

Abstract
Similar to the incredible advances in DNA sequencing, the de novo synthesis of DNA is subject to innovations and fast progress in terms of synthesis speed and cost. We will discuss novel techniques that are expected to enable high‐throughput synthesis of oligonucleotides on microarrays and the subsequent assembly into longer fragments, up to whole genomes. Especially, the inherent disadvantages of microarray‐derived oligonucleotide pools for gene synthesis will be discussed in detail, and also the different approaches to still render these oligonucleotides useful for gene assembly. These so‐called next‐generation techniques will lead to a significant cost reduction of gene synthesis and to the possibility of much larger projects, such as whole genome synthesis.
Keywords: Cloning, Gene synthesis, Genome synthesis, Microarrays, Microfluidics, Oligonucleotides
Abbreviations
- bp
base pair
- CPEC
circular polymerase extension cloning
- NGS
next generation sequencing
- oligo
oligonucleotide
- TypeIIS‐RE
type IIS restriction enzyme
- USER
uracil‐specific excision reagent
1. Introduction
A major prerequisite for modern biotechnology and synthetic biology, in particular, is the availability of low‐cost de novo gene synthesis 1. This not only allows liberalization from natural sources of genetic material and unrestricted flexible design of DNA sequences, but also adapting coding sequences to the genetic requirements of the target organism. Today, most biotechnological projects make good use of gene synthesis and profit on safety, availability, reliability, throughput, flexibility, and last but not least, total cost 2. Its application not only completely changed the way in which scientists think, when designing appropriate DNA constructs, but also facilitates outsourcing of related experimental steps in order to concentrate on less trivial scientific operations. Researchers can electronically access DNA sequences through comprehensive databases, redesign constructs in silico to specifically fit given requirements, and order the designed genes online being synthesized and shipped within a matter of days 2.
Current protocols for gene synthesis are based on the ability of chemically synthesized single‐stranded oligonucleotides (oligos) to self‐assemble into larger contiguous sequences (see Fig. 1). The assembly process itself is usually a multiplex primer extension reaction under controlled temperature cycling conditions.
Figure 1.

Comparison of traditional and next generation gene synthesis workflows (for <1 kb genes/fragments). In the traditional gene synthesis, one synthesizer produces oligonucleotides for about 3–5 fragments, while a microarray synthesis can yield hundreds of fragments. All the numbers and steps are examples and do not represent a certain workflow by one company or by one single publication.
Unfortunately, template‐less chemical production of oligos is still a significant cost factor. Herein, we will discuss new techniques that are expected to enable high‐throughput synthesis of oligo and the subsequent assembly into fragments. We will further discuss new methods for assembly and cloning to further assemble these fragments into longer constructs, up to whole genomes. These so‐called next‐generation techniques will lead to a significant cost reduction of gene synthesis and will therefore pave the way for the field of synthetic biology and for whole genome synthesis.
2. Oligonucleotide synthesis and microarrays
The phosphoramidite method, which was introduced in the 1980s by Caruthers and coworkers 3, 4, is still commonly used to synthesize oligos in synthesis columns, which is a major bottle neck in gene synthesis, with respect to costs and throughput.
Because the number of different oligos is too low for high‐throughput gene synthesis and each oligo is produced in vast excess, a switch from columns to microarrays would be very attractive in terms of costs and parallelization (Fig. 1).
Microarrays, based on mask‐based lithography, were developed for the parallel analysis of DNA sequences by Fodor et al. in the early 1990s 5, 6 , starting the success of Affymetrix. The flexibility of this process was later improved by Nimblegen by using a digital micro‐mirror array 7. Later, Gao et al. used photogenerated acid to synthesize microarrays with standard phosphoramidites 8, which was commercialized by LC Sciences, and the Southern group developed a similar technique based on electrochemical deprotection on microchips 9, 10, today available by Customarray.
In general, all of the above methods control the deprotection step and have the disadvantage that a full cycle for all four amidites is necessary to couple one base, leading to longer synthesis time and higher error rates. For this reason, controlling the sequence by adding the correct amidite (in analogy to the standard column‐based synthesis) is favorable for speed and quality of the synthesis. Miniaturization of this process was achieved by adapting ink‐jet technology 11 and was later commercialized by Agilent 12.
Even with all these improvements over the last 25 years, microarray‐based oligo synthesis still comes with several disadvantages. Microarrays yield a low‐concentrated pool of thousands of oligos and are usually prone to more errors than column‐synthesized oligos, due to edge effects including escaping protons, light beam drift, or droplet misalignment 13. Additionally, a quality control of the single oligos is nearly impossible, and definitely impracticable with current techniques, which can lead to oligos being over‐ or underrepresented in the pool 14. Because of these shortcomings, microarray‐derived oligos cannot be used for gene synthesis without adapting the workflow, or the microarray.
3. Microarrays for gene synthesis
Many different strategies and methods have been developed during the last years to overcome these problems and pave the way for a truly next generation technique. Although all different disadvantages typically have to be solved to render a certain approach fully functional, we will discuss them separately here, i.e. concentration, pool complexity, and error rates.
3.1. Low concentration of oligonucleotides
The concentrations of oligo mixtures from microarrays are typically 1–20 pmol for the whole pool, which means that only atto‐ to femtomol quantities of every oligo are available for gene assembly reactions 15. Unfortunately, this is several orders of magnitudes less than the required concentration for a successful gene assembly in standard volumes (1 pmol) 16.
The most obvious solution to this shortcoming would certainly be to simply synthesize oligos in higher quantities. Lee et al. showed that when columns of silica particles are integrated on a microfluidic chip, different oligos can be synthesized in almost nanomolar quantities for direct assembly into genes 17. However, this approach was limited to 16 oligos, and is not easily scalable to thousands of synthesis columns.
Another way to achieve higher concentrations is to miniaturize the volume of the assembly reaction. It was shown that assembly is possible in microfluidic chambers of 500 nL volumes with no negative influence on assembly efficiency 18. To be truly compatible with microarray yields, the volume would need to be further decreased to about 10 nL, which would render the retrieval of the assembled genes very difficult, as these small volumes are difficult to handle without loss and a subsequent amplification of the assembled gene would be needed. As discussed by the authors, all of this would only be possible if the chamber encapsulates the microarray to avoid loss of the synthesis product. The first step in this direction, although with larger volumes, was shown by Quan et al. 19, who used a custom‐built printer to synthesize oligos in microwells. Recently, Twist Bioscience has started to develop a similar approach by using microfabricated nanowells that capture oligos in small volumes 20.
However, the approach which is most commonly used by groups working with microarrays for gene synthesis is amplification of the oligos by PCR. It was first shown in 2004 in several publications 21, 22, 23, that a PCR of microarray‐derived oligos can yield concentrations that are useful for gene assembly. Although this method seems simple and is compatible with biomolecular workflows, it suffers from (i) amplification bias 24, leading to concentration differences in the amplified pool, and therefore to high percentages of unsuccessful assembly reactions; (ii) even higher error rates of oligos as primer binding sites need to be chemically synthesized, and errors correlate with length 25; and (iii) additional preparative steps for the removal of primer binding sites 16, 25. To overcome amplification bias, linear amplifications have been successfully implemented 19, but only with a fourfold concentration increase, and therefore a miniaturization of the assembly volume was still needed. Borovkov et al. proposed to amplify after an assembly of small fragments of less than 300 bp, which reduced amplification bias by tenfold in their experiments 15. As we will discuss in Section 3.2, direct assembly is limited to low complexity pools and therefore will not allow to completely exploit the full potential of commercial microarrays.
3.2. Complex mixtures of oligonucleotides
Current microarray systems are typically used to produce 10 k– 1 M oligos per chip for a wide range of different applications. The high density of synthesis positions available on commercial microarrays entails that the oligos can only be cleaved and eluted together in a highly complex pool. Hence, for DNA synthesis most users have focused on a density in the range of thousands different oligos per chip in an effort to balance yield per oligo (see above) and complexity. As all oligos are present, the probability for cross hybridizations among different fragments increases and chances of successful assembly decreases accordingly. It has been shown that the one‐pot assembly of many different genes is only possible for low‐complexity mixtures, and not for the amount of different oligos that are produced on commercial microarrays 15.
One possible solution for this problem is physical separation, i.e. to synthesize subsets of microarrays in different locations and/or volumes. This possibility was first discussed by Kong et al. 18, and was later shown experimentally by Quan et al. to be a viable option to keep oligos separated 19. Different subsets of oligos were synthesized in small pots that were embossed into plastic slides. Each pot contained all oligos belonging to one fragment and had no fluidic contact during the assembly with the other reactions. Unfortunately, commercial microarrays cannot be used for this method as they have been optimized for high density. Another disadvantage is that expensive microstructures need to be used (compared to the flat surface of a common microarray).
Other groups have therefore developed protocols to directly use the pools that can be derived from commercial microarrays. Tian et al. assembled a multitude of genes from one pool, which was also achieved by others at the same time 21, 22, 23. As discussed before, these approaches proved difficult to scale and could not use all the possible oligos on highly parallel arrays. Kosuri et al. showed that the selective amplification of barcoded oligos belonging to one fragment (subpool) can decrease the complexity of the pool and leads to more successful assemblies 25. Their approach, which was also used by Kim et al. 16, involved synthesizing additional primer binding sites barcoding different fragments, which could be used for amplification with the corresponding primer set. Both groups showed that the incomplete removal of primer sites by enzymatic cleavage leads to failures in assembly. In another experiment, Kosuri et al. preamplified larger mixtures of oligos before the sub‐pool amplification, with better assembly results. However, since they needed even longer oligos (200 mers) for this approach, the error rates were substantially higher (1/250 bp) in the assemblies 25. The advantage of selective amplification is that standard laboratory equipment and consumables can be used, and it works directly with pools from (commercial) microarrays. However, primers need to be designed very carefully to be orthogonal and to keep amplification bias to a minimum. As the removal of the primer sites needs an additional gel isolation, the process is difficult to automate and expensive. Klein et al. recently showed the multiplexed co‐assembly of up to 2271 small fragments, and omitted the enzymatic cleavage site by using uracil‐containing pool specific primers, which could later easily be removed by uracil specific excision reagents 14. In summary, all these approaches show very good potential to make use of microarrays, but significantly increase the time to synthesize a gene (see Fig. 1).
3.3. Error rate
Currently, conditions for mass production of genes are chosen to have a >95 % chance of picking at least one correct fragment with a single screen after transformation (Fig. 1), limiting the size of the initial fragment to <3 kb. Thus, to not further extend gene synthesis time, DNA fragments with low error rates are important to reduce the screening effort, as a resynthesis is needed if no correct clone can be found. Besides using more stringent assembly conditions 15, special procedures are necessary to compensate for the higher error rates of microarray‐derived oligos (Fig. 2); the most common ones being (i) the removal of errors by mismatch binding proteins or mismatch cleaving enzymes and (ii) Sequencing‐based methods to identify error free DNA with subsequent physical retrieval of correct DNA, or (iii) its selective amplification.
Figure 2.
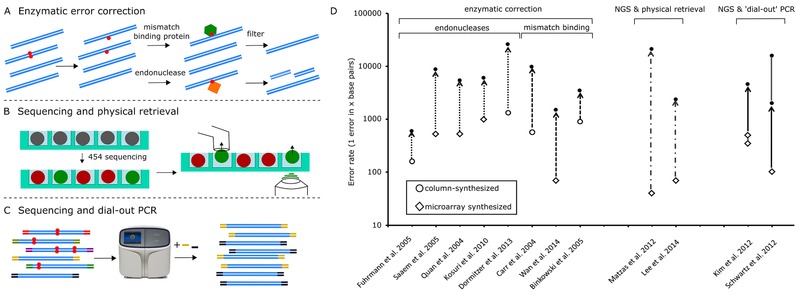
Different approaches for error correction. (A) Enzymatic error correction uses either mismatch binding proteins with filtration to remove DNA with errors, or enzymes that cleave erroneous DNA. (B) DNA that has been sequenced can be located and physically removed by micropipettes 31, or lasers 32. (C) Next generation sequencing of barcoded molecules can identify correct molecules that are then amplified with the barcode as a primer 33. (D) Comparison of the different error correction methods.
Removal of errors with error correcting enzymes, e.g. mismatch cleaving enzymes such as T7 endonuclease I, CEL1, T4 endonuclease VII, and Escherichia coli endonuclease V is probably the most commonly used approach 25, 26, 27. Mismatch cleaving enzymes recognize mismatched base pairs (bp) in double stranded DNA and cleave 2–5 bp downstream of the mismatch. The resulting overhangs, which contain the mismatched base, are then removed by 3′‐5′ exonucleases or 5′‐3′ exonuclease activity of proof reading polymerases. Error‐free fragments are then assembled via PCR 26. Mismatch binding proteins such as MutS capture error containing fragments and only the purified, error‐free DNA will be used for further assembly 28.
Error correction methods using only mismatch binding proteins are not efficient for error removal in long synthetic DNA fragments where virtually all fragments contain errors 29. For longer DNA fragments, a combination of restriction digestion with subsequent MutS‐filtration led to error‐reduced long genes 29. Unfortunately, not all types of errors are corrected equally, e.g. some enzymes preferentially correct insertions and deletions, but are less efficient for substitutions. Thus, enzyme choice depends on the expected error type in the oligos or synthetic DNA fragments.
In a very recent publication, solid‐state nanopores were used to discriminate between correct and incorrect fragments by detecting MutS binding 30. If nanopore‐based monitoring of DNA quality could be used to purify the error‐free DNA, it might lead to a novel strategy for high‐throughput preparation of synthetic DNA for gene synthesis 30.
Use of massive parallel sequencing enabled by next‐generation sequencing (NGS) platforms offers another approach to identify correct sequences rapidly and efficiently. These methods can be applied both to select correct oligos prior to assembly and error‐free larger fragments after assembly. This was first used to identify correct oligos with the so‐called “mega cloning” prior to assembly (Fig. 2B) 31. Here, oligos synthesized on a microarray platform were sequenced on a Roche 454 GS FLX platform. Single beads containing the correct oligos were selected from the picotiter plate, and transferred with a micropipette into a 96‐well plate. The sequence verified oligos were then amplified and used for the assembly of larger fragments resulting in a significantly reduced error rate.
In a faster and more automated approach, the so‐called “sniper cloning” 32, Lee et al. transferred the beads with a laser pulse from the sequencing plate to a 96‐well plate. Both retrieval methods require highly specialized equipment and are not compatible with all NGS platforms.
An NGS‐based method that does not require special equipment and can be implemented on any NGS‐sequencer is tag‐directed retrieval, also termed dial‐out PCR 33. It is also applicable for complex pools of DNA such as libraries, multiplex assembly reactions, or error‐rich sequences, where enzymatic error correction methods are often not sufficient 27, 33, 34. In dial‐out PCR, either single molecules of oligonucleotides 33 or up to 300 bp 14, 34 are barcoded with primer tags (see Fig. 2B). Correct fragments are then identified by NGS, and amplified using PCR primers specifically recognizing the tags. The sequence verified fragments can then be used to assemble larger constructs 34. Ninety percent error‐free fragments of up to 252 bp in length could be retrieved from a 250plex assembly reaction by using this technique 15. Due to steadily decreasing costs for NGS, dial‐out PCR is a very cost‐effective method, but also adds significantly to the overall gene synthesis time (Fig. 1) 33.
4. Assembly into longer constructs
After assembly and error reduction, the linear gene synthesis product may be used as such, e.g. directly for in vitro transcription/translation, or is inserted into a vector using classical restriction endonuclease cloning, seamless assembly techniques (Fig. 1) 35, 36, 37 or any other cloning method. As discussed before, compiling synthetic constructs exceeding 3 kb requires advanced error reduction protocols and/or sequence‐independent and scar‐less assembly of cloned and sequence‐verified building blocks.
We will thus discuss novel assembly techniques that enable scar‐less assembly of several fragments in one step. They can broadly be classified into TypeIIS restriction enzyme (TypeIIS‐RE) based methods and homology‐based methods.
TypeIIS‐RE based assembly methods take advantage of the fact that TypeIIS‐REs cut indiscriminately at a defined distance from their recognition site creating an overhang which is not part of the recognition sequence. Appended to the sequence ends in the correct orientation, they can be used to create any desired overhang at any position without leaving behind the recognition sequence. The Golden Gate method uses TypeIIS restriction sites at the ends of fragments and vector to generate unique overhangs for the scar‐less assembly of several fragments in a defined order 38. Golden Gate cloning allows the assembly of up to nine fragments in one reaction but is limited by the necessity to avoid or remove the relevant TypeIIS restriction sites within the fragments 39. MASTER (methylation‐assisted tailorable ends rational 40) removes this limitation through the use of a methylation‐dependent TypeIIS‐RE and PCR fragments generated with methylated primers such that only restriction sites at the fragment ends are recognized and cut.
Homology‐based assembly methods require the incorporation of short stretches of homologous sequence at the fragment and vector ends. Methods of this type were first described in 1990 41, 42, 43 but only recently protocols were developed that enable highly efficient, sequence independent assembly of multiple fragments.
These protocols create single stranded overhangs from the homologous stretches; assembly is then achieved by annealing complementary single strands. In sequence and ligase independent cloning (SLIC 44), the 3′ exonuclease activity of T4 DNA polymerase in the absence of dNTPs is used to chew‐back one strand from 3′ to 5′. Exonuclease activity is halted by the addition of dCTP, and the overhangs are used to assemble up to five fragments into a construct with single‐stranded gaps. These are repaired in vivo after transformation into E. coli. Gibson assembly 45 uses T5 exonuclease for 5′ to 3′ chew‐back, Phusion DNA polymerase for gap filling, and a ligase to seal the nicks, resulting in a covalently closed vector from a one‐cup in vitro reaction. Five fragments are efficiently assembled using Gibson assembly, and constructs up to 318 kb can be generated in multiple steps. Single‐stranded overhangs can also be formed through the activity of uracil‐N‐glycosylase and endonuclease VIII on DNA fragments that were generated using uracil‐containing primers as in uracil‐specific excision reagent (USER) cloning and USER fusion 46. USER fusion assembles up to three fragments in a single reaction.
Circular polymerase extension cloning (CPEC 47) utilizes a high‐fidelity DNA polymerase and temperature cycling to allow the homologous ends to anneal to and extend each other, forming the nicked target construct. CPEC has been used to assemble four fragments with a total size of 8.4 kb but entails possible introduction of sequence errors by the DNA polymerase. CPEC and USER cloning require in vivo repair in E. coli to generate the resulting plasmid, analogous to SLIC.
Several methods use the intrinsic homologous recombination activity of cells for ex vivo or in vivo assembly: Seamless Ligation Cloning Extract (SLiCE 48) employs a bacterial cell extract to assemble up to six fragments of the same type as the in vitro approaches described above.
The budding yeast Saccharomyces cerevisiae possesses considerable homologous recombination activity, which has been used for the assembly of large constructs from overlapping fragments for decades, e.g. by transformation associated recombination 49 or the DNA‐assembler method 50. Saccharomyces cerevisiae has been used to assemble 25 fragments in vivo 51, and assembly of constructs up to 1.1 Mb has been achieved 51. More recently, it has been shown that also certain E. coli strains possess sufficient homologous recombination activity to allow for transformation associated recombination of up to six fragments that create a 6.5 kb plasmid by simple transformation of fragments and vector 52.
In combination with microarray‐based next‐generation gene synthesis workflows that provide sequence‐correct DNA fragments at very low cost, these techniques allow for the efficient and cost‐effective production of large to very large constructs, up to the synthesis of complete genomes from scratch. In 2010, Gibson et al. published the first completely synthetic bacterial genome of 1.1 Mbps that made use of a hierarchical combination of seamless assembly methods, i.e. Gibson assembly and homologous recombination in yeast 53. More recently, the first synthetic eukaryotic yeast chromosome was published by the Sc2.0 consortium 54.
The goal here is to eventually build a completely synthetic variant of the yeast genome of ∼12 Mb including first approaches to redesign the natural wild‐type sequence, therefore, expanding the engineering to the content and architecture of the genome. Further, in 2016 Boeke et al. announced the human “Genome Project‐Write,” with the aim of synthesizing the entire human genome of ∼3000 Mb 55. With this undertaking it is to be expected that the development of high‐throughput next generation gene synthesis technology is further propelled and consequently gene synthesis costs will significantly drop over the next years. In addition, new and innovative DNA assembly and DNA propagation techniques will be needed and are likely to emerge from this ambitious moonshot project.
5. Concluding remarks
The progress of writing DNA has been truly impressive since the first automatic oligo synthesizer was developed in the 1980s 56. The herein discussed processes show the potential for a massive parallelization and cost reduction. The reliability could be greatly improved by coupling them with conventional techniques as a backup for complex genes and when the new approach fails. This all assumes that a scientist ordering a gene is willing to sacrifice fast delivery times for decreasing costs. For researchers with very large orders, e.g. working on whole genome synthesis, this is probably necessary to keep within their budgets. For customers ordering an individual gene, which is crucial to advance the project in time, it might be more difficult to accept.
Nonetheless, many reports have already shown that microarray‐derived genes can advance the understanding of analysis of promoter and enhancer functions in an unprecedented multiplexed manner 57, 58. More exciting experiments can be expected as large projects become a routine tool in biomolecular research.
As a result, it will be even more important that the according bioinformatics tools are developed to design DNA. In analogy to the design of computer chips, where the individual transistor is usually not designed anymore, new levels of abstraction will help to design, and maybe simulate, parts that will later be synthesized 59.
In either setting, tangible DNA molecules, genes, and genomes, are needed to build and create Synthetic Biology's innovations and makings. Like in any other engineering discipline, easy access to these building blocks is a major driver of the advancement of the technology itself. Simple, low‐cost access to synthetic DNA therefore will accelerate biotechnology and life‐science research and open new opportunities in the new era of a bio‐based economy.
The authors are all employees within Thermo Fisher Scientific/Geneart GmbH.
Acknowledgments
We would like to thank Axel Trefzer, Thomas Pöhmerer, and Melanie Horlacher for helpful discussions and proof reading of this manuscript.
6 References
- 1. Cameron, D. E. , Bashor, C. M. , Collins, J. J. , A brief history of synthetic biology. Nat. Rev. Microbiol. 2014, 12, 381–390. [DOI] [PubMed] [Google Scholar]
- 2. Notka, F. , Liss, M. Wagner, R. , Industrial scale gene synthesis. Methods Enzymol. 2011, 498, 247–275. [DOI] [PubMed] [Google Scholar]
- 3. Beaucage, S. L. , Caruthers, M. H., Deoxynucleoside phosphoramidites—a new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedr. Lett. 1981, 22, 1859–1862. [Google Scholar]
- 4. Caruthers, M. H. , Beaucage, S. L. , Becker, C. , Efcavitch, J. W. et al., Deoxyoligonucleotide synthesis via the phosphoramidite method. Gene Amplif. Anal. 1983, 3, 1–26. [PubMed] [Google Scholar]
- 5. Fodor, S. P. , Read, J. L. , Pirrung, M. C. , Stryer, L. et al., Light‐directed, spatially addressable parallel chemical synthesis. Science 1991, 251, 767–773. [DOI] [PubMed] [Google Scholar]
- 6. Pease, A. C. , Solas, D. , Sullivan, E. J. , Cronin, M. T. et al., Light‐generated oligonucleotide arrays for rapid DNA sequence analysis. Proc. Natl. Acad. Sci. U S A 1994, 91, 5022–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singh‐Gasson, S. , Green, R. D. , Yue, Y. , Nelson, C. , Blattner, F. et al., Maskless fabrication of light‐directed oligonucleotide microarrays using a digital micromirror array. Nat. Biotechnol. 1999, 17, 974–978. [DOI] [PubMed] [Google Scholar]
- 8. Gao, X. , LeProust, E. , Zhang, H. , Srivannavit, O. et al., A flexible light‐directed DNA chip synthesis gated by deprotection using solution photogenerated acids. Nucleic Acids Res. 2001, 29, 4744–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Egeland, R. , Marken, F. , Southern E., An electrochemical redox couple activitated by microelectrodes for confined chemical patterning of surfaces. Anal. Chem. 2002, 74, 1590–1596. [DOI] [PubMed] [Google Scholar]
- 10. Ghindilis, A. L. , Smith, M. W. , Schwarzkopf, K. R. , Roth, K. M. et al., CombiMatrix oligonucleotide arrays: Genotyping and gene expression assays employing electrochemical detection. Biosens. Bioelectron. 2007, 22, 1853–1860. [DOI] [PubMed] [Google Scholar]
- 11. Blanchard, A. P. , Kaiser, R. J. , Hood, L. E., High‐density oligonucleotide arrays. Biosens. Bioelectron. 1996, 11, 687–690. [Google Scholar]
- 12. Hughes, T. R. , Mao, M. , Jones, A. R. , Burchard, J. et al., Expression profiling using microarrays fabricated by an ink‐jet oligonucleotide synthesizer. Nat. Biotechnol. 2001, 19, 342–347. [DOI] [PubMed] [Google Scholar]
- 13. Saaem, I. , Ma, K. S. , Marchi, A. N. , LaBean, T. H. , Tian, J. D. , In situ synthesis of DNA microarray on functionalized cyclic olefin copolymer substrate. ACS Appl. Mater. Interface 2010, 2, 491–497. [DOI] [PubMed] [Google Scholar]
- 14. Klein, J. C. , Lajoie, M. J. , Schwartz, J. J. , Strauch, E. M. et al., Multiplex pairwise assembly of array‐derived DNA oligonucleotides. Nucleic Acids Res. 2016, 44, e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Borovkov, A. Y. , Loskutov, A. V. , Robida, M. D. , Day, K. M. et al., High‐quality gene assembly directly from unpurified mixtures of microarray‐synthesized oligonucleotides. Nucleic Acids Res. 2010, 38, e180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim, H. , Jeong, J. , Bang, D., Hierarchical gene synthesis using DNA microchip oligonucleotides, J. Biotechnol. 2011, 151, 319–324. [DOI] [PubMed] [Google Scholar]
- 17. Lee, C. C. , Snyder, T. M. , Quake, S. R., A microfluidic oligonucleotide synthesizer. Nucleic Acid Res. 2010, 38, 2514–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kong, D. S. , Carr, P. A. , Chen, L. , Zhang, S. , Jacobson, J. M., Parallel gene synthesis in a microfluidic device. Nucleic Acid Res. 2007, 35, e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quan, J. , Saaem, I. , Tang, N. , Ma, S. et al., Parallel on‐chip gene synthesis and application to optimization of protein expression. Nat. Biotechnol. 2011, 29, 449–452. [DOI] [PubMed] [Google Scholar]
- 20. Banyai, W. , Peck, B. J. , Fernadez, A. , Chen, S. , Indermuhle, P. , De novo synthesized gene libraries. US Patent Application 2015/0038373, 2015.
- 21. Tian, J. D. , Gong, H. , Sheng, N. J. , Zhou, X. C. , Gulari, E. et al., Accurate multiplex gene synthesis from programmable DNA microchips. Nature 2004, 432, 1050–1054. [DOI] [PubMed] [Google Scholar]
- 22. Richmond, K. E. , Li, M. H. , Rodesch, M. J. , Patel, M. et al., Amplification and assembly of chip‐eluted DNA (AACED): A method for high‐throughput gene synthesis. Nucleic Acids Res. 2004, 32, 5011–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou, X. , Cai, S. , Hong, A. , You, Q. et al., Microfluidic PicoArray synthesis of oligodeoxynucleotides and simultaneous assembling of multiple DNA sequences. Nucleic Acids Res. 2004, 32, 5409–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Polz, M. F. , Cavanaugh, C. M. , Bias in template‐to‐product ratios in multitemplate PCR. Appl. Environ. Microbiol. 1998, 64, 3724–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kosuri, S. , Eroshenko, N. , Leproust, E. M. , Super, M. et al., Scalable gene synthesis by selective amplification of DNA pools from high‐fidelity microchips. Nat. Biotechnol. 2010, 28, 1295–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fuhrmann, M. , Oertel, W. , Berthold, P. , Hegemann, P. , Removal of mismatched bases from synthetic genes by enzymatic mismatch cleavage. Nucleic Acids Res. 2005, 33, e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saaem, I. , Ma, S. , Quan, J. , Tian, J. , Error correction of microchip synthesized genes using Surveyor nuclease. Nucleic Acids Res. 2012, 40, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wan, W. , Li, L. , Xu, Q. , Wang, Z. , Yao, Y. et al., Error removal in microchip‐synthesized DNA using immobilized MutS. Nucleic Acids Res. 2014, 42, e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Binkowski, B. F. , Richmond, K. E. , Kaysen, J. , Sussman, M. R. , Belshaw, P. J. , Correcting errors in synthetic DNA through consensus shuffling. Nucleic Acids Res. 2005, 33, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carson, S. , Wick, S. T. , Carr, P. A. , Wanunu, M. , Aguilar, C. A. , Direct analysis of gene synthesis reactions using solid‐state nanopores. ACS Nano 2015, 9, 12417–12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matzas, M. , Stahler, P. F. , Kefer, N. , Siebelt, N. et al., High‐fidelity gene synthesis by retrieval of sequence‐verified DNA identified using high‐throughput pyrosequencing. Nat. Biotechnol. 2010, 28, 1291–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee, H. , Kim, H. , Kim, S. , Ryu, T. et al., A high‐throughput optomechanical retrieval method for sequence‐verified clonal DNA from the NGS platform. Nat. Commun. 2015, 6, 6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwartz, J. J. , Lee, C. , Shendure, J. , Accurate gene synthesis with tag‐directed retrieval of sequence‐verified DNA molecules. Nat. Methods 2012, 9, 913–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim, H. , Han, H. , Ahn, J. , Lee, J. et al., ‘Shotgun DNA synthesis’ for the high‐throughput construction of large DNA molecules. Nucleic Acids Res. 2012, 40, e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Padgett, K. A. , Sorge, J. A., Creating seamless junctions independent of restriction sites in PCR cloning. Gene 1996, 168, 31–35. [DOI] [PubMed] [Google Scholar]
- 36. Dietmaier, W. , Fabry, S. , Schmitt, R., DISEC‐TRISEC: Di‐ and trinucleotide‐sticky‐end cloning of PCR‐amplified DNA. Nucleic Acids Res. 1993, 21, 3603–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gibson, D. G. , Benders, G. A. , Axelrod, K. C. , Zaveri, J. et al., One‐step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome. Proc. Natl. Acad. Sci. U S A 2008, 105, 20404–20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Engler, C. , Kandzia, R., Marillonnet , S., A one pot, one step, precision cloning method with high throughput capability. PLoS One 2008, 3, e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Engler C, Marillonnet S., Golden gate cloning. Methods Mol. Biol. 2014, 1116, 119–131. [DOI] [PubMed] [Google Scholar]
- 40. Chen, W. H. , Qin, Z. J. , Wang, J. , Zhao, G. P., The MASTER (methylation‐assisted tailorable ends rational) ligation method for seamless DNA assembly. Nucleic Acids Res. 2013, 41, e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aslanidis, C. , de Jong, P. J., Ligation‐independent cloning of PCR products (LIC‐PCR). Nucleic Acids Res. 1990, 18, 6069–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones, D. H. , Sakamoto, K. , Vorce, R. L. , Howard, B. H., DNA mutagenesis and recombination. Nature 1990, 344, 793–794. [DOI] [PubMed] [Google Scholar]
- 43. Shuldiner, A. R. , Scott, L. A. , Roth, J., PCR‐induced (ligase‐free) subcloning: A rapid reliable method to subclone polymerase chain reaction (PCR) products. Nucleic Acids Res. 1990, 18, 1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li, M. Z. , Elledge, S. J., Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 2007, 4, 251–256. [DOI] [PubMed] [Google Scholar]
- 45. Gibson, D. G. , Young, L. , Chuang, R. Y. , Venter, R. C. et al., Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [DOI] [PubMed] [Google Scholar]
- 46. Geu‐Flores, F. , Nour‐Eldin, H. H. , Nielsen, M. T. , Halkier, B. A., USER fusion: A rapid and efficient method for simultaneous fusion and cloning of multiple PCR products. Nucleic Acids Res. 2007, 35, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Quan, J. , Tian, J., Circular polymerase extension cloning of complex gene libraries and pathways. PLoS One 2009, 4, e6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang, Y. , Werling, U. , Edelmann, W. , SLiCE: A novel bacterial cell extract‐based DNA cloning method. Nucleic Acids Res. 2012, 40, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma, H. , Kunes, S. , Schatz, P. J. , Botstein, D., Plasmid construction by homologous recombination in yeast. Gene 1987, 58, 201–216. [DOI] [PubMed] [Google Scholar]
- 50. Shao, Z. , Zhao, H. , Zhao, H. , DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 2009, 37, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gibson, D. G. , Benders G. A., Andrews‐Pfannkoch, C. , Denisova, E. A. et al., Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 2008, 319, 1215–1220. [DOI] [PubMed] [Google Scholar]
- 52. Kostylev, M. , Otwell, A. E. , Richardson, R. E. , Suzuki, Y., Cloning should be simple: Escherichia coli DH5α‐mediated assembly of multiple DNA fragments with short end homologies. PLoS One 2015, 10, e013746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gibson, D. G. , Glass, J. I. , Lartigue, C. , Noskov, V. N. et al., Creation of a bacterial cell controlled by a chemically synthesized genome. Science 2010, 329, 52–56. [DOI] [PubMed] [Google Scholar]
- 54. Annaluru, N. , Muller, H. , Mitchell, L. A. , Ramalingam, S. et al., Total synthesis of a functional designer eukaryotic chromosome. Science 2014, 344, 55–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Boeke, J. , Chruch, G. , Hessel, A. , Kelley, N. et al., The Genome Project–Write. Science 2016, 353, 126–127. [DOI] [PubMed] [Google Scholar]
- 56. Alvarado‐Urbina, G. , Sathe, G. M. , Liu, W. C. , Gillen, M. F. et al., Automated synthesis of gene fragments. Science 1981, 214, 270–274 [DOI] [PubMed] [Google Scholar]
- 57. Patwardhan, R. P. , Lee, C. , Litvin, O. , Young, D. et al., High‐resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 2009, 27, 1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Smith, R. P. , Taher, L. , Patwardhan, R. P. , Kim, M. J. et al., Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat. Genet. 2013, 45, 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nielsen, A. A. K. , Der, B. S. , Shin, J. , Vaidyanathan, P . et al., Programming circuitry for synthetic biology, Science 2016, 352, 48–50. [Google Scholar]