wide identification of the key genes 17, 18, 19, 20. In this study, we employed high‐throughput RNA sequencing (RNA‐seq) and isobaric tags for relative and absolute quantitation (iTRAQ) methods to illustrate global transcriptomic and proteomic profiling of the mutants, respectively. The mutant strain M18 was chosen as the hydrolyzate‐tolerant strain for transcriptomic and proteomic analysis because it exhibited the highest tolerance to the inhibitory compounds compared to other mutants (M11 and M14) 15. Theoretically, we can obtain the maximized significant difference of transcriptomic and proteomic results from M18. Then the genes with significantly changed transcriptional levels and the enhanced pathways, which possibly were involved in the tolerance to the inhibitory hydrolyzate could be illustrated.
2. Materials and methods
2.1. Strain and culture conditions
Rhodosporidium toruloides ACCC 20341 used as the wild‐type strain in this study was reserved in our lab. M18 isolated from our previous works was used as mutant strain since it had the higher tolerance for the inhibitory lignocellulosic hydrolyzates than the other mutants 15. Both of them are haploid strains. Culture conditions of R. toruloides have been described in detail previously 15. Both of the strains were grown in YEPD liquid medium (yeast extract 10 g/L, glucose 20 g/L, peptone 20 g/L) at pH 6.0, 28–30°C, and 200 rpm in flasks. Then the culture pellets of 12, 48, and 96 h were collected, washed, frozen, and stored at –80°C for further RNA isolation and total protein extraction.
2.2. RNA isolation, RNA‐seq library preparation and sequencing
RNA extraction and RNA‐seq library preparation were described in detail in our previous works 21. Total RNA was isolated and purified using RNA purification kit (Amresco Inc., USA) according to the instructions. Agilent 2100 bioanalyzer was utilized for examination of the quality of total RNA (rRNA 28 s/18 s). All the RNA samples were performed for RNA‐seq library preparation and direct‐sequencing on the Illumina platform. Less than 10 μg total RNA from lag phase (12 h), logarithmic phase (48 h), and stationary phase (96 h) captured by magnetic oligo (dT) beads was used for cDNA synthesis. The random hexamer primers and reverse transcriptase were utilized for first‐strand cDNA generation, and the second‐strand cDNA was synthesized using DNA polymerase I. After that, RNA libraries of single and paired‐end were constructed. The whole procedure was following Illumina's standard protocols.
2.3. Annotation and data analysis
Several alignment programs and database specifically for annotation of the yeast transcriptomic data were considered for this study, including KEGG, COG, and SOAP. The essential Gene Ontology (GO) terms were especially assigned by Blast2GO 22 through a search of the NR database. The gene transcriptional patterns and the abundance of a particular transcript relative to controls are our desired information in this study. Hierarchical analysis of the RNA sequencing data was described previously 23 using the software Cluster 3.0. The RNA‐seq results were estimated by Cluster 3.0 according to the comparison of transcriptional profiling of genes to the correlative controls. The software Java TreeView was used for display of cluster results of each gene.
2.4. Quantitative real‐time RT‐PCR analysis
The RNA samples for quantitative real‐time RT‐PCR analysis were collected from cells grown under the same growth condition as described in RNA‐seq transcriptomic analysis. The RT‐qPCR reaction was performed in each reaction containing 25 μL of 2× SYBR Premix Ex Taq (TaKaRa, Japan), 4 μL of each cDNA samples, 0.5 mL of primers at 5 mM, and dissolved in sterile distilled water. GAPDH gene of R. toruloides was selected as control for normalizing expression of the samples. All the quantitative PCR reactions were carried out employing the Mx3000P (Agilent Technologies, US) with SYBR fluorescence signal detection. Data from the assay were converted to the formula as fold changes , while Ct represents the threshold cycle 24, 25. Data were determined through triplicate independent experiments and statistical significance was considered at p < 0.05.
2.5. Total protein extraction and iTRAQ sample preparation
Total protein of R. toruloides cells of each phase was extracted in lysis buffer containing 2 M thiourea, 7 M urea, 4% CHAPS at pH 8.5, coupled with PMSF, 2 mM EDTA, and 10 mM DTT. The suspension was sonicated at 200 W for 25 min and then centrifuged at 4°C, 30 000 × g for 15 min. The supernatant was mixed well with chilled acetone containing 10% (v/v) TCA. After centrifugation, pellets were re‐suspended in iTRAQ dissolution buffer (0.5 M TEAB) containing 0.5% RapiGest (Waters, USA) and the total protein concentrations were determined using the BCA Protein Assay Kit (Pierce, USA) 26. In this process, each sample of three phases was digested at 37°C for 20 h, and peptides were reconstituted in 0.5M TEAB according to the protocol for 8‐plex iTRAQ reagent. After that, the strong‐cation exchange (SCX) chromatography was employed with LC20AB HPLC Pump (Shimadzu, Japan) system. Then peptides were eluted through a gradient of buffer (10, 30, 1 min for A, 15–60% B containing 1 M KCl and 25 mM NaH2PO4 dissolved in 25% acetonitrile at pH 2.7, and 60–100% B, respectively). Strata X C18 column (Phenomenex, USA) was employed for the desalting process of the eluted peptides, which were pooled into 18–20 fractions.
2.6. Mass spectrometric analysis
Triple‐TOF 5600 System equipped with a Nanospray III source (AB SCIEX, ON) was utilized for mass spectrometry analysis. Data were acquired using an ion spray of 2.5 kV (30 psi curtain gas and 15 psi nebulizer gas). Triplicate independent LC‐ESI‐MS/MS injections were performed for all samples. 35 ± 5 eV of the sweeping collision energy coupled with iTRAQ rolling collision energy was employed to all the precursor ions. Proteins identification was performed by using MASCOT search engine (Matrix Science, UK). To reduce the identification of false peptides, only samples with scores (≥20) at the 99% confidence interval by MASCOT probability analysis greater than “identity” were counted as identified. It was acceptable that one protein correlative to at least two unique peptides. The quantitative ratios of protein were analyzed and normalized by the median ratio in MASCOT. We used ratios with p‐values <0.05, and fold changes of ≥2 were considered as significant. Large quantity of information such as details about genomes, proteins, and metabolic pathways could be reconstructed and analyzed using in silico modeling 8. Here the functional annotations of the proteins were conducted using Blast2GO program against the nonredundant protein database (NR; NCBI). The KEGG database (http://www.genome.jp/kegg/) and the COG database (http://www.ncbi.nlm.nih.gov/COG/) were used to classify and group these identified proteins.
3. Results and discussion
3.1. RNA sequencing and annotation
The transcriptome of wild type R. toruloides and the mutant hydrolyzate‐tolerant strain M18 at 12, 48, and 96 h were sequenced using RNA‐seq by Illumina platform. A total of more than 76 million raw sequencing short reads was obtained from the RNA‐seq transcriptomics analysis from six samples of each strain. After data filtering process of eliminating reads with low‐quality bases (such as reads shorter than 20 bp or over 40 bp) and the reads mapped to noncoding RNA of R. toruloides, it was found that about 82% of the sequences were available to be identified. The numbers of genome‐mapped reads of wild‐type R. toruloides at 12, 48, and 96 h were 1 646 486, 1 726 750, and 1 503 797, compared to numbers of M18 of 1 159 192, 1 314 876, and 1 872 243. With each sequence annotation and transcriptional profiling analysis, all of these samples have the mapped ratio larger than 50% (Supporting Information Table S1). Reproducibility between biological replicates of wild‐type and the mutant strain samples at three time points were performed and it was demonstrated that good quality of cDNA reads had been obtained with the correlation coefficient between 0.96 and 0.99. Sequencing saturation analysis that was used for estimating the transcriptome coverage for the six datasets was carried out subsequently (Supporting Information Fig. S1). Using RNA‐seq alignment combined with de novo gene prediction, we found that the sequence reads matched to all 7898 coding genes, indicating that the RNA sequencing of each unique gene is enough to cover almost all the identified transcripts in R. toruloides cells.
Six thousand four hundred and seventy six coding genes, about 82% of all the identified genes, have been annotated using the GO categories. On the basis of the different global transcriptional profiling of the two strains and gene annotation by GO, the key genes that influenced resistance of the mutant R. toruloides strain M18 to hydrolyzate could be illustrated. In order to avoid the transcriptional changes caused by the response to growth phases, we performed expression analysis of R. toruloides cultured at lag (12 h), exponential (48 h), and stationary phases (96 h). The abundance of the filtered genes in the dataset was determined according to the number of transcripts per million (TPM) of distinct clean tags from the RNA sequencing data. The transcriptional quantification and identification were confirmed by mapping the clean tags to the unique genes and the results demonstrated that transcript level of 507 genes were significantly changed, including 181 upregulated and 326 downregulated, respectively (Fig. 1). The false discovery rates (FDR) <0.01 and the value of fold change ≥2 were used to determine the statistical significance of gene transcription.
Figure 1.
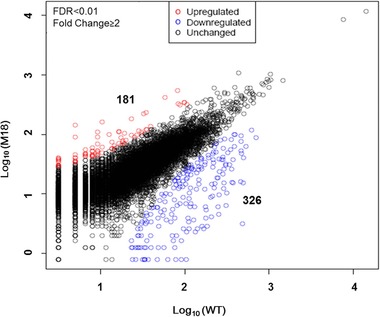
Global profiling of the genes with changed transcription. Five hundred and seven genes were significantly changed, including 181 upregulated and 326 downregulated, respectively.
3.2. Transcriptomics analysis
Using a threshold of two‐fold change and the GO categories, we determined that 65 and 56 genes of the mutant M18 with identified specific functions were significantly up‐ and downregulated compared to the wild‐type R. toruloides, respectively (Tables 1 and 2). These genes with significantly changed transcript levels were found involved in metabolism (glycolysis, sucrose, and amino acid metabolism), cellular processes (RNA splicing and processing, oxidation‐reduction, oxidative phosphorylation, and cell division), DNA repair, response to stress, and so on. The up‐ and downregulated genes were divided into seven and six categories, respectively, according to their cellular processes or molecular functions. An overview of the transcriptional profiling of each category indicated that the alterations of transcript levels at logarithmic phase were much higher than the other two phases. This is consistent with several previous studies 19, 21, 27. Although the initial transcription abundance was not high, 87.7% of the upregulated genes and 64.3% of the downregulated genes of M18 at logarithmic phase were found expressed at least two times higher than the wild‐type. The gene coding an MFS general substrate transporter was upregulated 63.1‐fold, with the highest expression of all other genes; the lowest expressed gene, which was identified as being involved in starch and sucrose metabolism was downregulated 42.33‐fold according to the GO categories.
Table 1.
The genes with significantly upregulated transcription levels of R. toruloides mutants compared with wild‐type studied in this work
| Gene | Fold change of expression levels | Functional description | ||
|---|---|---|---|---|
| Lag phase | Logarithmic phase | Stationary phase | ||
| DNA repair | ||||
| Mo06397 | 5.7 | 46.63 | 32.1 | DNA repair; response to DNA damage |
| HTP7 | 1.41 | 21.26 | 10.45 | DNA primase activity |
| RAD5 | 7.22 | 17.03 | 10.9 | DNA binding; helicase activity |
| APE2 | 3.8 | 42.12 | 19.23 | Exodeoxyribonuclease III |
| APE1 | 2.17 | 6.36 | 8.93 | Exodeoxyribonuclease III |
| polα | 6.67 | 14.79 | 8.09 | Subunit α of DNA polymerase |
| polδ | 9.03 | 10.18 | 8.89 | Subunit δ of DNA polymerase |
| PCNA | 5.77 | 41.52 | 33.86 | Proliferating cell nuclear antigen |
| Fen1 | 7.18 | 30.46 | 36.8 | Flap endonuclease‐1 |
| Lig1 | 5.17 | 30.34 | 20.91 | DNA ligase |
| Lig | 4.11 | 9.19 | 17.49 | DNA ligase |
| Transferase | ||||
| MFS | 2.69 | 63.1 | 20.66 | MFS general substrate transporter |
| MFS2 | 6.05 | 28.97 | 14.5 | Transmembrane transport |
| MFS3 | 1.59 | 22.87 | 27.8 | Transferase activity |
| HTP12 | 7.79 | 22.69 | 10.03 | Transmembrane transport |
| HTP5 | 4.38 | 19.44 | 6.67 | Transferase activity |
| NUP | 2.56 | 18.45 | 11.2 | Transmembrane transport |
| MFS4 | 9.14 | 17.03 | 26.1 | Transmembrane transport |
| MFS5 | 2.06 | 14.73 | 16 | Spermidine transport |
| GTF | 2.56 | 4.86 | 4.33 | Glycosyltransferase family |
| TPP | 4.11 | 8.55 | 10.4 | Intracellular protein transport |
| MFS9 | 9.92 | 12.99 | 12.12 | Transmembrane transport |
| Glycolysis and citrate cycle | ||||
| PGM | 14.23 | 12.93 | 5.56 | Phosphoglucomutase |
| ALDH | 9.5 | 19.52 | 30.5 | Aldehyde dehydrogenase family 7 member |
| Mo02457 | 7 | 18.19 | 5.98 | Catalytic activity |
| PI12 | 8.1 | 16.16 | 13.33 | Hydrolase activity |
| GPI8 | 6.45 | 13.24 | 2.21 | Cysteine‐type peptidase activity |
| Pck1 | 3.71 | 12.65 | 11.56 | Phosphoenolpyruvate carboxykinase |
| ADH | 4.25 | 4.98 | 5.5 | Alcohol dehydrogenase |
| MAPK signaling pathway | ||||
| PKC | 1.67 | 36.33 | 25.64 | Protein kinase activity |
| Ste20 | 11.29 | 14.3 | 6.76 | P21‐activated kinase 1 |
| HTP23 | 8.4 | 21.51 | 9.53 | Ras GTPase activator activity |
| PPG4 | 3.12 | 7.05 | 7.72 | Protein tyrosine/serine/threonine phosphatase activity |
| Ssk2 | 10.28 | 19.77 | 15.54 | Cytochrome c oxidase assembly protein COX19 |
| Cell division | ||||
| HTP9 | 7.05 | 30.09 | 7.86 | Cell adhesion |
| HEC | 3.12 | 26.6 | 22.2 | Attachment of spindle involved in homologous chromosome segregation |
| FA58C | 5.01 | 18.83 | 12.65 | Spindle assembly involved in mitosis |
| SPC | 6.2 | 18.57 | 4.73 | C2H2 zinc finger domain binding |
| HTP19 | 3.39 | 13.33 | 15.26 | Nucleosome assembly |
| RNA splicing and processing | ||||
| UPP2 | 7.94 | 45.5 | 26.3 | RNA splicing |
| Snu13 | 4.83 | 7.68 | 7.72 | U4/U6 small nuclear ribonucleo‐protein |
| Lsm | 2.35 | 21.28 | 13.75 | U6 snRNA‐associated Sm‐like protein |
| Mo01536 | 6.26 | 16.34 | 15.76 | mRNA processing |
| Prp18 | 7.1 | 15.86 | 5.94 | pre‐mRNA‐splicing ATP‐dependent RNA helicase |
| LIGA | 9.02 | 28.22 | 9.18 | Nuclear mRNA splicing, via spliceosome |
| TAR | 5.82 | 38.54 | 30.2 | Transcript antisense to ribosomal RNA protein |
| Prp28 | 14.53 | 13.18 | 3.7 | pre‐mRNA ‐splicing ATP‐dependent RNA helicase |
| HTP2 | 6.56 | 20.53 | 8.82 | Regulation of RNA splicing; RNA binding |
| LOC | 2.98 | 18.56 | 24.5 | Nuclear speck; RNA splicing |
| PPG4 | 9.94 | 13.87 | 9.95 | mRNA processing |
| CSAF | 7.09 | 9.54 | 19.56 | C2H2 zinc finger domain binding |
| PPG6 | 4.32 | 19.92 | 11.27 | RNA splicing |
| SF3b | 11.63 | 19.02 | 15.1 | Pre‐mRNA‐splicing factor ini1 |
| Response to stress | ||||
| SMC5 | 8.59 | 28.59 | 20.14 | Response to DNA damage stimulus |
| ART | 12.03 | 24.99 | 18.63 | Oxidoreductase activity, acting on CH‐OH group of donors |
| LDS | 6.85 | 10.81 | 12.36 | Response to oxidative stress |
| ESR | 17.24 | 16.95 | 9.97 | Steroid biosynthetic process |
| UPP | 2.27 | 15.54 | 19.1 | Invasive growth in response to glucose limitation |
| PSD | 9.18 | 12.88 | 6.2 | Phosphatidylserine decarboxylase |
| ACD | 8.85 | 12.87 | 10.31 | Acyl‐CoA dehydrogenase |
| EXP | 2.03 | 5.98 | 4.41 | Expressed protein |
| Mo03213 | 9.28 | 13.3 | 15.24 | Cellular response to stress |
Table 2.
The genes with significantly downregulated transcription levels of R. toruloides mutants compared with wild‐type studied in this work
| Gene | Fold change of expression levels | Functional description | ||
|---|---|---|---|---|
| Log phase | Stationary phase | Logarithmic phase | ||
| Oxidation‐reduction | ||||
| MNSOD | 20.92 | 40.56 | 40.23 | Oxidoreductase activity |
| CYTB | 12.32 | 32.89 | 38.35 | Respiratory electron transport chain |
| Mo01889 | 4.67 | 30.2 | 13.82 | Oxidoreductase activity, acting on the CH‐CH group of donors |
| ND1 | 12.12 | 27.63 | 10.6 | Oxidoreductase activity |
| HTP6 | 6.07 | 15.61 | 27.6 | Oxidoreductase activity |
| COX1 | 10.36 | 25.49 | 12.8 | Electron carrier activity; oxidoreductase activity |
| MGN | 8.16 | 9.96 | 18.12 | Monooxygenase activity; oxidoreductase activity |
| COX14 | 2.54 | 8.02 | 7.34 | Electron carrier activity; Oxidoreductase activity |
| MGN6 | 3.34 | 11.53 | 14.59 | Oxidoreductase activity |
| RBCS | 3.21 | 19.1 | 10.2 | Ribulose‐bisphosphate carboxylase activity |
| SCD | 2.89 | 15.69 | 9.44 | 3‐oxoacyl‐acyl‐carrier‐protein reductase (NADPH) activity |
| NBP | 8.35 | 13.2 | 18.2 | Oxidoreductase activity |
| Mo01112 | 2.16 | 10.3 | 11.5 | Oxidoreductase activity |
| NBP | 2.98 | 9.19 | 20.9 | Oxidoreductase activity |
| OMO | 3.92 | 8.64 | 6.26 | Oxidoreductase activity |
| HTP15 | 4.37 | 12.86 | 4.31 | O‐methylsterigmatocystin oxidoreductase |
| NAH | 3.54 | 3.15 | 2.77 | Oxidoreductase activity |
| Starch and sucrose metabolism | ||||
| PGM2 | 2.08 | 42.33 | 23.1 | Phosphoglucomutase activity |
| FSG | 1.59 | 13.16 | 16.5 | β‐fructofuranosidase, glycoside hydrolase family |
| TIF2 | 24.70 | 25.2 | 12.57 | Catalytic activity |
| BGS | 5.81 | 11.7 | 16.2 | 1,3‐beta‐glucan synthase |
| ACT | 2.16 | 17.9 | 14.47 | Amino acid transmembrane transporter activity |
| CRP | 10.43 | 12.3 | 20.2 | Inorganic cation transmembrane transporter activity |
| CAT | 3.17 | 5.27 | 8.4 | Lactate transmembrane transporter activity |
| HTP4 | 7.44 | 8.53 | 3.26 | Actin cytoskeleton organization |
| Amino acid metabolism | ||||
| HisF | 2.39 | 4.34 | 19.5 | Glutamine amidotransferase |
| PCI4 | 5.22 | 20.02 | 16.2 | Carboxamide isomerase |
| PPAP41 | 3.4 | 12.23 | 17 | Phosphoribosyl‐ATP pyrophosphohydrolase |
| ASL | 1.71 | 4.69 | 9.56 | Argininosuccinate lyase |
| GDD1 | 2.65 | 8.85 | 6.13 | Glutamate dehydrogenase |
| SAFC2 | 6.63 | 7.24 | 3.9 | Aspartate aminotransferase, cytoplasmic |
| PIPA | 4.67 | 3.03 | 5.2 | Proline iminopeptidase |
| DAT | 2.13 | 3.74 | 4.85 | Diamine N‐acetyltransferase |
| TAF1 | 2.4 | 4.98 | 2.33 | Tyrosine aminotransferase |
| Oxidative phosphorylation | ||||
| Mo06061 | 4.78 | 20.4 | 12.3 | Establishment of mitochondrion localization |
| RTG2 | 2.12 | 18.5 | 11.2 | Protein phosphorylation |
| ATPI | 5.26 | 14.9 | 9.39 | ATP synthesis coupled proton transport |
| ATP9 | 4.32 | 12.5 | 5.86 | Hydrogen ion transmembrane transporter activity |
| ATPE | 12.41 | 8.33 | 6.12 | ATP synthesis coupled proton transport |
| STA | 6.76 | 6.2 | 3.13 | Ras GTPase activator activity |
| Protein binding and processing | ||||
| PPG3 | 4.39 | 28.23 | 22.3 | Protein binding |
| ALG | 7.75 | 25.5 | 21.54 | Calnexin, protein processing |
| GFA | 2.51 | 14.1 | 13.78 | Formaldehyde‐activating enzyme |
| SUR7 | 2.2 | 6.85 | 11.52 | Protein binding; |
| RNA binding & transcription | ||||
| PPG4 | 5.89 | 20.33 | 12.09 | mRNA processing |
| RPS7 | 6.5 | 17.56 | 7.4 | RNA binding |
| GRP | 9.45 | 11.9 | 9.22 | Nucleic acid binding |
| RPL2 | 1.34 | 8.83 | 4.52 | RNA binding |
| PMAR | 10.2 | 5.98 | 7.06 | RNA binding |
| PPG47 | 9.11 | 3.77 | 4.54 | C2H2 zinc finger domain binding |
In comparison of transcriptional patterns of the wild‐type R. toruloides and the mutant M18, all of the 121 significantly changed genes have been clustered with average linkage using Cluster 3.0 (Fig. 2). Transcription dynamic relations of these clustered genes respond to ARTP mutagenesis were illustrated by the hierarchical clustering analysis. It was found that highly expressed genes ALDH, Pck1, and PGM related to glycolysis shared the significant similar transcriptional patterns with CSAF, HTP2, SF3b, and LOC which are involved in RNA splicing and processing. Figure 2 shows that transcriptome alterations in RNA splicing and processing are much higher than the former. Thus RNA splicing and processing appears to predominantly affect expression levels of glycolysis. Genes of response to stress (ESR, PSD, SMC5, and EXP) and DNA repair also shared significant similarities in patterns of transcription, suggesting their relevance for the development of the resistance to cell injury caused by ARTP. However, most highly expressed genes classified as response to stress according to GO categories are relevant to anti‐oxidative, oxidation‐reduction process or carbon source limitation. Gasch et al. have identified many genes in Saccharomyces cerevisiae that are highly activated or repressed in response to a few oxidants using genome‐wide surveys of changes in transcripts and proteins 28. Therefore it is understandable to find that 17 genes involved in oxidation‐reduction process were significantly downregulated in M18 strain (Table 2).
Figure 2.

Hierarchical cluster analyses for comparison of the transcriptional profiling of R. toruloides and the mutant M18 strains. For each gene, we calculated the logarithm (base 2) of the ratio between wild‐type and M18 at 12, 48, and 96 h. Using a threshold of two‐fold change and the GO categories, 65 and 56 genes of the mutant M18 with identified functions were significantly up‐ and downregulated compared to the wild‐type R. toruloides, respectively. The bottom color bar represents the transcript level of each gene, which red and blue denote to upregulated and downregulated, respectively.
In addition, transcriptional levels of the genes related to oxidative phosphorylation were also remarkably downregulated, demonstrating that the cellular respiration probably had been repressed after ARTP treatment. In yeast, toxic chemical compounds or mutagens always affect cell growth through inhibiting respiration activity, which sometimes leads to negative influences on the oxidation‐reduction process to a certain extent 29. Compared with other clustered downregulated genes, PGM2, FSG, BGS, TIF2, and CAT had the most relevant and key roles in starch and sucrose metabolism. They had the similarity in transcriptional patterns with the genes (e.g. PCI4, ASL, and SAFC2) involved in amino acids metabolism. It is illustrated that carbohydrate metabolism and amino acid metabolic processes of Saccharomyces pastorianus had been downregulated during autolytic process 30. These results indicate that genes involved in amino acid metabolism or carbohydrate (starch and sucrose) metabolism possibly play a role on cell death or autolytic process as exposed to various stresses (e.g. mutagens, oxidative stress, salt, and osmotic stress).
To validate the high‐throughput RNA sequencing data, 20 genes were selected at random for quantitative real‐time PCR analysis (Supporting Information Fig. S2). qPCR analysis was performed for the genes between the mutant M18 and wild‐type strain as control for all three phases. We observed the transcriptional levels of 85% genes tested by RNA‐Seq were higher than that of qPCR analysis. However, the results showed good concordance between qPCR and RNA‐Seq transcriptomics data (with correlation coefficient of 0.78).
3.3. Comparative analyses of transcriptomic and proteomic profiling
Proteomic analysis of R. toruloides cell samples by 2D‐LC–MS/MS approach has been used in numerous studies concerning about lipid accumulation 31, 32, 33 and lipid droplets formation and evolution 34. Zhu et al. used comparative proteomic analysis to illustrate the nitrogen metabolism and lipid accumulation under different nitrogen limitation conditions at the lipid‐production stage in R. toruloides 31. Chen's group demonstrated the essential proteins of R. toruloides correlated to lipid synthesis and regulation using 2D‐LC–MS/MS approach has been applied for protein profiling together with iTRAQ labeling method 33. It is obtained some insights into the process of lipid accumulation profile during different stages in the cell cycle using the strategy of proteomic analysis and comparative study of R. toruloides 32, 34.
In this study, 4081 proteins of R. toruloides have been identified and annotated by proteomic COG analysis. These identified proteins are classified as biological process and molecular function including 19 and 8 categories, respectively. Each category and its number of proteins are illustrated in Fig. 3A. According to the genomic data of R. toruloides, 47.9% of the annotated genes could be confirmed at the proteomic level. Under the “≥2 fold‐change rule”of transcriptomic and proteomic analysis, there are 507 genes and 366 proteins with significantly changed transcriptional and expression levels, respectively (Fig. 3B). However, the results showed that more than 50% genes matched and correlated well with their corresponding proteins, and transcriptional levels of 39 genes are significantly changed (≥5 fold‐change), among which 29 proteins are upregulated and 10 are downregulated. It is well known that RNA transcription and protein abundance are not always correlated well 35, and the potential reasons for this inconsistency are assumed that translational regulation, difference in protein half‐lives in vivo, or significant levels of experimental error including differences with respect to the experimental conditions being compared led to the lack of a strong correlation between transcriptomic and proteomic datasets 36, 37. Thus it is probably that these identified and correlated genes obtained by multi‐omics analysis play a key role in cellular resistance of mutant M18 to the inhibition of lignocellulose hydrolyzate. On the basis of COG database and KEGG pathway analysis, we reconstructed the four cellular processes (i.e. nucleotide excision repair, glycolysis, spliceosome assembly, and MAPK signaling) in which most well‐correlated proteins on transcriptomic and proteomic analysis are involved, and highlighted the significantly highly expressed proteins in these processes (Fig. 4).
Figure 3.

Illustration of differential proteomic profiling of R. toruloides wild‐type and M18 strains. (A) GO enrichment and COG analysis of differentially expressed proteins, which are classified as biological process and molecular function. (B) Venn diagram showing the shared 29 upregulated and 10 downregulated genes are matched and correlated well with their corresponding proteins.
Figure 4.

On the basis of COG database and KEGG pathway analysis, four cellular processes (i.e. nucleotide excision repair, glycolysis, spliceosome assembly, and MAPK signaling) which are likely related to mechanism of hydrolyzate‐tolerance of M18, have been identified and illustrated. The key significantly highly expressed proteins are highlighted in these processes. PGM, phosphoglucomutase; Pck1, phosphoenolpyruvate carboxykinase; ALDH: aldehyde dehydrogenase family 7 member; ADH, alcohol dehydrogenase; Sm, small nuclear ribonucleoprotein E; Lsm, U6 snRNA‐associated Sm‐like protein; SF3b, pre‐mRNA‐splicing factor ini1; Snu13, U4/U6 small nuclear ribonucleoprotein; Prp16, 18, 28, pre‐mRNA‐splicing ATP‐dependent RNA helicase; APEX, Exodeoxyribonuclease III; PCNA, proliferating cell nuclear antigen; Pol α, β, δ, subunits of DNA polymerase; Fen1, flap endonuclease‐1; Lig, DNA ligase; Ste20, p21‐activated kinase 1; Ssk2, cytochrome c oxidase assembly protein COX19.
Expression of 11 proteins related to nucleotide excision repair in M18 strain were upregulated at least ten times higher than the wild‐type, showing that these proteins are probably utilized for cell renovation from DNA damage after ARTP mutagenesis. The expression levels of these proteins were consistent with the corresponding transcriptional data of the genes involved in “DNA repair” in Table 1. In addition to DNA ligase (Lig and Lig1) and endonuclease‐1 (Fen1), several subunits of DNA polymerase (polα and polδ) and exodeoxyribonuclease III (APEI and APEII) were also highly expressed. It is easy to understand that enhanced expression of these DNA repair genes was the direct reactions of cells exposed to environmental mutagens. Actually transcription‐coupled DNA repair has been reported as an effective approach of nucleotide excision repair dedicated to removal of DNA damage in the actively transcribed genes in yeast strains 38. A number of anti‐oxidant and DNA repair processes have been characterized by isolating mutants affected by anti‐oxidants and resistant to stress, or genes that confer resistance when overexpressed 39, suggesting that overexpression of some key genes of nucleotide excision repair process could improve the response to stress.
Four proteins, phosphoglucomutase (PGM), phosphoenolpyruvate carboxykinase (Pck1), aldehyde dehydrogenase (ALDH), and alcohol dehydrogenase (ADH), which are related to glycolysis and citric acid cycle have been upregulated at least five times in the M18 strain (Fig. 4). The glucose 1‐phosphate is converted to glucose 6‐phosphate by PGM before glucose enters the glycolysis. Although PGM catalyzes a reversible reaction, it would facilitate the interconversion of glucose 6‐phosphate from glucose 1‐phosphate according to the KEGG pathway analysis of M18 in this study. Actually, metabolic fate of glucose 6‐phosphate depends on the needs of the cell at the time it is generated. If the cell is low on energy, glucose 6‐phosphate will travel down the glycolytic pathway, eventually yielding two ATP molecules 40. It is concluded that the conversion of glucose 1‐phosphate to glucose 6‐phosphate by PGM would be enhanced to require more energy to relieve the stress when cell is exposed to the inhibition of the toxic hydrolyzate. Similar to the PGM, phosphoenolpyruvate carboxy kinase 1 (Pck1) is a key regulatory enzyme driving gluconeogenesis and converting oxaloacetate into phosphoenolpyruvate and carbon dioxide, whereas Pck1 catalyze the opposite direction in M18 on the basis of KEGG analysis. Obviously the result shows here that the additional catabolic flux conducted into the citric acid cycle was catalyzed by Pck1, in contrast with its normal role in gluconeogenesis. Therefore, the citric acid cycle was enhanced, resulting in producing more energy to improve the stress tolerance and lesions caused by chemicals or mutagens. Actually under natural or stressful circumstances, the active citric acid cycle may enhance the fitness of microbe 41. The results and analysis suggest that the Pck1 possibly has an essential role in stress tolerance of M18 mutant strain. It is very interesting to find that aldehyde dehydrogenase (ALDH) and alcohol dehydrogenase (ADH) have been highly expressed. ALDH catalyzes the oxidation of acetate, while ADH facilitates the interconversion of aldehyde to alcohol with the reduction of NAD+ to NADH (Fig. 4). However, these two proteins are not involved in either glycolysis or citric acid cycle, indicating that they might play a role in a different pathway compared with PGM and Pck1. Acetic acid (in form of acetate in hydrolyzate) has been identified as one of the major inhibitors which could decrease lipid productivity and even change the fatty acid composition of the lipids from R. toruloides 15. Thus it seems that high of ALDH and ADH has been utilized to relieve the inhibition of the acetate in lignocellulose hydrolyzate. If this hypothesis is true, the upregulated transcription and expression of ALDH and ADH are not caused by ARTP mutagenesis, but induced by the stressful environments.
The third cellular process which most involved proteins are well‐correlated to the transcriptional data is spliceosome assembly including seven proteins (i.e. Prp16, Prp18, Prp28, Sm, Lsm, SF3b, and Snu13). These highly expressed proteins are corresponding to the genes functioning as mRNA splicing (Fig. 4). In higher eukaryotes, the process of splicing is utilized for regulation of both qualitative and quantitative aspects of gene expression 42. In fact, regulation of gene expression of mRNA splicing was always associated with cellular response to stress (e.g. exposure to toxic levels of ethanol), but different mechanisms seem to inhibit constitutive mRNA splicing and to affect alternative splicing regulation, which could be important to regulate gene expression during recovery from environmental stress 43. Alteration of the transcriptional levels of the genes involved in mRNA splicing of R. toruloides M18 might be caused by activation of the inhibitors in the hydrolyzate. Another explanation for the stress tolerance of the mutant strain is RNA processing has emerged as a novel pathway that probably contributes to maintain genome stability. Recently, there are reports that have uncovered how DNA damage induces RNA splicing changes that give rise to mRNA variants encoding different protein isoforms with the potential to affect the cellular response and the cell fate 44. An example that the DNA damage can alter RNA splicing came from the observation that Drosophila S2 cells treated with camptothecin (a topoisomerase I inhibitor) lead to replication forks arrest 45.Another example is that the S2 cells exposed to mutagen expressed splicing variants of the transcription factor TAF1 that was shown to control the G2/M transition 45. On the basis of these reports, the activation of the DNA damage might modify RNA splicing by affecting the expression or inducing some post‐translational modifications of RNA splicing factors. Thus, RNA splicing process emerges as integration of cellular stresses of lignocellulose hydrolyzate and ARTP mutagenesis for elucidating the mechanism of hydrolyzate‐tolerant R. toruloides M18 strain.
MAPK signaling pathway is assumed to be the fourth candidate cellular process that participates in regulation of the hydrolyzate‐tolerance of M18 strain (Fig. 4). The p21‐activated kinase I (Ste20 or Pak1) that modifies cell morphology in most eukaryotic cells 46, and the COX19 (Ssk2) that is a subunit of transmembrane protein of cytochrome c oxidase involved in cytochrome c oxidase assembly in bacteria and eukaryotes 47 are the only two proteins highly expressed related to MAPK signaling pathway in M18. Ste20 has been implicated in a wide range of biological activities, while the essential role of Ssk2 was demonstrated as a tight control over oxidative phosphorylation process 47. MAPK signaling pathways have been illustrated as the important regulator of transcriptional and proteomic responses to extracellular stress signals 48. In the budding yeast Saccharomyces cerevisiae, the MAPK pathways control diverse cellular processes, for instance, the high osmolarity glycerol‐1 (Hog1) pathway is activated to upregulate expression of the enzymes required for the osmolyte glycerol synthesis in response to stress of increased osmolarity 49. Thus in the R. toruloides mutant strain M18, highly expressed Ste20 and Ssk2 seemed to function in the central regulations of the MAPK signaling pathways responding to the oxidation and high osmolarity of hydrolyzate, suggesting that upregulated proteins involved in MAPK signaling pathways are likely activated by the stressful environments, not by the ARTP mutagenesis.
4. Concluding remarks
In this study, the hydrolyzate‐resistant mechanism of the mutant R. toruloides M18 strain has been investigated using transcriptomic and proteomic approaches. The multi‐omics technologies including RNA‐seq and iTRAQ enabled rapid high‐throughput analysis of mRNA sequencing and proteins quantification at different cellular levels. However, it is impossible to exactly characterize and demonstrate the complex mechanism of the stress tolerance of M18 through any single approach because RNA transcription and protein expression are not always correlated well. We combined both of the transcriptomic and proteomic analysis to illustrate that 39 genes are significantly changed on both transcription and expression levels (≥5 fold changes). Our quantitative analyses suggest that regulatory mechanisms of the key genes might act positively or negatively occur in R. toruloides strains. The 39 key genes are involved in four cellular processes such as nucleotide excision repair, glycolysis, spliceosome assembly, and MAPK signaling, suggesting these processes are likely related to the mechanism of tolerance of M18 strain. The results demonstrated that enhancement of expression of the key genes could improve the cellular response to stress of lignocellulosic hydrolyzate. However, on the basis of expression patterns, functions, and KEGG analyses, enhanced expression of genes involved in DNA repair are probably due to ARTP mutagenesis, while the other highly expressed genes related to glycolysis, mRNA splicing and MAPK signaling pathway are likely induced by stressful environments of hydrolyzate. In our recent study, we constructed the Ste20 knockout strain of M18, and results showed that Ste20 knockout individually caused 2–4% loss of the acetate tolerance but the utilization of glucose and xylose are not affected. We suppose that if the transcriptional enhanced key genes identified in the study such as pgm, pck1, adh, or prp28 can be overexpressed in the M18 strain, the cellular stress tolerance to lignocellulosic hydrolyzate of R. toruloides could be probably reinforced. This work significantly enriches the knowledge of the molecular basis of hydrolyzate‐tolerance of the R. toruloides mutant. Furthermore, it will facilitate the engineering of key gene targets of R. toruloides or other microorganisms for achieving high‐tolerance hosts and improvement of robustness and efficiency of the microbial lipids.
Practical application
In this study, the hydrolyzate‐resistant mechanism of the mutant R. toruloides M18 strain has been investigated using transcriptomic and proteomic approaches. We have previously reported a mutant strain M18 that had strong tolerance for the stress environments of hydrolyzate. Here, we analyzed the global metabolic responses to the stress in hydrolyzate of R. toruloides and elucidate the tolerant mechanism of the M18. Thirty nine key genes that are involved in four cellular processes are likely related to the mechanism of tolerance of the mutant strain. This work enriches the knowledge of the molecular basis of hydrolyzate‐tolerance of the R. toruloides mutant and could facilitate the engineering of key genes of R. toruloides for achieving high‐tolerance hosts to lignocellulosic hydrolyzate.
The authors have declared no conflict of interest.
Supporting information
Table S1. Sequencing quality evaluationof the cDNA samples ofR. toruloidesculturedatdifferentphases. Figure S1. Sequencing saturation analysis of all the reads. Figure S2. Examination and verification of the transcriptional levels of the clustered genes with real‐time RT‐PCR method.Data were determined through triplicate independent experiments and the error bars represent standard deviation.
Acknowledgments
This work was financially supported by National Natural Science Foundation of China (number 21406130) and Tsinghua Scientific Research Funding (numbers 2012Z02295 and 2012Z98148).
5 References
- 1. Latif, H. , Zeidan, A. A. , Nielsen, A. T. , Zengler, K. , Trash to treasure: Production of biofuels and commodity chemicals via syngas fermenting microorganisms. Curr. Opin. Biotechnol. 2014, 27, 79–87. [DOI] [PubMed] [Google Scholar]
- 2. Hwang, H. T. , Qi, F. , Yuan, C. , Zhao, X. et al., Lipase‐ catalyzed process for biodiesel production: Protein engineering and lipase production. Biotechnol. Bioeng. 2014, 111, 639–653. [DOI] [PubMed] [Google Scholar]
- 3. Vasudevan, P. T. , Briggs, M. , Biodiesel production – current state of the art and challenges. J. Ind. Microbiol. Biotechnol. 2008, 35, 421–430. [DOI] [PubMed] [Google Scholar]
- 4. Subramaniam, R. , Dufreche, S. , Zappi, M. , Bajpai, R. , Microbial lipids from renewable resources: Production and characterization. J. Ind. Microbiol. Biotechnol. 2010, 37, 1271–1287. [DOI] [PubMed] [Google Scholar]
- 5. Fjerbaek, L. , Christensen, K. V. , Norddahl, B. , A review of the current state of biodiesel production using enzymatic transesterification. Biotechnol. Bioeng. 2009, 102, 1298–1315. [DOI] [PubMed] [Google Scholar]
- 6. Li, Q. , Du, W. , Liu, D. , Perspectives of microbial oils for biodiesel production. Appl. Microbiol. Biotechnol. 2008, 80, 749–756. [DOI] [PubMed] [Google Scholar]
- 7. Li, Y. , Zhao, Z. , Bai, F. , High‐density cultivation of oleaginous yeast Rhodosporidium toruloides Y4 in fed‐batch culture. Enzyme. Microbiol. Technol. 2007, 41, 312–317. [Google Scholar]
- 8. Bommareddy, R. R. , Sabra, W. , Maheshwari, G. , Zeng, A. P. , Metabolic network analysis and experimental study of lipid production in Rhodosporidium toruloides grown on single and mixed substrates. Microb. Cell. Fact. 2015, 14, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tchakouteu, S. S. , Kalantzi, O. , Gardeli, C. , Koutinas, A. A. et al., Lipid production by yeasts growing on biodiesel‐derived crude glycerol: Strain selection and impact of substrate concentration on the fermentation efficiency. J. Appl. Microbiol. 2015, 118, 911–927. [DOI] [PubMed] [Google Scholar]
- 10. Leiva‐Candia, D. E. , Tsakona, S. , Kopsahelis, N. , García, I. L. et al., Biorefining of by‐product streams from sunflower‐based biodiesel production plants for integrated synthesis of microbial oil and value‐added co‐products. Bioresour. Technol. 2015, 190, 57–65. [DOI] [PubMed] [Google Scholar]
- 11. Wang, Q. , Guo, F. J. , Rong, Y. J. , Chi, Z. M. , Lipid production from hydrolysate of cassava starch by Rhodosporidium toruloides 21167 for biodiesel making. Renewable Energy 2012, 46, 164–168. [Google Scholar]
- 12. Zhou, W. , Wang, W. , Li, Y. , Zhang, Y. , Lipid production by Rhodosporidium toruloides Y2 in bioethanol wastewater and evaluation of biomass energetic yield. Bioresour. Technol. 2013, 127, 435–440. [DOI] [PubMed] [Google Scholar]
- 13. Zhao, X. , Peng, F. , Du, W. , Liu, C. et al., Effects of some inhibitors on the growth and lipid accumulation of oleaginous yeast Rhodosporidium toruloides and preparation of biodiesel by enzymatic transesterification of the lipid. Bioprocess. Biosyst. Eng. 2012, 35, 993–1004. [DOI] [PubMed] [Google Scholar]
- 14. Huang, C. , Zong, M. H. , Wu, H. , Liu, Q. P. , Microbial oil production from rice straw hydrolysate by Trichosporon fermentans. Bioresour Technol. 2009, 19, 4535–4538. [DOI] [PubMed] [Google Scholar]
- 15. Qi, F. , Kitahara, Y. , Wang, Z. T. , Zhao, X. B. et al., Novel mutant strains of Rhodosporidium toruloides by plasma mutagenesis approach and their tolerance for inhibitors in lignocellulosic hydrolyzate. J. Chem. Technol. Biot. 2014, 89, 735–742. [Google Scholar]
- 16. Okada, N. , Ogawa, J. , Shima, J. , Comprehensive analysis of genes involved in the oxidative stress tolerance using yeast heterozygous deletion collection. FEMS Yeast. Res. 2014, 14, 425–434. [DOI] [PubMed] [Google Scholar]
- 17. Thorpe, G. W. , Fong, C. S. , Alic, N. , Higgins, V. J. et al., Cells have distinct mechanisms to maintain protection against different reactive oxygen species: Oxidative‐stress‐response genes. Proc. Natl. Acad. Sci. USA 2004, 101, 6564–6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hirasawa, T. , Yoshikawa, K. , Nakakura, Y. , Nagahisa, K. et al., Identification of target genes conferring ethanol stress tolerance to Saccharomyces cerevisiae based on DNA microarray data analysis. J. Biotechnol. 2007, 131, 34–44. [DOI] [PubMed] [Google Scholar]
- 19. Ma, M. , Liu, L. Z. , Quantitative transcription dynamic analysis reveals candidate genes and key regulators for ethanol tolerance in Saccharomyces cerevisiae . BMC Microbiol. 2010, 10, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alexandre, H. , Ansanay‐Galeote, V. , Dequin, S. , Blondin, S. , Global gene expression during short‐term ethanol stress in Saccharomyces cerevisiae . FEBS Lett. 2001, 498, 98–103. [DOI] [PubMed] [Google Scholar]
- 21. Qi, F. , Wang, C. , Liu, Y. , Kaleem, I. et al., Transcriptional profiling of protein expression related genes of Pichia pastoris under simulated microgravity. Plos One 2011, 6, e26613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aparicio, G. , Gotz, S. , Conesa, A. , Segrelles, D. et al., Blast2GO goes grid: Developing a grid‐enabled prototype for functional genomics analysis. Stud. Health. Technol. Inform. 2006, 120, 194–204. [PubMed] [Google Scholar]
- 23. Eisen, M. B. , Spellman, P. T. , Brown, P. O. , Botstein, D. Cluster analysis and display of genome‐wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee, E. J. , Schmittgen, T. D. , Comparison of RNA assay methods used to normalize cDNA for quantitative real‐time PCR. Anal. Biochem. 2006, 357, 299–301. [DOI] [PubMed] [Google Scholar]
- 25. Livak, K. J. , Schmittgen, T. D. , Analysis of relative gene expression data using real‐time quantitative PCR and the 2‐ΔΔCt method. Methods, 2001, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 26. Gan, C. S. , Chong, P. K. , Pham, T. K. , Wright, P. C. , Technical, experimental, and biological variations in isobaric tags for relative and absolute quantitation (iTRAQ). J. Proteome. Res. 2007, 6, 821–827. [DOI] [PubMed] [Google Scholar]
- 27. Liu, Z. L. , Ma, M. , Song, M. , Evolutionarily engineered ethanologenic yeast detoxifies lignocellulosic biomass conversion inhibitors by reprogrammed pathways. Mol. Genet. Genomics 2009, 282, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gasch, A. P. , Spellman, P. T. , Kao, C. M. , Carmel‐Harel, O. et al., Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000, 11, 4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chambers, P. , Issaka, A. , Palecek, S. P. , Saccharomyces cerevisiae JEN1 promoter activity is inversely related to concentration of repressing sugar. Appl. Environ. Microbiol. 2004, 70, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu, W. , Wang, J. , Li, Q. , Comparative proteome and transcriptome analysis of lager brewer's yeast in the autolysis process. FEMS Yeast. Res. 2014, 14, 1273–1285. [DOI] [PubMed] [Google Scholar]
- 31. Zhu, Z. , Zhang, S. , Liu, H. , Shen, H. et al., A multi‐omic map of the lipid‐producing yeast Rhodosporidium toruloides. Nat. Commun. 2012, 3, 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu, H. , Zhao, X. , Wang, F. , Li, Y. et al., Comparative proteomic analysis of Rhodosporidium toruloides during lipid accumulation. Yeast 2009, 26, 553–566. [DOI] [PubMed] [Google Scholar]
- 33. Shi, J. , Feng, H. , Lee, J. , Ning, Chen. W. , Comparative proteomics profile of lipid‐cumulating oleaginous yeast: an iTRAQ‐coupled 2‐D LC‐MS/MS analysis. PLoS One 2013, 26, e85532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhu, Z. , Ding, Y. , Gong, Z. , Yang, L. et al., Dynamics of the lipid droplet proteome of the oleaginous yeast Rhodosporidium toruloides . Eukaryot. Cell. 2015, 14, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nie, L. , Wu, G. , Culley, D. E. , Scholten, J. C. et al., Integrative analysis of transcriptomic and proteomic data: challenges, solutions and applications. Crit. Rev. Biotechnol. 2007, 27, 63–75. [DOI] [PubMed] [Google Scholar]
- 36. Greenbaum, D. , Colangelo, C. , Williams, K. , Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome. Biol. 2003, 4, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nie, L. , Wu, G. , Brockman, F. J. , Zhang, W. , Integrated analysis of transcriptomic and proteomic data of Desulfovibrio vulgaris: zero‐inflated Poisson regression models to predict abundance of undetected proteins. Bioinformatics. 2006, 22, 1641–1647. [DOI] [PubMed] [Google Scholar]
- 38. Li, W. , Selvam, K. , Ko, T. , Li, S. Transcription bypass of DNA lesions enhances cell survival but attenuates transcription coupled DNA repair. Nucleic. Acids. Res. 2014, 42, 13242–13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krems, B. , Charizanis, C. , Entian, K. D. Mutants of Saccharomyces cerevisiae sensitive to oxidative and osmotic stress. Curr. Genet. 1995, 27, 427–434. [DOI] [PubMed] [Google Scholar]
- 40. Rhyu, G. I. , Ray, W. J. , Markley, J. L. Enzyme‐bound intermediates in the conversion of glucose 1‐phosphate to glucose 6‐phosphate by phosphoglucomutase. Phosphorus NMR studies. Biochemistry. 1984, 23, 252–260. [DOI] [PubMed] [Google Scholar]
- 41. Thomas, V. C. , Kinkead, L. C. , Janssen, A. , Schaeffer, C. R. et al., A Dysfunctional tricarboxylic acid cycle enhances fitness of Staphylococcus epidermidis during β‐Lactam Stress. MBio. 2013, 20, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Black, D. L. , Protein diversity from alternative splicing: a challenge for bioinformatics and post‐genome biology. Cell, 2000, 103, 367–370. [DOI] [PubMed] [Google Scholar]
- 43. Biamonti, G. , Caceres, J. F. , Cellular stress and RNA splicing. Trends. Biochem. Sci. 2009, 34, 146–153. [DOI] [PubMed] [Google Scholar]
- 44. Lenzken, S. C. , Loffreda, A. , Barabino, S. M. , RNA Splicing: a new player in the DNA damage response. Int. J. Cell. Biol. 2013, 2013, 153634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Katzenberger, R. J. , Marengo, M. S. , Wassarman, D. A. , ATM and ATR pathways signal alternative splicing of Drosophila TAF1 pre‐mRNA in response to DNA damage. Mol. Cell. Biol. 2006, 26, 9256–9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sells, M. A. , Boyd, J. T. , Chernoff, J. , p21‐activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J. Cell. Biol. 1999,145, 837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li, Y. , Park, J. S. , Deng, J. H. , Bai, Y. , Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J. Bioenerg. Biomembr. 2006, 38, 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hazzalin, C. A. , Mahadevan, L. C. , MAPK‐regulated transcription: a continuously variable gene switch? Nat. Rev. Mol. Cell. Biol. 2002, 3, 30–40. [DOI] [PubMed] [Google Scholar]
- 49. Gustin, M. C. , Albertyn, J. , Alexander, M. , Davenport, K. , MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1998, 62, 1264–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequencing quality evaluationof the cDNA samples ofR. toruloidesculturedatdifferentphases. Figure S1. Sequencing saturation analysis of all the reads. Figure S2. Examination and verification of the transcriptional levels of the clustered genes with real‐time RT‐PCR method.Data were determined through triplicate independent experiments and the error bars represent standard deviation.