

Abstract
The hydroxylation of tryptophan is an important reaction in the biosynthesis of natural products. 5‐Hydroxytryptophan (5HTP) is not only an important compound for its pharmaceutical value but also because it is the precursor of other molecules, such as serotonin. In this study, we have extended the metabolism of an E. coli strain to produce 5HTP. Aromatic amino acid hydroxylase from Cupriavidus taiwanensis (CtAAAH) was selected using an in silico structure‐based approach. We have predicted and selected several substrate‐determining residues using sequence, phylogenetic and functional divergence analyses; we also did rational design on CtAAAH to shift the enzyme preference from phenylalanine to tryptophan. Whole cell bioconversion assays were used to show the effect of predicted sites. In general, all of them decreased the preference toward phenylalanine and increased the tryptophan synthesis activity. The best performer, CtAAAH‐W192F, was transformed into a strain that had the tryptophanase gene disrupted and carried a human tetrahydrobiopterin (BH4) regeneration pathway. The resulting strain was capable of synthesizing 2.5 mM 5HTP after 24 hours. This work demonstrates the application of computational approaches for protein engineering and further coupling with the bacterial metabolism.
Keywords: 5‐Hydroxytryptophan, Aromatic amino acid hydroxylase, Protein engineering, Protein in silico screening, Synthetic pathway
Abbreviations
- AAAH
Aromatic amino acid hydroxylase
- FDI
Functional diverse type‐I
- FDII
Functional diverse type‐II
- 5HTP
5‐hydroxytrptophan
- ML
Maximum Likelihood
- NJ
Neighbor‐Joining
1. Introduction
5‐Hydroxytryptophan (5HTP) is a natural non‐canonical amino acid, and it is the precursor of the neurotransmitter serotonin. 5HTP has been used for clinical purposes for over 30 years since is effective for the treatment of a variety of conditions (e.g. depression, insomnia, chronic headaches, binge eating associated with obesity). Unlike serotonin, 5HTP is well absorbed from oral doses and can easily cross the blood–brain barrier 1. In terms of volume demand, 5HTP stood at 136.4 tons in 2012, which corresponds to a value of 31.7 million US dollars (http://www.transparencymarketresearch.com). A key challenge for its manufacture stands in the fact that the production depends on the extraction from seeds of the African plant Griffonia simplicifolia, and therefore the supply of the raw material is linked to seasonal and regional conditions. Chemical synthesis of 5HTP has been reported 2; however, this is not economically feasible in large scale. Therefore, the biotechnological production provides a promising alternative for 5HTP production, especially because microorganisms have simpler genetic background and metabolic network than the native producer.
To date, Escherichia coli remains as the dominant industrial microorganism producer of many complex compounds 3, and as the prime prokaryotic genetic model, E. coli also has been engineered to produce 5HTP 4, 5. In the past two decades, E. coli aromatic amino acid producers have been significantly improved with the advent of rational metabolic engineering 6, 7. The construction of tryptophan producers has driven the deep research and engineering of its pathways, a condition that entails the emergence of new and effective strategies for the production of tryptophan derivatives. Unlike natural amino acids, the biosynthesis of non‐canonical amino acids from simple sugars often requires the incorporation of an artificial metabolic pathway to expand the cell's capabilities, the optimization of this pathway usually require extensive protein engineering 8, 9.
Efforts have been made to engineer E. coli strains capable of converting tryptophan to 5HTP 4, 5. Mammalian tryptophan 5‐hydroxylase is capable of synthesizing 5HTP via tryptophan hydroxylation, but it has low activity and poor stability when expressed in prokaryotes 10; therefore, this method is not suitable for production purposes. Often, metabolic engineering uses enzymes with a non‐native substrate to perform transformations within the novel synthetic pathways. Pioneering studies have used bacterial aromatic amino acid hydroxylases (AAAH), which has a strong preference toward phenylalanine, and shifted the substrate preference for tryptophan, based on sequence comparison and modification 5, 11. Protein engineering can facilitate the tailoring of the biocatalysts by shifting the substrate specificity with high yields of the desired molecules.
The combination of crystallographic studies and the continuous progress in molecular modeling methods have opened new perspectives for structure‐based protein engineering, further comparison of the sequences and functions of related proteins can be used to identify important residues for substrate specificity 12, 13. The development of these areas allows for example the design and optimization of the binding pocket conformation guided by the features of the ligands. In the same way, point mutations could also be rationally designed and the preference of different substrates could be evaluated in silico, reducing significantly the number of mutants to be tested in the laboratory.
In this work, we aimed to expand the tryptophan pathway for the production of 5HTP. We performed protein engineering on Cupriavidus taiwanensis AAAH based on structural models generated in silico. Using sequence, phylogenetic and functional divergence analyses, we identified important residues responsible for the substrate specificity and compared the protein selectivity at both in silico and in vivo levels. The best candidate was used to elongate the tryptophan pathway in an E. coli strain which has a cofactor recycling pathway.
2. Materials and methods
2.1. Computational analyses
To identify members of the AAAH superfamily the keyword “aromatic amino acid hydroxylase” was used as a query to search against the NCBI protein reference sequence database. Hypothetical and predicted sequences were eliminated, as well as redundant entries. Amino acid sequences were aligned using MUSCLE 14, and refinements within the alignments were done manually.
Phylogenetic analyses employing the Neighbor‐Joining (NJ) method with 10 000 bootstrap values were done in MEGA v7 15. NJ trees were compared with Maximum Likelihood (ML) analysis, using the best‐fit models predicted with ProtTest v2.4 16 and constructing the tree in MEGA using a bootstrap of 1000 to assess the reliability of interior branches. Functional divergence type‐I and type‐II (FDI and FDII, respectively) analyses were done using DIVERGE v3.0 17.
Tertiary structures of bacterial AAAH were generated with the I‐TASSER server 18 and compared with homologous validated protein structures. A rotamer library was used during the structure determination 19, and side chains were refined using the Protein Database (http://www.rcsb.org) entries 1MMK and 3E2T as guides. Molecular docking analysis of tryptophan and phenylalanine using the generated AAAH (wild type and variants) were performed using AutoDock Vina v1.1.2 20. The ligands were manually placed in the active site using the substrate position in other AAAHs crystal structures as a guide, for these analyses we used a grid box of 15 × 15 × 15 Å and the level of exhaustiveness set to 10. Molecular graphics and analyses of all the structures were performed with the UCSF Chimera package 21.
2.2. DNA manipulation
Chromosomal DNA from C. taiwanensis was prepared using NucleoSpin® Tissue (Macherey‐Nagel, Germany) according to the manufacturer's instructions. The aaah gene from C. taiwanensis was amplified by PCR using the following primers (5′ to 3′): CtAAAH‐forward, TATGGATCCATGCGCATTATATGCATGATCTATCGAAATC and CtAAAH‐reverse, ATCAAGCTTTCAGATGTCTTCGGTATCGGCC. The generated fragment was cloned within the BamHI and HindIII sites of the pACYCDuet vector to yield the plasmid pCtAAAH.
Site‐direct mutagenesis experiments were carried out using QuickChange site‐directed mutagenesis kit (Stratagene, USA). The resulting plasmids were transformed separately into NEB10ß strains by CaCl2 method 22. All mutations were verified by DNA sequencing. The obtained mutant plasmids were introduced into E. coli BL21(DE3)∆tnaA. Tryptophanase A gene was knocked out using the λ recombination system 23. Details of the strains and plasmids used in this study are listed in Table 1.
Table 1.
Bacterial strains and plasmids used in this study
| Characteristics | Source | |
|---|---|---|
| Strains | ||
| C. taiwanensis | DSM No.: 17343, type strain | DMSZ |
| E. coli BL21(DE3) | fhuA2 [lon] ompT gal (λDE3) [dcm] ∆hsdS λDE3 = λsBamHIo ∆EcoRI‐B int::(lacI::PlacUV5::T7 gene1) i21 ∆nin5 | NEB |
| E. coli BL21(DE3)∆tnaA | BL21(DE3)∆tnaA | This study |
| E. coli BL21 cofactor | BL21(DE3)∆tnaA pBbE1k‐2 | This study |
| Plasmids | ||
| pACYCDuet | Novagen | |
| pBbE1k‐2 | pBbE1K inserted with DHPR, PCD | 32 |
| pCtAAAH | pACYCDuet inserted with C. taiwanensis AAAH | This study |
| pCtAAAH L113Y | pACYCDuet inserted with C. taiwanensis AAAHL113Y | This study |
| pCtAAAH W192F | pACYCDuet inserted with C. taiwanensis AAAHW192F | This study |
| pCtAAAH Y222H | pACYCDuet inserted with C. taiwanensis AAAHY222H | This study |
| pCtAAAH S223C | pACYCDuet inserted with C. taiwanensis AAAHS223C | This study |
| pCtAAAH P229A | pACYCDuet inserted with C. taiwanensis AAAHP229C | This study |
| pCtAAAH Y244C | pACYCDuet inserted with C. taiwanensis AAAHY244C | This study |
2.3. In vivo enzyme assays
The in vivo assays were carried out to evaluate the activity of CtAAAH (wild types and variants) toward phenylalanine and tryptophan. E. coli BL21(DE3)∆tnaA harboring different plasmids were cultivated in LB medium containing 50 μg/mL ampicillin. Isopropyl‐ß‐D‐thiogalactopyranoside (IPTG) to a final concentration of 0.4 mM was added to the culture when OD600 reached 0.4. Cells were then cultivated at 25°C with constant agitation at 220 rpm. After a 16 h induction period, cells were harvested by centrifugation at 3300 x g and washed twice with 100 mM HEPES‐NaOH buffer (pH 8.0).
5HTP and tyrosine were produced in a reaction mixture containing 100 mM HEPES‐NaOH buffer (pH 8.0), 4 mM L‐tryptophan or L‐phenylalanine, 4 mM BH4, 0.1 mM FeSO4, 50 mM D‐glucose, 1% (v/v) Triton X‐100 and E. coli whole cells (OD600 between 15 and 20) in a total volume of 500 μL. The reaction was performed for 30 min at 30°C. After the reaction, cells were removed by centrifugation at 12 000 x g for 10 min at 4°C, the supernatant was filtrated and stored at 4°C before HPLC analysis.
2.4. Microbial production of 5HTP
E. coli BL21(DE3)∆tnaA strain was transformed with corresponding plasmids. Batch fermentations were carried out in 300 mL baffled shake flasks with a total volume of 30 mL. Overnight LB cultures were inoculated in Seed Medium to an initial OD600 of 0.1 and grew at 37°C for 8–10 h with constant agitation. Seed Medium contained (per liter): glucose (12 g), MgSO4·7H2O (0.5 g), KH2PO4 (2 g), (NH4)2SO4 (4 g), yeast extract (5 g), monosodium citrate dehydrate (2 g), biotin (0.1 mg), DL‐calcium pantothenate (0.5 mg), ascorbic acid (176 mg) and 10 mL of 100× stock trace elements. The stock trace elements solution was composed of (per liter in 0.1 N HCl): Na2MoO4·2H2O (2.5 g), AlCl3·6H2O (2.5 g), FeSO4·7H2O (10 g), CoCl2·6H2O (1.75 g), CaCl2·2H2O (10 g), ZnSO4·7H2O (0.5 g), CuCl2·2H2O (0.25 g), H3BO3 (0.125 g), Na2MoO4·2H2O (0.5 g). Afterward, the cells were transferred to Fermentation Medium to an initial OD600 of 0.05 and grew at 30°C with constant agitation. Fermentation Medium was the same as Seed Medium, except for 1 g/L of yeast extract instead of 5 g/L, and 1 g/L of tryptophan. The pH of both media were adjusted at 6.7 using a 5M KOH solution.
2.5. Analytics
Aromatic amino acids and 5HTP were analyzed by HPLC as described in da Luz et al. 24. Samples were derivate using the AccQ‐tag method (Waters, USA) and fluorescence was detected as described in the manufacturer's manual. Measurements were done on an Ultimate‐3000 HPLC system (ThermoFisher, Germany) with a binary gradient where eluent “A” was 140 mM sodium acetate with 0.1% v/v ACN and eluent “B” was 60% v/v ACN. The gradient was as follows: at 0 min 0% B; at 0.5 min 1% B; at 21 min 12% B; at 26 min 17% B; at 55 min 33% B; and remained constant from 62 until 80 min at 100% B. All gradient changes were linear between the points given above. Separation was done using a Kinetex RP column (2.6 μm, C18, 100 × 4.6 mm, Phenomenex, USA) at 45°C, and an injection volume of 10 μL was used.
3. Results and discussion
3.1. Computational analysis and selection of an AAAH prototype
A total of 507 AAAH protein sequences were collected after the database search and selection process. The results revealed the presence of AAAH homologs in different taxa: 284 eukaryotic sequences of which the mayor group, 235 sequences, belonged to the Metazoan taxon (Animal Kingdom), the other 49 sequences were distributed among the Fungi, Viridiplantae (mosses and green algae), and Protista groups. For the Prokaryote group, 223 sequences were retrieved from the database, these sequences were distributed among different taxa: Proteobacteria, Bacteroidetes, Actinobacteria, Firmicutes and so on (Supporting Information Table 1).
To explore the phylogenetic relationship among AAAH paralogous proteins, a NJ phylogenetic tree with the 507 sequences was inferred from the amino acid sequences. A ML tree was also constructed under the best‐fit model, Jones‐Taylor‐Thornton (JTT), with discrete gamma distribution in three categories (gamma shape = 3,163). The tree topologies assessed by ML and NJ methods were substantially similar. The phylogenetic analysis placed the prokaryotic AAAH at the base of the tree. The eukaryotic taxon showed three distinct clusters: phenylalanine (PAH), tryptophan (TPH), and tyrosine (TH) hydroxylase (Supporting Information Fig. 1). These groups were unambiguously separated from protists, fungi and lower plants (mosses/green algae). The results are consistent with previous phylogenetic studies of AAAHs 5, 25.
Functional diverse analyses among prokaryotic and eukaryotic clusters appear to be of great interest, given that with these we could identify potential sites that differ between the groups of enzymes that have phenylalanine or tryptophan preference. Such sites could be helpful in designing novel enzymes for the production of 5HTP. The FDI analysis for these clusters revealed significant θI values between the bacterial and eukaryotic AAAH, indicating that diversification has taken place during evolution (Table 2). We identified three different groups of residues in our analysis: (i) sites that are conserved within all the clusters, many of these amino acids are involved in the enzyme activity, e.g. conserved motifs and functionally important residues such as the 2‐His‐1‐caboxylate facial triad which is involved in the coordination of the iron molecule in the active pocket of all hydroxylases 26; (ii) sites that are conserved within Eukaryotes and Prokaryotes taxa, but that are different between them; and (iii) sites that seem to be related with specific substrate preference (Fig. 1). The FDII analysis showed a low θII value of the TPH/PAH pair (Table 2.). Since there is compiling evidence regarding of the substrate specificity of these two groups, the FDII analysis confirms that the predicted substrate‐determining residues between the latter group might have relevant functions specifically related to phenylalanine or tryptophan (activity, inhibition, activation, etc.). To gain more insight into the possible function of these predicted FDII sites, we mapped residues with high posterior probability (Qk) values in the modeled AAAHs structures (Fig. 2).
Table 2.
Functional divergence between eukaryotic TPH/PAH and prokaryotic paralogous proteins
| Group 1 | Group 2 | Type‐I | Type‐II | ||
|---|---|---|---|---|---|
| θI a | Qk>0.9b | θII a | Qk>0.9b | ||
| AAAH | PAH | 0.664 ± 0.090 | 37 | 0.374 ± 0.160 | 66 |
| AAAH | TPH | 0.747 ± 0.094 | 34 | 0.445 ± 0.161 | 78 |
| TPH | PAH | 0.201 ± 0.060 | 0 | 0.059 ± 0.095 | 15 |
θI and θII, correspond to the functional divergence Type‐I and Type‐II coefficient, respectively. Coefficients were estimated using DIVERGE v3.0 17.
Qk, posterior probability.
Figure 1.
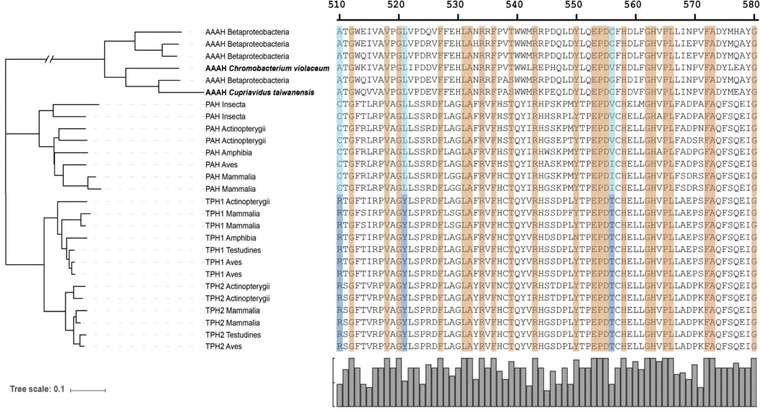
Protein tree and alignment for the AAAH superfamily: unrooted protein distance tree and alignment for 30 aromatic amino acid hydroxylases with phenylalanine (AAAH and PAH) or tryptophan (TPH1 and TPH2) affinity. Amino acids in the alignment are mainly present in the active pocket, amino acids colored in orange indicate conserve residues, amino acids highlighted with blue and light blue are strongly related to the substrate preference. A consensus graph is present below the alignment.
Figure 2.
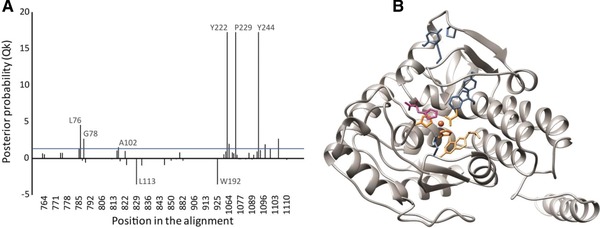
Prediction of substrate‐determining residues based on functional divergence type‐II analysis of tryptophan and phenylalanine hydroxylase, and their position in Cupriavidus taiwanensis modeled structure: (A) posterior probability (Qk) of the homologous sites, a threshold line of Qk = 0.9 is indicated in the graph; (B) predicted amino acids (blue) are mapped in the structure, phenylalanine (magenta) is present at the active site, as well as the cofactor, 2‐His‐1‐caboxylate facial triad and the iron molecule (orange).
By combining phylogenetic evidence with the FDI and FDII analyses, we hypothesized that even after a long‐term evolution process, prokaryotic AAAH and eukaryotic PAH still share some conserved amino acids that determine their substrate preference. Furthermore, the low divergence between animal PAH and TPH (and high sequence similarity) indicate that the substrate preference may involve only a small number of residues. Based on this hypothesis we selected six residues and did site‐directed mutagenesis of the protein.
Before exploring the substrate‐determining amino acids, we generated protein structure models of the AAAHs of 10 bacteria: Chromobacterium violaceum, Pseudomonas aeruginosa, Xanthomonas campestris, Streptomyces kanamyceticus, Echinicola vietnamensis, Pseudoalteromonas atlantica, Pseudomonas putida, Mesorhizobium plurifarium, Archangium violaceum and Cupriavidus taiwanensis. Some of the crystal structures of these proteins have been already determined 26, 27, and we use these, as well as others eukaryotic AAAHs for verification and comparison purposes. We performed phenylalanine and tryptophan docking analyses using the generated structures. AAAH from C. taiwanensis (CtAAAH) performed better during the in silico tryptophan docking analysis, hence we decided to use this enzyme as a prototype for our further work. Moreover, when we used this model and created variants with homologous sites that have been reported to increase the tryptophan affinity for bacterial AAAHs, the docking results showed higher affinity toward tryptophan than the other structures. Relative binding energies of CtAAAH for phenylalanine and tryptophan are present in Table 3.
Table 3.
In silico relative docking binding energy and in vivo activity of Cupriavidus taiwanensis AAAH (wild‐type and mutants) using phenylalanine or tryptophan as substrate
| AAAH | in silico RBEa | Preference (Phe/Trp) | in vivo activityb | Preference (Phe/Trp) | ||
|---|---|---|---|---|---|---|
| Phe | Trp | Phe → Tyr | Trp → 5HTP | |||
| CtAAAH (wt) | 1.00 | 1.00 | 1.00 | 86.8 ± 5.7 | 10.5 ± 0.4 | 8.3 |
| wt L113Y | 0.83 | 0.84 | 0.99 | 73.8 ± 2.3 | 24.1 ± 0.9 | 3.1 |
| wt W192F | 0.75 | 0.86 | 0.87 | 79.1 ± 3.7 | 57.8 ± 1.2 | 1.4 |
| wt Y222H | 0.78 | 0.82 | 0.95 | 41.9 ± 2.9 | 25.0 ± 0.8 | 1.6 |
| wt S223C | 0.90 | 0.80 | 1.12 | 63.7 ± 3.1 | 21.6 ± 0.9 | 2.9 |
| wt P229A | 0.81 | 0.90 | 0.90 | 59.5 ± 3.5 | 35.4 ± 1.0 | 1.7 |
| wt Y244C | 0.81 | 0.84 | 0.96 | 48.8 ± 2.7 | 33.5 ± 1.2 | 1.5 |
RBE, relative binding energy estimated using AutoDock Vina v1.1.2 20.
Activity estimated as μM · h−1 · OD600 −1. All data are reported as the mean ± standard error of the mean (SEM) from three independent experiments.
3.2. Protein engineering for the modification of substrate preference
The design of synthetic metabolic pathways often requires different roles of the wild‐type proteins we would like to use. The CtAAAH enzyme has high similarity with other hydroxylase reported to have phenylalanine preference. We aimed to alter its substrate specificity and create a new catalyst with characteristics that fulfill our needs.
The aaah gene from C. taiwanensis was cloned into the low copy plasmid pACYCDuet under control of an isopropyl ß‐D‐1‐tiogalactopyranoside (IPTG)‐inducible T7 promoter. The resulting expression construction, pCtAAAH, was transformed in strain BL21(DE3)∆tnaA. Because tryptophanase encoded by tnaA has been reported to catalyze the degradation of tryptophan and 5HTP to pyruvate, ammonia, and indole or hydroxy‐indole, respectively 4, 28, we knocked out this gene from E. coli strain BL21(DE3).
The selectivity and activity of the selected model was evaluated using E. coli strains expressing CtAAAH wild‐type. The in vivo specific activity assay results showed that the enzyme has a strong preference toward phenylalanine (86.8 ± 4.3 μM · h−1 · OD600 −1) when compared with tryptophan (10.5 ± 0.4 μM · h−1 · OD600 −1). However, when the tryptophan activity was compared with other bacterial wild‐type AAAHs reported in the literature, CtAAAH showed around three‐fold higher activity toward tryptophan (X. campestris = 2.91 ± 0.21 μM · h−1 · OD600 −1) 5. These confirm the prediction of our initial computational screening.
The FDII analysis showed 15 residues with high Qk: large Qk values indicate high evolutionary rates or physiochemical differences between homologous amino acids of two clades. These sites were tracked within the alignments and the corresponding position of the C. taiwanensis sequence was compared with the sequence from the enzymes that have either phenylalanine or tryptophan preference. Six out of this 15 sites were selected because the C. taiwanensis amino acids were similar (belong to the same chemical group) to the sequences of the PAH cluster, and at the same time different from the TPH group (L113Y, W192F, Y222H, S223C, P229A, Y244C). Three amino acids L113, W192 and Y244 seem to be involved in the substrate and ligand stabilization and predicted mutations would be favorable for the tryptophan hydroxylation due to binding interactions and volume effect 29 (Fig. 2).
In the modeled CtAAAH·BH4·Trp complex, the pterin ring of BH4 π‐stacks with P119, the L113 residue is also involved in the binding since it is located within 3.5 Å from the cofactor. The mutation L113Y adds an extra π‐π stacking interaction (3.2 Å), so that the enzyme‐cofactor complex could be stabilized and orientated. Homologous mutations Y235L of human TPH decreased the K m of BH4 30, which in turn affected the apparent affinity for tryptophan. Similar results were reported by Kino et al. 2009 11 using DMPH4 as an analog cofactor. In our model tryptophan is docked in the structure and stabilized by ionic interaction with S215 and R135, and through π stacking interactions with H150, F197 and W192. TPHs present a phenylalanine in the W192 homologous site, in our W192F‐model this position seems to be involved in the binding of the indole ring of the L‐tryptophan. Also, volume‐effect may play an important role in the stabilization of the substrate, since the distance between the residue and the substrate (or an analog) is between 3.8 to 4.1 Å in different reported structures. In our case, when the wild‐type is docked with tryptophan the distance to the W192 residue is 3.6 Å, and it increases to 4.0 Å in the case of the W192F variant. Residue Y244 stabilizes the position W192 through a π‐ π interaction, a modification in this position could give more flexibility to the W192 residue, thus increasing the tryptophan preference due to a volume‐effect as mentioned before. The residues Y222, S223 and P229 are out from the binding pocket, predicted mutations could play a role in the interaction or stabilization of the flexible loop (242‐251).
To investigate the performance of the predicted substrate‐determining residues, enzyme assays were done using generated mutants. As a result, the W192F mutant exhibited a 5.5‐fold increase in tryptophan hydroxylation activity when compared with the wild type, meanwhile the activity toward phenylalanine decreased by ∼10%. The substrate preference for phenylalanine over tryptophan was shifted from 8.2 to 1.3. The other mutations, in general, presented a similar pattern: phenylalanine activity decreased, meanwhile the tryptophan activity increased. It is worth noting that although we did not observe a strong correlation between the in silico and in vivo Phe/Trp preference (R2 = 0.61), we did observe correspondence among the high and the low performers in terms of substrate selectivity: mutants W192F and P229A were predicted to have more preference toward tryptophan, meanwhile L113Y and S223C showed low Phe/Trp ratio (Table 3).
Advances have been made to predict mathematically favorable mutations for the creation of enzymes with wanted characteristics based on limited experimental data. This approach would facilitate protein engineering greatly, especially in the absence of high‐throughput screening methods. The example developed in this paper has great potential for the tailoring of enzymes with desired substrate specificity for synthetic biology and metabolic engineering. Nevertheless, final scope of this method should be prudently addressed, since we are aware that this is a specific example, and future experiments could bring different insights.
3.3. Pathway engineering for the production of 5HTP
If one aims to expand the tryptophan metabolism to produce 5HTP in E. coli, three important components are needed: the hydroxylase enzyme, a pterin cofactor, and a cofactor regeneration systems since E. coli lacks it. One of the issues of the expression of AAAHs in bacteria is the availability of the cofactor. The most common cofactor reported to participate in the hydroxylation of aromatic amino acids is tetrahydrobiopterin (BH4). While a few bacterial taxa, such as Cyanobacteria and Chlorobia, produce BH4, most of the prokaryotes cannot synthesize this cofactor. As aaah genes are present in different groups of bacteria, they must use a different cofactor available in prokaryotes. Pribat et al. 31 reported that tetrahydromonapterin (MH4), which is naturally produced in E. coli, is the cofactor of Pseudomonas AAAH and can also be used in eukaryotic tyrosine hydroxylase as well. Therefore, we explored the possibility that CtAAAH could use endogenous MH4 as a cofactor.
When compared E. coli BL21(DE3)ΔtnaA cultures harboring pCtAAAH, wild‐type and W192F, the latter showed a stagnation during cell growth and low 5HTP production (0.21 mM), whereas the wild‐type reached a maximum growth of OD600 = 7.6 (comparable to the control) and no 5HTP was detected. A similar effect has been observed in previous studies 5. This result could be explained because the enzyme might have been using MH4 as a cofactor and there is no recycling system in E. coli. To establish an artificial MH4 regeneration pathway we transformed plasmid pBbE1k‐2, which encodes for human pterin‐4a‐carbinolamine dehydratase (PCD) and dihydropteridine reductase (DHPR). PCD and DHPR participate in a well‐known BH4 recycling system in animals 32. After the tryptophan hydroxylation, the pterin cofactor is converted into pterin‐4a‐carbinoalamine, and PCD catalyzes its dehydration and produces dihydropteridine. The cycle is closed when DHPR reduces and reactivates the pterin (Fig. 3) 33.
Figure 3.
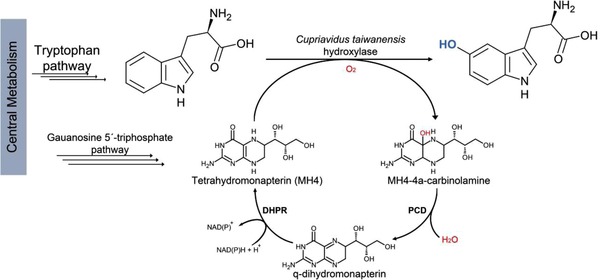
Synthetic pathway for the hydroxylation of tryptophan to 5‐hydroxytryptophan. CtAAAH hydroxylates tryptophan in the presence of oxygen and tetrahydromonapterin. In this reaction the cofactor is oxidated. Afterward, the pterin‐4a‐carbinolamine dehydratase (PCD) dehydrates MH‐4a‐carbinolamine, and dihydropteridine reductase (DHPR) reduces back the pterin cofactor.
There were no differences in the growth of BL21(DE3)∆tnaA when these cells were carrying the cofactor regeneration system; and as expected, when pCtAAAH‐W192F was cotransformed with pBbE1k‐2, cell viability dramatically improved, comparable with the values of the strains without the CtAAAH‐W192F. These cells grew in media supplied with 5 mM tryptophan, they produced up to 2.5 mM 5HTP in a 24 hs cultivation period (Fig. 4), three times more than previously reported by Lin et al. 2014 5. This result shows that the strain expressing the engineered CtAAAH‐W192F and the MH4 regeneration pathway genes successfully produces 5HTP.
Figure 4.
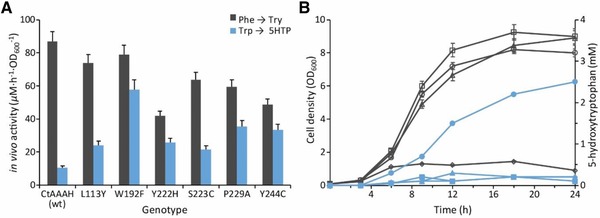
Modification of CtAAAH via protein engineering: (A) in vivo activity of wild‐type (wt) CtAAAH and its mutants, dark gray and blue bars indicates the activities toward phenylalanine and tryptophan, respectively. (B) Cell growth (dark gray) and 5HTP production (blue) during fermentation by fed‐batch using shaken flasks. BL21(DE3)∆tnaA (squares), BL21(DE3) pCtAAAH‐W192F (diamonds), BL21(DE3) cofactor (triangles), BL21(DE3) cofactor pCtAAAH‐W192F (circles). Bars present in the graphs correspond to the standards errors means (SEM) from three independent experiments.
4. Concluding remarks
The dramatic increase in healthcare cost has become a significant burden to the world. The biosynthesis of chiral drugs intermediates with environmentally friendly approaches help provide more affordable pharmaceuticals.
Here, we have expanded the natural metabolic capability of E. coli to produce the non‐canonical amino acid 5HTP using protein engineering added with a computational screening approach of a selection of a wild‐type enzyme and the prediction of key mutations. Initially, we generated 10 structures and selected CtAAAH as the prototype enzyme to work in the laboratory. Using sequence comparison, phylogenetic and functional diverge analyses we successfully identified relevant residues for phenylalanine/tryptophan substrate preference. All these sites were shown to increase the tryptophan preference of the enzyme at the expense of phenylalanine. We succeeded in introducing an artificial pathway for the L‐tryptophan oxidation into E. coli using the best generated mutant CtAAAH‐W192F, the endogenous MH4 cofactor, and the human proteins PDC and DHPR for the cofactor regeneration pathway.
This engineered strain is capable of producing 5HTP, and when combined with a tryptophan overproducing strain, a simple sugar could be used as a carbon source for the production, which will make the microbial system more economically attractive.
Practical applications
The selection of native enzymes is keynote before protein and metabolic engineering. In this work, we present a practical approach for the prediction of suitable enzymes based sequence and structure analysis. We identify key residues involved in the protein‐ligand interactions and based on these we selected a fit enzyme as starting material for protein engineering. The paper details the elongation of the tryptophan pathway for the biosynthesis of 5‐hydroxytryptophan (5HTP) via the incorporation of a modified AAAH and a cofactor regeneration system. These results can help to reduce production costs 5HTP, which is used by the pharmaceutical industry and serves as a chemical platform to produce other derivatives. Based on computational analyses, we also identified novel sites involved in the AAAH's phenylalanine/tryptophan selectivity. A further combination of these mutations could increase the selectivity and/or activity toward tryptophan.
The authors have declared no conflict of interest.
Supporting information
Supporting Information Figure S1. Phylogenetic tree of 729 aromatic amino acid hydroxylases. Branches of eukaryontes and prokaryontes are distinguishable by color, black and gray respectively. Stripes around the tree designate the type of hydroxylase which corresponds to each specie. Duplication events are marked within each clade. Tree was inferred using the Neighbor‐Joining method. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. Evolutionary distances were computed using Poisson correction method. All positions with less than 2% site coverage were eliminate, a total of 495 positions were in the final dataset. Evolutionary analysis was conducted in MEGA9.
Supporting Information
Acknowledgments
The authors would like to acknowledge the Deutscher Akademischer Austauschdienst (DAAD), the Ministerio de Ciencia, Tecnología y Telecomunicaciones (MICITT) and Consejo Nacional para Investigaciones Científicas y Tecnológicas (CONICIT) from Costa Rica for financial support.
5 References
- 1. Birdsall, T. C. , 5‐Hydroxytryptophan: a clinically‐effective serotonin precursor. Altern. Med. Rev. 1998, 3, 271–280. [PubMed] [Google Scholar]
- 2. Frangatos, G. , Chubb, F. L. , A new synthesis of 5‐hydroxytryptophan. Can. J. Chem. 1959, 37, 1374–1376. [Google Scholar]
- 3. Chen, X. , Zhou, L. , Tian, K. , Kumar, A. et al., Metabolic engineering of Escherichia coli: a sustainable industrial platform for bio‐based chemical production. Biotechnol. Adv. 2013, 31, 1200–1223. [DOI] [PubMed] [Google Scholar]
- 4. Hara, R. , Kino, K. , Enhanced synthesis of 5‐hydroxy‐l‐tryptophan through tetrahydropterin regeneration. AMB Express 2013, 3, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lin, Y. , Sun, X. , Yuan, Q. , Yan, Y. , Engineering bacterial phenylalanine 4‐hydroxylase for microbial synthesis of human neurotransmitter precursor 5‐hydroxytryptophan. ACS Synth. Biol. 2014, 3, 497–505. [DOI] [PubMed] [Google Scholar]
- 6. Yakandawala, N. , Romeo, T. , Friesen, A. D. , Madhyastha, S. , Metabolic engineering of Escherichia coli to enhance phenylalanine production. Appl. Microbiol. Biotechnol. 2008, 78, 283–291. [DOI] [PubMed] [Google Scholar]
- 7. Chen, L. , Zeng, A. P. , Rational design and metabolic analysis of Escherichia coli for effective production of L‐tryptophan at high concentration. Appl. Microbiol. Biotechnol. 2016, 101, 559–568. [DOI] [PubMed] [Google Scholar]
- 8. Arnold, F. H. , Combinatorial and computational challenges for biocatalyst design. Nature 2001, 409, 253–257. [DOI] [PubMed] [Google Scholar]
- 9. Chen, Z. , Zeng, A. P. , Protein engineering approaches to chemical biotechnology. Curr. Opin. Biotechnol. 2016, 42, 198–205. [DOI] [PubMed] [Google Scholar]
- 10. Wang, L. , Erlandsen, H. , Haavik, J. , Knappskog, P. M. et al., Three‐dimensional structure of human tryptophan hydroxylase and its implications for the biosynthesis of the neurotransmitters serotonin and melatonin. Biochemistry 2002, 41, 12569–12574. [DOI] [PubMed] [Google Scholar]
- 11. Kino, K. , Hara, R. , Nozawa, A. , Enhancement of l‐tryptophan 5‐hydroxylation activity by structure‐based modification of l‐phenylalanine 4‐hydroxylase from Chromobacterium violaceum . J. Biosci. Bioeng. 2009, 108, 184–189. [DOI] [PubMed] [Google Scholar]
- 12. Chen, Z. , Zeng, A. P. , Protein design in systems metabolic engineering for industrial strain development. Biotechnol. J. 2013, 8, 523–533. [DOI] [PubMed] [Google Scholar]
- 13. Chen, Z. , Wilmanns, M. , Zeng, A. P. , Structural synthetic biotechnology: from molecular structure to predictable design for industrial strain development. Trends Biotechnol. 2010, 28, 534–542. [DOI] [PubMed] [Google Scholar]
- 14. Edgar, R. C. , MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumar, S. , Stecher, G. , Tamura, K. , MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abascal, F. , Zardoya, R. , Posada, D. , ProtTest: selection of best‐fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [DOI] [PubMed] [Google Scholar]
- 17. Gu, X. , Statistical methods for testing functional divergence after gene duplication. Mol. Biol. Evol. 1999, 16, 1664–1674. [DOI] [PubMed] [Google Scholar]
- 18. Roy, A. , Kucukural, A. , Zhang, Y. , I‐TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shapovalov, M. V. , Dunbrack, R. L. , A smoothed backbone‐dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trott, O. , Olson, A. J. , Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pettersen, E. F. , Goddard, T. D. , Huang, C. C. , Couch, G. S. et al., UCSF Chimera ‐ A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 22. Sambrook, J. , Russell, D. W. , Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York: 2000. [Google Scholar]
- 23. Datsenko, K. A. , Wanner, B. L. , One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. da Luz, J. A. , Hans, E. , Zeng, A. P. , Automated fast filtration and on‐filter quenching improve the intracellular metabolite analysis of microorganisms. Eng. Life Sci. 2014, 14, 135–142. [Google Scholar]
- 25. McKinney, J. A. , Turel, B. , Winge, I. , Knappskog, P. M. et al., Functional properties of missense variants of human tryptophan hydroxylase 2. Hum. Mutat. 2009, 30, 787–794. [DOI] [PubMed] [Google Scholar]
- 26. Erlandsen, H. , Kim, J. Y. , Patch, M. G. , Han, A. et al., Structural comparison of bacterial and human iron‐dependent phenylalanine hydroxylases: similar fold, different stability and reaction rates. J. Mol. Biol. 2002, 320, 645–661. [DOI] [PubMed] [Google Scholar]
- 27. Ekström, F. , Stier, G. , Eaton, J. T. , Sauer, U. H. , Crystallization and X‐ray analysis of a bacterial non‐haem iron‐containing phenylalanine hydroxylase from the Gram‐negative opportunistic pathogen Pseudomonas aeruginosa . Acta Crystallogr. ‐ Sect. D Biol. Crystallogr. 2003, 59, 1310–1312. [DOI] [PubMed] [Google Scholar]
- 28. Gong, F. , Ito, K. , Nakamura, Y. , Yanofsky, C. , The mechanism of tryptophan induction of tryptophanase operon expression: tryptophan inhibits release factor‐mediated cleavage of TnaC‐peptidyl‐tRNA(Pro). Proc. Natl. Acad. Sci. USA 2001, 98, 8997–9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daubner, S. C. , Moran, G. R. , Fitzpatrick, P. F. , Role of tryptophan hydroxylase phe313 in determining substrate specificity. Biochem. Biophys. Res. Commun. 2002, 292, 639–41. [DOI] [PubMed] [Google Scholar]
- 30. McKinney, J. , Teigen, K. , Frøystein, N. A. , Salaün, C. et al., Conformation of the substrate and pterin cofactor bound to human tryptophan hydroxylase. Important role of Phe313 in substrate specificity. Biochemistry 2001, 40, 15591–15601. [DOI] [PubMed] [Google Scholar]
- 31. Pribat, A. , Blaby, I. K. , Lara‐Núñez, A. , Gregory, J. F. et al., FolX and FolM are essential for tetrahydromonapterin synthesis in Escherichia coli and Pseudomonas aeruginosa . J. Bacteriol. 2010, 192, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Satoh, Y. , Tajima, K. , Munekata, M. , Keasling, J. D. et al., Engineering of l‐tyrosine oxidation in Escherichia coli and microbial production of hydroxytyrosol. Metab. Eng. 2012, 14, 603–610. [DOI] [PubMed] [Google Scholar]
- 33. Davis, M. D. , Kaufman, S. , Milstien, S. , Conversion of 6‐substituted tetrahydropterins to 7‐isomers via phenylalanine hydroxylase‐generated intermediates. Proc. Natl. Acad. Sci. USA 1991, 88, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure S1. Phylogenetic tree of 729 aromatic amino acid hydroxylases. Branches of eukaryontes and prokaryontes are distinguishable by color, black and gray respectively. Stripes around the tree designate the type of hydroxylase which corresponds to each specie. Duplication events are marked within each clade. Tree was inferred using the Neighbor‐Joining method. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. Evolutionary distances were computed using Poisson correction method. All positions with less than 2% site coverage were eliminate, a total of 495 positions were in the final dataset. Evolutionary analysis was conducted in MEGA9.
Supporting Information