

Abstract
Terpene synthase catalyses acyclic diphosphate farnesyl diphosphate into desired sesquiterpenes. In this study, a fusion enzyme was constructed by linking Santalum album farnesyl pyrophosphate synthase (SaFPPS) individually with terpene synthase and Artemisia annua Epi‐cedrol synthase (AaECS). The stop codon at the N‐terminus of SaFPPS was removed and replaced by a short peptide (GSGGS) to introduce a linker between the two open reading frames. This fusion clone was expressed in Escherichia coli Rosseta DE3 cells. The fusion enzyme FPPS‐ECS produced sesquiterpene 8‐epi‐cedrol from substrates isopentenyl pyrophosphate and dimethylallyl pyrophosphate through sequential reactions. The K m values for FPPS‐ECS for isopentyl diphosphate was 4.71 µM. The fusion enzyme carried out the efficient conversion of IPP to epi‐cedrol, in comparison to single enzymes SaFPPS and AaECS when combined together in enzyme assay over time. Further, the recombinant E. coli BL21 strain harbouring fusion plasmid successfully produced epi‐cedrol in fermentation medium. The strain having fusion plasmid (pET32a‐FPPS‐ECS) produced 1.084 ± 0.09 mg/L epi‐cedrol, while the strain harbouring mixed plasmid (pRSETB‐FPPS and pET28a‐ECS) showed 1.002 ± 0.07 mg/L titre in fermentation medium by overexpression and MEP pathway utilization. Structural analysis was done by I‐TASSER server and docking was done by AutoDock Vina software, which suggested that secondary structure of the N‐ C terminal domain and their relative positions to functional domains of the fusion enzyme was greatly significant to the catalytic properties of the fusion enzymatic complex than individual enzymes.
Keywords: epi‐cedrol synthase, fusion enzymes, recombinant expression
Abbreviations
- DMAPP
dimethylallyl pyrophosphate
- DTT
dithiothreitol
- DXP
deoxy‐D‐xylulose‐5‐phosphate
- ECS
epi‐cedrol synthase
- FPP
farnesyl diphosphate
- FPPS
farnesyl pyrophosphate synthase
- GPP
geranyl diphosphate
- IPP
isopentyl diphosphate
- IPTG
isopropyl thio‐β‐D‐thiogalactopyranoside
- MEP
methyl D‐erythritol 4‐phosphate
- MVA
mevalonate
1. INTRODUCTION
Sesquiterpenes (C15) are chemically and structurally diverse class of terpenes which have found vast commercial applications in chemical, flavour, fragrance, pharmaceutical and, nutraceutical industries 1, 2. However, these are produced in very low concentrations by mevalonate (MVA) and methyl D‐erythritol 4‐phosphate (MEP) or deoxy‐D‐xylulose‐5‐phosphate (DXP) pathaway in natural sources, hampering industrially viable commercial production. Therefore, biotechnological intervention is considered as a key solution to this issue 3. For the same, a detailed outline of the biosynthetic pathway of terpenoids in source plant, including genes and enzymes involved in the pathway is mandatory 4.
Since last two decades, several hundreds of genes encoding sesquiterpene synthase have been isolated from a different plant source. Many of these genes or pathways have been transferred from their natural producer organisms to well characterized microbial host. Further, additional engineering of these genes and pathway has aided to the process of achieving higher product titre 5. Fusion of different enzymes catalyzing sequential reactions of a metabolic pathway is an emerging strategy in the field of metabolic engineering 6, 7, 8. The fusion protein, produced by fusing sequences of two genes together under a single promoter, offers the advantage of producing a larger protein with two or more functions which may catalyse consecutive steps more efficiently than a simple mixture of the individual free enzymes in a metabolic pathway 7, 8, 9, 10. Possibly, the proximity of fusion enzymes influences the efficiency of sequential reactions 9. Use of fusion proteins also reduces the number of vectors in a heterologous expression system. The construction of bi‐functional enzyme also can reduce the cost of recombinant protein purification and enhance the cofactor regeneration rate, which simplifies the reconstitution of metabolic pathways 6, 11. Fusion protein strategy has also been explored earlier for metabolic engineering of sesquiterpenoids, such as fusion of patchoulol synthase (PTS) and farnesyl diphosphate synthase (FPPS), which channeled more metabolic flux to patchoulol production 12, FPPS‐epi‐aristolochene synthase (eAS) for the production of epi‐aristolochene 13, and fusion of (S)‐linalool synthase (AaLS1) which led to enhancement in the production of linalool in yeast host 14.
Sesquiterpenes from Artemisia annua L have received significant interest on account of wide variety of pharmacological properties like antibacterial, antiviral, antifungal, insecticide, anticancer activity, etc. 15, 16, 17. Amongst these metabolites, artemisinin, a sesquiterpene lactone is most widely used as an antimalarial drug 18. Immense commercial interest has brought about many investigations to explore sesquiterpene biosynthesis in A. annua and its biotechnological manipulation. As a result, artemisinin has been successfully produced at industrially desired scales in alternative microbial host via metabolic engineering of artemisinin biosynthesis related sesquiterpene synthase genes 19, 20, 21. In due course of time, exploration of A. annua sesquiterpene biosynthesis inevitably led to many sesquiterpene synthase genes being cloned and characterized from this plant including those producing epi‐cedrol (ECS), β‐caryophyllene, β‐copaene, germacrene, etc. 22. Among these sesquiterpenes, epi‐cedrol remains least explored metabolite with high untapped commercial potential. Epi‐cedrol is potential precursor to cedrenes which are being used as important source materials for generation of advanced high density jet and diesel biofuels currently under development 23. Further, cedrol is widely used in pharmaceutical industries for sedative, anti‐inflammatory, and cytotoxic activities 24, 25, 26. Therefore, epi‐cedrol yield enhancement signals industrial benefit, and biotechnological solution should be ventured upon.
PRACTICAL APPLICATION
The fusion enzyme approach and its applications in natural product production and multi‐enzyme reactions in single step: the construction of fusion enzymes with small linker peptides is cost effective for recombinant protein purification and enhances the reaction rate compared to individual enzymes. Our work demonstrates that the small Gly and Ser rich peptide between unnatural multifunctional fusion enzyme FPPS‐ECS enhances the heterologous production of plant derived sesquiterpene epi‐cedrol by MEP pathway. This study shall be useful to those who are interested in heterologous expression and production of natural products. Also, this work may be extended to other fusion or bi‐enzyme systems for the chemo‐enzymatically biosynthesis of various products.
Fusion protein strategy can be implemented with sesquiterpene synthase genes for a yield enhancement by fusion of FPPS‐with a sesquiterpene synthase gene. Such a fusion protein would catalyse one step production of corresponding sesquiterpenes from isopentyl diphosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) with enhancement in FPPS precursor pool towards sesquiterpene synthase genes via mevalonate (MVA) and non‐mevalonate (MEP) pathways that operate in the cytosol and plastid, respectively 27.
In our previous studies, we have cloned and functionally characterized sesquiterpene synthase gene epi‐cedrol synthase (AaECS) from A. annua and farnesyl pyrophosphate synthase (SaFPPS) from S. album in the bacterial host 28, 29. In this study, we have constructed fusion proteins by linking SaFPPS gene with sesquiterpenoid synthase gene AaECS with the help of short Gly‐Ser‐Gly‐Gly‐Ser linker and expressed in E. coli, then characterized for single step biosynthesis of 8‐epi‐cedrol in maximum production by metabolic engineering.
2. MATERIALS AND METHODS
2.1. Materials
The E. coli strains Rosetta (DE3) and BL21(DE3), as well as the Polymerase AccuPrime Pfx Supermix were purchased from Invitrogen‐Thermo Fischer. T4 DNA ligase and high‐Fidelity (HF) Restriction enzymes were purchased from New England Biolabs, [1‐14C] IPP (55 Ci۔mmol−1), geranyl diphosphate (GPP), and [1‐3H], FPP (16 Ci۔mmol−1) were synthesized according to previous reports 30. Isopropyl thio‐β‐D‐thiogalactopyranoside (IPTG), DMAPP, antibiotics, and GenElute™ PCR Clean‐Up kits were purchased from Sigma‐Aldrich.
2.2. PCR cloning of FPPS from S. album into pET 32a
A previously constructed cloned cDNA of FPPS 29 (GenBank Acc. No. KF011939) from S. album was used to amplify wild‐type FPPS without a stop codon and linker fragment (Gly‐Ser‐Gly‐Gly‐Gly‐Ser) at 3′ end including Bam HI site which encodes for glycine and serine. Wild‐type FPPS was initially amplified by using blunt P1 and P2 primer and further amplified using P3 (forward) and P4 (reverse) containing Nco I and Bam HI restriction sites, respectively (Table 1). PCR was carried out in a total volume of 50 µL with the following reagents: 45 µL Accuprime Pfx polymerase (Invitrogen), 20 pmol of each primer, and 10 ng of cloned plasmid. PCR cycling was: one cycle of 95°C (5 min); 35 cycles of 95°C (30 s), 54°C (30 s), 68°C (1.1 min); 68°C (5 min). The FPPS‐wild‐type fragment was digested with Nco I and Bam HI. The bacterial expression vector pET32a (Novagen) was cleaved with the same enzymes and treated with alkaline phosphatase (NEB). Vector and the fragment were purified by using the GenElute PCR Clean‐Up kit (Sigma‐Aldrich). Ligation of the fragment in frame with a multifunctional His‐tag in the vector was carried out using T4 DNA ligase (Thermo Fisher Scientific). The ligation mixture of pET32a FPPS was transformed into E. coli Mach1 T1R (Thermo Fisher Scientific). Colonies were analyzed by PCR using P3 primer and T7 vector specific reverse primer to confirm the presence of the FPPS gene. Positive colonies were grown in Luria‐Bertani medium containing ampicillin (100 µg/mL) and the plasmid was purified using the plasmid GenElute Plasmid Miniprep kit (Sigma‐Aldrich) and used as a template for insertion of another gene for constructing fusion.
Table 1.
Primers used in cloning
| Primer | Sequence (5′‐3′) | Template | RE* |
|---|---|---|---|
| P1 | ATGGGCGATCGGAAAACCAAA | SaFPPS F | ‐ |
| P2 | CTTCTGCCGCTTGTATATCTTCGC | SaFPPS R | ‐ |
| P3 | GAGACCATGGGCGATCGGAAAACC | SaFPPS F | Nco I |
| P4 | AAGGATCC GCCGCTGCCCTTCTGCCGCTTGTATA | SaFPPSLnk R | Bam HI |
| P5 | AGCCTGATTGTTGAAGATGTTATTCGTCCG | AaECS F | ‐ |
| P6 | TTAGGTGATGATGGCATCCACAAACAG | AaECS R | ‐ |
| P7 | ACGGGATCCAGCCTGATTGTTGAAGATGTTA | AaECS F | Bam HI |
| P8 | AAGCGGCCGCTTAGGTGATGATGGCATC | AaECS R | Not I |
Restrictions sites are underlined and linker sequences are in bold.
2.3. Cloning of ECS from A. annua into FPPS‐pET32a
For fusion of FPPS and ECS, ECS gene fragment without start codon was amplified by using blunt P5 and P6 primer from a cloned plasmid 28 (GenBank No. AF157059) and further amplified using P7 (forward) and P8 (reverse) containing a Bam HI and Not I restriction sites, respectively (Table 1). PCR was carried out in a total volume of 50 µL with the following reagents: 45 µL Accuprime Pfx polymerase, 20 pmol of each primer and 10 ng of cloned plasmid. PCR cycling was: 1 cycles of 95°C (5 min); 35 cycles of 95°C (30 s), 54°C (30 s), 68°C (1.30 min); 68°C (5 min). The ECS‐wild‐type fragment was digested with Bam HI and Not I. The newly constructed FPPS‐pET32a cloned bacterial expression vector was digested with the same enzymes and treated with alkaline phosphatase for inhibiting self‐ligation. Vector and the fragment were kit purified and ligated by using T4 DNA ligase (Invitrogen) as mentioned above. Transformed colonies were analyzed by using PCR with P7 forward primer and T7 vector specific reverse primer to confirm the presence of the wild‐type ECS gene. Positive colonies were grown in Luria–Bertani medium containing ampicillin (100 µg/mL) and the plasmid was purified using the plasmid GenElute Plasmid Mini‐prep kit (Sigma‐Aldrich). This fusion plasmid was used for protein expression.
2.4. Optimization of expression and purification of the recombinant proteins
The plasmids were transferred into E. coli Rosetta DE3α cells. The cells carrying the plasmids FPPS‐pRSETB 29, ECS‐pET28a 28, and FPPS‐ECS‐pET32a were grown in 1 L Terrific broth (Hi‐media, India), with ampicillin (100 µg/mL) and chloramphenicol (34 µg/mL), at 37°C. At OD600 of ∼0.8, IPTG was added to the final concentration of 1 mM. The cells were harvested after overnight incubation at 16°C by centrifugation at 6500 × g for 10 min at 16°C, and pellets were re‐suspended in 15 mL lysis buffer (50 mM Tris/HCl pH 7.8, containing 300 mM NaCl and 10% glycerol, 5 mM imidazole, 1 mM dithiothreitol), lysozyme (1 mg/mL), CHAPS (0.01%), and Triton X 100 (0.1%) were added. The cells were disrupted by sonication (Braun‐Sonic 2000 microprobe at maximum power for 10 × 30 s bursts with a 30 s chilling period on ice between bursts). The sonicated crude was centrifuged at 10 000 × g for 15 min at 4°C. The supernatant was collected and used for His‐Tag Ni2+‐NTA affinity purification. 25 mM Imidazole concentration was used for washing undesired proteins and 250 mM imidazole was used to elute the final pure proteins. Fractions of 0.5 mL were collected and checked by using 10% SDS‐PAGE analysis and further desalted by using GE‐ÄKTA instrument. Desalted proteins were stored at −80°C. Protein concentrations and purified recombinant enzymes were determined according to Bradford using BSA as standard 31.
Optimization of expression was carried out for FPPS‐ECS‐pET32a fusion proteins constructs. The parameters investigated were IPTG concentration (0.2, 0.4, 0.6, 0.8, and 1.0 mM) used for induction, induction temperature (16, 25, and 30°C), and time of harvest after induction (16, 12, and 8 h).
2.5. Enzyme assays and kinetics
Enzyme activity of single FPPS and ECS was assayed as per our previous reports using IPP, GPP as a substrate for FPPS and FPP as a substrate for ECS 28, 29. The newly constructed fusion protein was monitored by measuring the amount of sesquiterpene 8‐epi‐cedrol from IPP, DMAPP and IPP, GPP by the enzymatic reaction (Figure 1). For FPPS‐ECS fusion, 400 µL enzymatic reaction mixture containing assay buffer pH 8.5 (25 mM Tris‐HCl, 5 mM dithiothreitol, 10 mM MgCl2, 20% glycerol), 15 µL purified fusion protein (25 µg), 100 µM IPP, 50 µM DMAPP (for control 50 µM GPP + 50 µM IPP, or 100 µM FPP used) were incubated in shaker bath incubator (Brunswick, Eppendorf) at 30°C for 1 h at 70 rpm. The enzymatic reaction was stopped by an addition of 10 µl absolute ethanol (95%) followed by vortexing for 30s. All the products were extracted with hexane (3 × 0.5 mL). The organic layer containing sesquiterpene products were dried over anhydrous Na2SO4 and reduced to ∼50 µL with a stream of dry nitrogen. The samples were identified by GC‐MS analysis by using Agilent Technology 5975–7890 GC‐MS system with a HP‐5MS capillary column (30 m × 0.25 mm × 0.25 µ coating of 5% phenyl methyl siloxane). Injections were cooled on‐column at 40°C with oven programming from 40°C (50 K/min) to 50°C (5‐min hold), then 10 K/min to 250°C, then 50 K/min to 300°C. Separations were made under a constant flow of 1 mL He y min. Mass spectral data were collected at 70 eV and analyzed by using MSD Chem station software.
Figure 1.
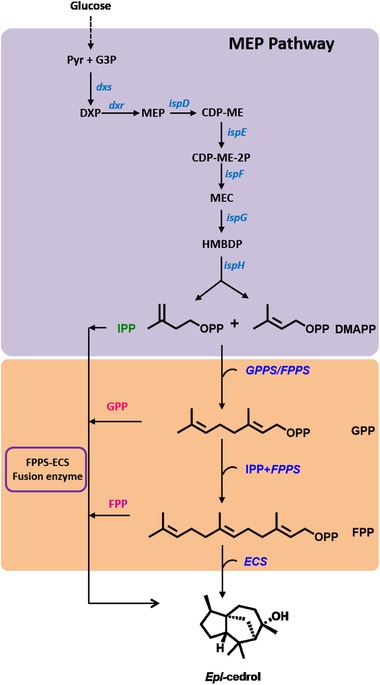
Schematic diagram of MEP pathway (blue), and epi‐cedrol biosynthetic pathway in recombinant E. coli. G3P, glyceraldehyde 3‐phosphate; 3Pyr, pyruvate; DXP, 1‐deoxy‐D‐ xylulose‐5‐phosphate; MEP, 2C‐methyl‐D‐erythritol‐4‐phosphate; CDP‐ME, 4‐diphosphocytidyl‐2C‐methyl‐D‐erythritol; CDP‐ME‐2P, 4‐diphosphocytidyl‐2C‐ methyl‐D‐erythritol‐2‐phosphate; MEC, 2C‐methyl‐D‐erythritol 2,4‐cyclodiphosphate; HMBDP, 1‐hydroxy‐2‐methyl‐2‐(E)‐butenyl‐4‐diphosphate; IPP, isopentenyl pyrophosphate; DMPP, dimethylallyl pyrophosphate; FPP, farnesyl pyrophosphate; GPPS, geranyl pyrophosphate synthase; FPPS, farnesyl pyrophosphate synthase; ECS, epi‐cedrol synthase
For the analysis of substrate channeling effect of fusion enzyme (FPPS‐ECS) as compared to mixed enzymes (FPPS+ECS) together, time‐dependent enzyme assay was carried out with the IPP and DMAPP as a substrate. In a typical 400 µL enzymatic reactions containing equimolar concentrations of FPPS+ECS and FPPS‐ECS enzymes (1.5 nM), 100 µM IPP and 50 µM DMAPP substrates were used. The assay products were harvested in different time of interval (30, 60, 90, 120, 150, 180 min) and quantified by GC‐MS analysis as per above mentioned method and program. The experiments were carried out in three replicates.
For the enzyme kinetic study, the substrate IPP 1–50 µM and DMAPP 0.5–25 µM were used for the conversion into epi‐cedrol by fusion enzyme and individual enzymes. The enzyme concentration was 1.5 nM for all the reactions and the geraniol was used as a standard. The comparative study and the coupled activity were done by relative peak percentage area of respected sesquiterpenes in the GC‐MS analysis.
2.6. Epi‐cedrol production
The recombinant E. coli BL21 strain individually harbouring pRSETB‐FPPS, pET28a‐ECS, and pET32a‐FPPS‐ECS fusion plasmids were cultured in 1000 mL TB medium supplemented with appropriate antibiotics. Working concentrations of ampicillin and kanamycin were 100 and 50 µg/mL, respectively. Starter cultures were grown at 37ºC and induced with 1 mM IPTG (OD600∼0.8) and allowed to continously grow at 30ºC, for 20 h. An amount of 50 mL dodecane was overlaid on medium to capture volatile compounds. Hundred millilitre of samples (cells) were harvested after 2 h of time interval up to 20 h after induction. OD measurements were conducted on a UV–vis spectrophotometer (Thermo‐Fisher Scientific Evolution 200) operating at 600 nm. Supernatant broth medium was used for the extraction of epi‐cedrol by four washes of 100 mL petroleum ether. Solvent was evaporated by rota‐evaporator (Heidolph, Germany). Samples were analysed and quantified by using GC‐MS analysis as per mentioned method. Farnesol was used as internal standard. The experiments were performed as well as analysed in triplicates.
2.7. Protein modelling and docking simulations
Homology modeling of FPPS‐ECS fusion protein was carried out utilizing the Swiss Model automated comparative protein modeling server (http://swissmodel.expasy.org) 32. According to the amino acid sequence homology, FPPS (AsFPPS) from A. spiciformis (PDB ID: 4KK2, sequence identity: 78.87%) for SaFPPS, and α‐Bisabolol synthase (AaBOS) from A. annua (PDB ID.: 4GAX, sequence identity: 53.65%) for ECS were selected as potential structural templates to obtain the homology based structural models. The three‐dimensional (3‐D) structural models of both the fusion enzyme complex with linker incorporation were built up using the web‐based I‐TASSER server 33. I‐TASSER server gives the C‐score which is a confidence score for estimating the quality of predicted models by I‐TASSER and calculated based on the significance of threading template alignments and the convergence parameters of the structure assembly simulations. It also provides TM‐score and RMSD values, which are known standards for measuring the structural similarity between two structures which are usually used to measure the accuracy of structure modeling when the native structure is known. The server predicted the cluster density, which is defined as the number of structure decoys at a unit space in the SPICKER cluster. A higher cluster density means the structure occurs more often in the simulation trajectory and, therefore, signifies a better quality model 33. Molecular graphics software program PyMOL 2.0.0 was used for illustration.
For docking simulations, initially the substrate binding pockets in FPPS‐ECS fusion protein model were identified using CASTp, Pocket‐Finder, and QSite Finder. Further, docking simulations were carried using AutoDock Vina (v4.2) with all the three substrates (IPP, DMAPP, and FPP) taken one at a time with grid box spacing at 1 Å. The docking parameters for FPPS‐ECS were set with grid box size of (64 × 84 × 110), (84 × 84 × 90) and center at and x = 87.528, y = 82.500, z = 83.944. For single enzymes, the docking parameters for FPPS with IPP and DMAPP were set with grid box size of (50 × 92 × 80), and centre at and x = 64.889, y = 58.472, z = 64.833, whereas the docking parameters of ECS with FPP with the grid box spacing at 1 Å was set at (64 × 72 × 86), and centre at and x = 75.806, y = 61.462, z = 69.722. The ligand protein interactions were analysed and visualized using Discovery Studio 4.5 Visualizer and Pymol v2.0.0 34.
3. RESULTS AND DISCUSSION
For expression of recombinant fused enzyme, the bacterial expression vector pET32a was used (cloning shown in Supporting Information Figures S1–S2), whereas, for the production of single enzymes, different expression vectors were used 28, 29. This selection was made because of increased protein solubility due to the presence of thioredoxin tag in the vector 35. The sequence also contains an enterokinase cleavage site for removal of the fusion tag. The three plasmids FPPS‐pRSETb, ECS‐pET28a, and FPPS‐ECS‐pET32a were isolated and transferred into E. coli Rosetta (DE3) for production of the recombinant enzymes. The transformed bacteria were grown and induced with IPTG (1 mM), and purified by Ni+2‐NTA affinity column and evaluated for FPPS and/or ECS activity. The recombinant multifunctional enzyme showed both activities, as the linker GSGGS is of ideal size to permit the two enzymes to fold properly. When fusing two multimeric enzymes without linker or short linker could lead to the formation of multimeric protein network which cause steric hindrance of the active domain, whereas, if a linker is too long it may be sensitive to proteolytic attack and also lead to reduced channeling of substrates between two active sites 7, 36.
Optimization of expression was carried out for the new fusion construct as mentioned in Section 2, 1 mM IPTG induction and 16ºC overnight (12–14 h) incubation showed the optimum level of expression in SDS gel (optimization shown in Supporting Information Figure S3), same conditions were applied for other constructs also. Fused enzyme was expressed as a fusion with the thioredoxin protein tag, which helps to improve the solubility of expressed proteins 35, 37. SDS‐PAGE images of all fractions of FPPS‐ECS fusion protein are shown in Figure 2(I). All the proteins (including single) were produced in large scale (2× 1000 mL TB broth) using the condition mentioned above and purified by Ni+2NTA affinity chromatography. For better and enhanced purification of fusion proteins, SEC was used. Partially purified protein was passed through a SEC (See Section 2). The characterization of recombinant protein was carried out with thioredoxin‐Stag‐His‐tag as N‐terminal fusion.
Figure 2.
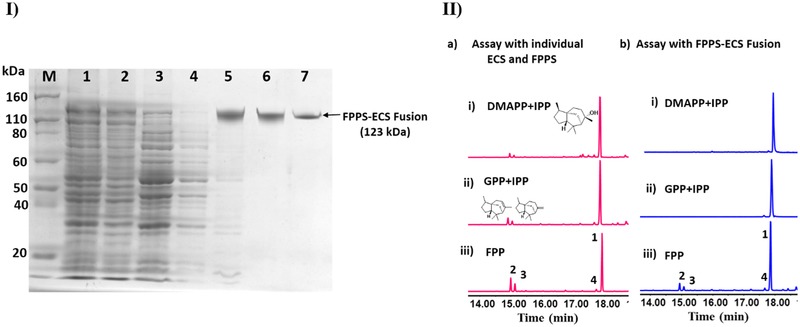
(I) SDS‐PAGE gel image of FPPS‐ECS fusion protein in which (1) crude fraction, (2) pellet, (3) supernatant, (4) 25 mM imidazole wash, (5–7) elution fractions (Purified), and (M) protein ladder (Thermo‐Fischer scientific). (II) GC‐FID chromatogram for the assay extracts: a) individual enzymes assay with substrate (i) 2×IPP+DMAPP, (ii) GPP+IPP, (iii) FPP; b) FPPS‐ECS fusion enzyme assay with substrate (i) 2xIPP+DMAPP (ii) GPP+IPP, and (iii) FPP. Products were epi‐cedrol (1), α‐cedrene (2), β‐cedrene (3), cedrol (4). Pink colour chromatograms represent the individual enzyme assay whereas blue colour chromatograms represent fusion enzyme assay
3.1. Kinetic properties of recombinant enzymes
The SEC‐purified pure enzymes were used to determine K m value according to standard techniques 29. The K m value of ECS for FPP was 3.74 µM. The K m values of fusion enzymes FPPS‐ECS for IPP were calculated to be 4.71 µM. A slightly higher K m value was observed for the single enzymes together than the fused enzyme for IPP as a substrate. K m value for DMAPP was half of the K m for IPP as the half concentration of DMAPP was used with the IPP in the kinetics study (kinetics shown in Supporting Information Figure S4). Table 2 reveals the catalytic potential of fused enzyme over single enzymes together. In case of fusion enzyme, K cat /K m ratio significantly increased compared to the combination of single enzymes which shows the fused enzyme has the catalytic efficiency more than individual enzymes together. Overall, these results correlated to those previously reported fusion proteins for FPPS and sesquiterpene synthases from other sources 12, 13, 14.
Table 2.
Kinetic parameters of free enzymes and fusion enzymes
| Enzymes | K m (µM) | V max (µM/min) | K cat (sec−1) | K cat /K m (×106 M−1S−1) |
|---|---|---|---|---|
| For IPP | ||||
| FPPS+ECS | 5.64 ± 0.2 | 0.81 ± 0.1 | 1.4 ± 0.1 | 0.24 ± 0.1 |
| FPPS‐ECS Fusion | 4.71 ± 0.1 | 0.92 ± 0.1 | 1.88 ± 0.1 | 0.40 ± 0.1 |
| For DMAPP | ||||
| FPPS+ECS | 2.82 ± 0.2 | 0.81 ± 0.1 | 1.4 ± 0.1 | 0.49 ± 0.1 |
| FPPS‐ECS Fusion | 2.35 ± 0.1 | 0.92 ± 0.1 | 1.88 ± 0.1 | 0.80 ± 0.1 |
| For FPP | ||||
| ECS | 3.74 ± 0.2 | 1.23 ± 0.1 | 0.95 ± 0.1 | 0.25 ± 0.1 |
Reaction conditions: 1.5 nM of free enzymes (added together) and fusion enzymes were used for the measurement of kinetic parameters against IPP. The reaction was performed as described in the method section using 1–30 µM IPP and 0.5‐15 µM dimethylallyl pyrophosphate respectively. For ECS, FPP was used as a substrate
ECS, epi‐cedrol synthase; FPP, farnesyl diphosphate; FPPS, farnesyl pyrophosphate synthase; IPP, isopentyl diphosphate.
3.2. Coupled enzyme activity
The fused enzyme FPPS‐ECS converts DMAPP and IPP or GPP or FPP to epi‐cedrol as a major product (Figure 1). The fusion assay was kept for 1 h, as the longer incubation time affects the activity of the fusion protein and the stability of the sesquiterpenes. In case of FPPS‐ECS fusion, the enzyme assay with DMAPP/IPP or GPP, the side products such as α‐cedrene, β‐cedrene were found in trace amount as compared to individual assay with FPP, but in the fusion enzyme assay with FPP, these products were observed with certainty (Figure 2II, Table 3). Although the ratios were different, but the relative amount of sesquiterpenoids formed from fusion enzyme was higher than the individually produced by two single enzymes with various substrate combinations. Same results were obtained in the time‐dependant enzymatic assay with fusion enzyme and free enzymes together. The relative amount of sesquiterpenoids formed from fusion enzyme was higher than the individually produced two single enzymes together over different time of interval with IPP and DMAPP as a substrate, as indicated by relative peak percent area (Figure 3A). This is because of protein‐protein interactions may also increase metabolic efficiency either by channeling intermediates between enzymes or by localizing two active sites in close proximity 17. The larger portion of the FPP produced by the FPPS part of the enzyme is converted to final product by ECS. This may be expected as the building‐up of the intermediate FPP is more rapid at higher amounts of FPPS and the subsequent ECS experiences a higher substrate concentration, i.e. a steady‐state condition is approached. As per previous reports, the orientation of the two enzymes does not affect the activity as well as K m value of the fusion enzymes 13. Finally, the performance of the multifunctional enzymes, the level of sesquiterpenoid produced from IPP by FPPS‐ECS was compared with that produced by corresponding amounts of two single enzymes (equal molar concentrations) over time. As FPPS has active domains for GPP synthesis from DMAPP and IPP, further GPP with IPP converted into FPP by this enzyme. The amount of FPP was higher in fusion enzyme than individual enzymes in minimum time of interval. Therefore, the amount of epi‐cedrol produced was considerably higher for the fusion enzyme than for the mixture of single enzymes, respectively. The amount of epi‐cedrol produced was considerably 1.66 fold higher for the fusion enzyme than for the mixed enzymes together after 1 h of incubation (Figure 3A). Fusion enzymes consume maximum amount of substrates present in the buffer leading to increase in the amount of desired metabolites. This result shows that the FPP produced by the first enzyme is transferred to the active site of ECS with limited diffusion into surrounding solution. A proximity effect and substrate channeling operates in the fusion enzymes and increases the overall catalytic activity of the enzymatic reaction. Similar substrate channeling has also been observed in previously constructed artificial bifunctional enzymes 7, 38.
Table 3.
Comparisons between (%) ratios of sesquiterpenes (%) formed from various substrates (DMAPP/IPP/GPP/FPP) with enzyme assay with individual enzymes (FPPS and ECS) and fusion enzyme (FPPS‐ECS)
| Enzyme assay with individual enzymes | Enzyme assay with fusion enzyme | |||||
|---|---|---|---|---|---|---|
| Substrates | DMAPP + IPP (%) | GPP + IPP (%) | FPP (%) | DMAPP + IPP (%) | GPP + IPP (%) | FPP (%) |
| Enzymatic products | ||||||
| epi‐Cedrol | 88.2 ± 2.3 | 85.1 ± 2.1 | 80.7 ± 3.5 | 97.6 ± 0.9 | 97.8 ± 1.2 | 83.2 ± 2.1 |
| α‐Cedrene | 5.8 ± 0.9 | 8.5 ± 1.1 | 12.8 ± 2.1 | 1.1 ± 0.3 | 0.3 ± 0.3 | 8.7 ± 2.1 |
| β‐Cedrene | 3.1 ± 0.6 | 4.2 ± 0.8 | 4.9 ± 1.3 | 0.4 ± 0.1 | 0.2 ± 0.1 | 4.6 ± 0.6 |
| Cedrol | 2.6 ± 0.3 | 2.1 ± 0.2 | 1.5 ± 0.4 | 0.8 ± 0.2 | 1.7 ± 0.4 | 3.5 ± 1.3 |
Enzyme assay condition: a 400 µL enzymatic reaction mixture containing Tris/Hepes assay buffer (pH 8.5), 15 µL purified protein (25–100 µg), 100 µM IPP, 50 µM DMAPP, 50 µM GPP and 100 µM FPP was incubated at 30ºC for 60 min.
DMAPP, dimethylallyl pyrophosphate; FPP, farnesyl diphosphate; GPP, geranyl diphosphate; IPP, isopentyl diphosphate.
Figure 3.
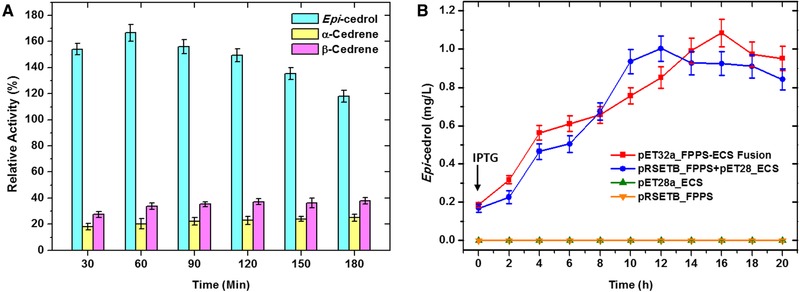
(A) Time dependant enzyme activity of the FPPS‐ECS fusion enzyme and FPPS+ECS free enzymes. The relative activity expressed in comparison of the percentage of the activity of free enzymes, i.e FPPS+ECS with IPP and DMAPP substrates only. (B) Production of epicedrol over time by E. coli BL21 strains harbouring pET32a‐FPPS‐ECS fusion, pRSET‐B‐FPPS+pET28a‐ECS mixed, pRSET‐B‐FPPS, and pET28a‐ECS plasmids (Error bars represent SD (n = 3))
3.3. Epi‐cedrol production
Biosynthesis of terpenoid production in E. coli has been carried out by the MEP or DXP pathway (Figure 1). This pathway consists of seven enzymatic steps that convert glyceraldehyde‐3‐phosphate (G3P) and pyruvate to IPP and DMAPP in a ratio of 5:1 39. It initiates with enzyme DXP synthase (DXS) which catalyzes the condensation of G3P and pyruvate to form DXP. These total seven genes (DXS, DXR, and Isp‐DEFGH) encoding the MEP/DXP pathway in the E. coli genome (Figure 1) are regulated by various promoters 40. Overexpression of terpene synthases leads to the biosynthesis of respective isoprenoids. This pathway is affected by various aspects of the intracellular metabolites including available carbon, ATP, the reducing power, and accumulation of intermediates of the pathway 41.
Production of epi‐cedrol was carried out by transferring all plasmid individually as well as mixed together into the E. coli BL21 cells. Batch culture was initiated and recombinant cells were harvested over different time of interval, epi‐cedrol was extracted from fermentation media and analyzed by GC–MS as mentioned in Section 2. Out of the four recombinant strains, only FPPS‐ECS fusion and FPPS+ECS mixed plasmids showed the good titre of epi‐cedrol in the fermentation medium. Strain harbouring pRSETB‐FPPS plasmid produced only FPPS which was utilized for the production of FPP in the cells. However, single pET28‐ECS plasmid harbouring strain produced negligible amount of epi‐cedrol after 12–14 h of induction (Figure 3B). For comparing activity, mixed plasmids together in E. coli showed good amount of epi‐cedrol titre in batch fermentation although maintaining multiple plasmids may increase the metabolic loads on the cell from DNA, RNA, and protein synthesis as well as the various antibiotic resistance proteins the cell must produce 42, and leads to affect the production of the desired final products 43. But the expression levels of two plasmids may vary in the cell compared to single fusion plasmid. Overall results showed that there was not much difference between the mixed plasmid and the fusion plasmid harbouring cell. In case of mixed plasmids optimum titre of epi‐cedrol was 1.002 mg/L obtained after 12 h of incubation (post induction), while E. coli harbouring fusion plasmid produced maximum 1.084 mg/L epi‐cedrol after 16 h of incubation. This is because of the different expression levels of genes and the substrate channeling effects. The recombinant strain overexpressing fusion as well as mixed plasmids, were three times higher the titre of epi‐cedrol than previously reported in engineered yeast (0.370 mg/L) 44.
3.4. Protein modelling and docking simulations
The FPPS‐ECS fusion protein model was predicted by I‐TASSER and gave a C‐score of −1.31. In order to gain insight in the fusion protein and substrate binding, I‐TASSER predicted protein model was docked with various substrates by AutoDock Vina. The structures of fusion protein FPPS‐ECS and individual proteins (FPPS and ECS), homology based models were predicted. FPPS was identified using co‐ordinates of FPPS from Artemisia spiciformis and the sequence identity was 78.87%. In the current study, out of the three proteins, the crystal structure has been resolved only for plant FPPS; however, epi‐cedrol synthase structure have been predicted based on homology modelling 45. These structural models would help to gain insight in the role of synthetic linker in protein structure and function. Terpene synthases are a diverse class of enzyme family with their specificity and activity being highly dependent on small number of amino acids present inside or near the active site cleft. To further understand cyclization of epi‐cedrol, the amino acid residues present in and around the active site were identified using the co‐ordinates of α‐Bisabolol synthase as a template with Swiss Modeling server, having a 53.65% of sequence similarity of the amino acids, respectively. Numerous studies have revealed that even though terpene synthases vary in sequence similarity but their active sites are highly conserved 46. Recently reported FPPS protein sequence has seven highly conserved regions (region I‐VII) present, from which Region II (L, X4LDDxxDxxxxRRG) and VI (GxxFQxxDDxxD….GK) are the most significant regions due to aspartate‐rich motifs and are involved in binding of both homoallylic (IPP) and allylic diphosphate substrates (GPP and DMAPP) and the divalent Mg2+ metal binding at G104 and W108 45, 47. This FPPS are joined with a flexible linker (G343S344G345G346S347) as shown in Figure 4A (red colour) with ECS. In an analysis of ECS structure, the enzyme catalyzes the formation of multiple products including two minor, from a single substrate because of the consequences of the highly reactive series of carbocationic intermediates formed during the enzyme assay 48. FPP was synthesized from the active pocket of FPPS in fusion protein and easily channeled to an α‐helical loop over the active site that protects the carbocation from the water 28. This loop helps the hydrophobic interactions between W273 and Y527 47, 48. These are conserved loops in the fusion of ECS as F872 and W617. Loops were closed together by R606 and R786 near to each other. This possibly assists in ionization of the substrate diphosphate ester and helps to move diphosphate anion away from the generating carbocation 48. The common aspartate‐rich motif DDxxDD is located at 642–646 positions (Figure 4B–D).
Figure 4.
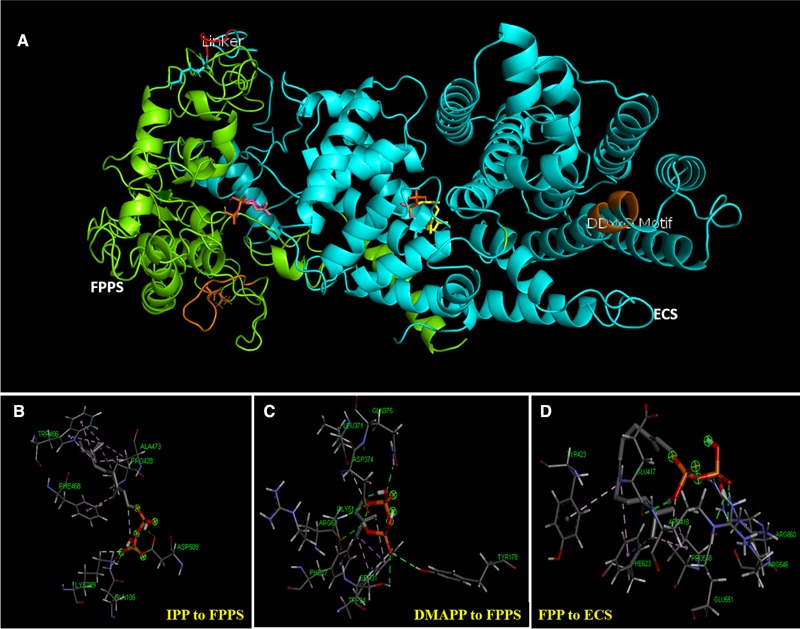
(A) Predicted structure of FPPS‐ECS fusion enzyme‐substrate complex by using I‐TASSER server and AutoDock software. FPPS and ECS were displayed in green and cyan color, respectively, while linker between two proteins was depicted in red. All the catalytic DDxxD motifs present in the proteins are displayed in orange. (B) Amino acid interactions in active pockets of FPPS enzyme with IPP; (C) FPPS enzyme with DMAPP, and (D) ECS enzyme with FPP of the fusion enzyme
The estimated TM‐score, RMSD, and cluster density for FPPS‐ECS fusion are 0.55 ± 0.15, 11.8 ± 4.5Å, and 0.0473 respectively, predicted by I‐TASSER server as details are mentioned in Section 2. On docking of fusion protein FPPS‐ECS, total 90 poses were generated for each ligand (IPP, DMAPP, and FPP) with protein and further analyzed according to minimum binding energy. The binding energies for fusion enzymes for substrates are mentioned in Table 4. These results indicated that the fusion protein bind to the substrates at a lower binding energy in comparison to their respective individual counterparts. This result is in concurrence with the experimental data, where the K m of Fusion enzyme for IPP was 4.71 whereas K m of mixed enzymes together for IPP was 5.64 (Table 2). This showed that individual enzymes have less binding energies toward respective substrates. The interactions of amino acids were also changed in case of individual enzymes (more models and individual interactions are shown in Supporting Information Figures S5–S6). Results, therefore, indicate that fusion of two enzymes may have led to their active pockets closing together and helped in the substrate channeling in the fusion enzymes leading to increase in the overall catalytic activity.
Table 4.
Binding energies between substrates and enzymes (FPPS, ECS, and FPPS‐ECS fusion)
| Substrates | FPPS | ECS | FPPS‐ECS fusion | |
|---|---|---|---|---|
| 1. | IPP | −5.6 | – | −6.2 kcal/mol |
| 2. | DMAPP | −5.6 | – | −6.2 kcal/mol |
| 3. | FPP | – | −7.0 | −7.2 kcal/mol |
ECS, epi‐cedrol synthase; FPPS, farnesyl pyrophosphate synthase.
4. CONCLUDING REMARKS
We have successfully constructed unnatural multifunctional fusion of epi‐cedrol synthase (sesquiterpene synthase) which produces a higher amount of epi‐cedrol from basic building block of isoprenoid biosynthesis pathway, i.e. IPP and DMAPP when compared to utilizing two different enzymes together. The results of this study clearly indicate that fusion protein strategy has a promising potential to improve product yield by using all substrates of the sesquiterpenoid pathways in heterologous expression. The fusion protein product was secreted in growth media indicating that an industrially friendly harvesting system for epi‐cedrol can be developed during downstream processing. It also reduces the time and cost of recombinant protein purification.
CONFLICT OF INTEREST
The authors have declared no conflict of interest.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
SSS gratefully acknowledges the Department of Science and Technology (DST), Government of India for the Fast Track Young scientist (SB/FT/CS‐042/2013) and Ramanujan fellowship (SR/S2/ RJN‐111/2012) scheme. GRN would like to thank CSIR‐Senior Research Fellowship (31/11(1026)/2018 EME I). PS would like to thank CSIR‐Research Associate Fellowship (31/11(953)/2017‐EMRI). SR is a recipient of DBT‐Research Associateship (January 2016). We are grateful to Director, CSIR‐National Chemical Laboratory, India for his constant support and encouragement.
Navale GR, Sharma P, Said MS, et al. Enhancing epi‐cedrol production in Escherichia coli by fusion expression of farnesyl pyrophosphate synthase and epi‐cedrol synthase. Eng Life Sci. 2019;19:606–616. 10.1002/elsc.201900103
Present Address
Dr. Sandip S. Shinde, Department Industrial and Engineering Chemistry, Institute of Chemical Technology Mumbai Marathwada Campus, Jalna, India 431213.
Contributor Information
H. V. Thulasiram, Email: hv.thulasiram@ncl.res.in.
Sandip S. Shinde, Email: shinde88@gmail.com.
REFERENCES
- 1. Fraga, B. M. , Huang, X. J. , Yang, J. S. , Yang, F. , et al., Natural sesquiterpenoids. Nat. Prod. Rep. 2013, 30, 1226–1264. [DOI] [PubMed] [Google Scholar]
- 2. Chappell, J. , Biochemistry and molecular biology of the isoprenoid biosynthetic pathway in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1995, 46, 521–547. [Google Scholar]
- 3. Daviet, L. , Schalk, M. , Factors affecting secondary metabolite production in plants: volatile components and essential oils. Flavour Fragr. J. 2010, 25, 123–127. [Google Scholar]
- 4. Pickens, L. B. , Tang, Y. , Chooi, Y. , Metabolic engineering for the production of natural products. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 211–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lange, B. M. , Ahkami, A. , Metabolic engineering of plant monoterpenes, sesquiterpenes and diterpenes–current status and future opportunities. Plant Biotechnol. J. 2013, 11, 169–196. [DOI] [PubMed] [Google Scholar]
- 6. Yu, K. , Liu, C. , Kim, B.‐G. , Lee, D.‐Y. , Synthetic fusion protein design and applications. Biotechnol. Adv. 2015, 33, 155–164. [DOI] [PubMed] [Google Scholar]
- 7. Elleuche, S. , Bringing functions together with fusion enzymes — from nature's inventions to biotechnological applications. Appl Microbiol Biotechnol. 2015, 99, 1545–1556. [DOI] [PubMed] [Google Scholar]
- 8. Orita, I. , Sakamoto, N. , Kato, N. , Yurimoto, H. , et al., Bifunctional enzyme fusion of 3‐hexulose‐6‐phosphate synthase and 6‐phospho‐3‐hexuloisomerase. Appl. Microbiol. Biotechnol. 2007, 76, 439–445. [DOI] [PubMed] [Google Scholar]
- 9. Conrado, R. J. , Varner, J. D. , Delisa, M. P. , Engineering the spatial organization of metabolic enzymes : mimicking nature's synergy. 2008, 492–499. [DOI] [PubMed]
- 10. Sarria, S. , Wong, B. , Martín, H. G. , Keasling, J. D. , et al., Microbial synthesis of pinene. ACS Synth. Biol. 2014, 3, 466–475. [DOI] [PubMed] [Google Scholar]
- 11. Zhang, Y. , Wang, Y. , Wang, S. , Fang, B. , Engineering bi‐functional enzyme complex of formate dehydrogenase and leucine dehydrogenase by peptide linker mediated fusion for accelerating cofactor regeneration. Eng. Life Sci. 2017, 17, 989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Albertsen, L. , Chen, Y. , Bach, L. S. , Rattleff, S. , et al., Diversion of flux toward sesquiterpene production in Saccharomyces cerevisiae by fusion of host and heterologous enzymes. Appl. Environ. Microbiol. 2011, 77, 1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brodelius, M. , Lundgren, A. , Mercke, P. , Brodelius, P. E. , Fusion of farnesyldiphosphate synthase and epi‐aristolochene synthase, a sesquiterpene cyclase involved in capsidiol biosynthesis in Nicotiana tabacum . 2002, 3577, 3570–3577. [DOI] [PubMed] [Google Scholar]
- 14. Deng, Y. , Sun, M. , Xu, S. , Zhou, J. , Enhanced (S)‐linalool production by fusion expression of farnesyl diphosphate synthase and linalool synthase in Saccharomyces cerevisiae . J. Appl. Microbiol. 2016, 121, 187–195. [DOI] [PubMed] [Google Scholar]
- 15. Ćavar, S. , Maksimović, M. , Vidic, D. , Parić, A. , Chemical composition and antioxidant and antimicrobial activity of essential oil of Artemisia annua L. from Bosnia. Ind. Crops Prod. 2012, 37, 479–485. [Google Scholar]
- 16. Garcia, L. C. , A review of Artemisia annua L.: its genetics, biochemical characteristics, and anti‐malarial efficacy. Int. J. Sci. Technol. 2015, 5, 38–46. [Google Scholar]
- 17. Abad, M. J. , Bedoya, L. M. , Apaza, L. , Bermejo, P. , The Artemisia L. genus: a review of bioactive essential oils. Molecules 2012, 17, 2542–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klayman, D. L. , Qinghaosu (artemisinin): an antimalarial drug from China. Science 1985, 228, 1049–1055. [DOI] [PubMed] [Google Scholar]
- 19. Westfall, P. J. , Pitera, D. J. , Lenihan, J. R. , Eng, D. , et al., Production of amorphadiene in yeast, and its conversion to dihydroartemisinic acid, precursor to the antimalarial agent artemisinin. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E111‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang, H. , Han, J. , Kanagarajan, S. , Lundgren, A. , et al., Studies on the expression of sesquiterpene synthases using promoter‐β‐glucuronidase fusions in transgenic Artemisia annua L. PLoS One 2013, 8, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paddon, C. J. , Westfall, P. J. , Pitera, D. J. , Benjamin, K. , et al., High‐level semi‐synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [DOI] [PubMed] [Google Scholar]
- 22. Shen, H. Y. , Li, Z. Q. , Wang, H. , Ma, L. Q. , et al., Advances in sesquiterpene synthases (cyclases) of Artemisia annua . Chin. J. Biotechnol. 2007, 23, 976–981. [DOI] [PubMed] [Google Scholar]
- 23. Harrison, K. W. , Harvey, B. G. , Renewable high density fuels containing tricyclic sesquiterpanes and alkyl diamondoids. Sustain. Energy Fuels 2017, 1, 467–473. [Google Scholar]
- 24. Bhatia, S. P. , McGinty, D. , Letizia, C. S. , Api, A. M. , Fragrance material review on cedrol. Food Chem. Toxicol. 2008, 46, S100–S102. [DOI] [PubMed] [Google Scholar]
- 25. Zhang, Y. , Han, L. , Chen, S.‐S. , Guan, J. , et al., Hair growth promoting activity of cedrol isolated from the leaves of Platycladus orientalis . Biomed. Pharmacother. 2016, 83, 641–647. [DOI] [PubMed] [Google Scholar]
- 26. Luo, F. , Ling, Y. , Li, Den. , Tang, T. , et al., Characterization of a sesquiterpene cyclase from the glandular trichomes of Leucosceptrum canum for sole production of cedrol in Escherichia coli and Nicotiana benthamiana . Phytochemistry 2019, 162, 121–128. [DOI] [PubMed] [Google Scholar]
- 27. Dubey, V. S. , Bhalla, R. , Luthra, R. , An overview of the non‐mevalonate pathway for terpenoid biosynthesis in plants. J. Biosci. 2003, 28, 637–646. [DOI] [PubMed] [Google Scholar]
- 28. Shinde, S. S. , Navale, G. R. , Said, M. S. , Thulasiram, H. V. , Stereoselective quenching of cedryl carbocation in epicedrol biosynthesis. Tetrahedron Lett. 2016, 57, 1161–1164. [Google Scholar]
- 29. Srivastava, P. L. , Daramwar, P. P. , Krithika, R. , Pandreka, A. , et al., Functional characterization of novel sesquiterpene synthases from Indian sandalwood, Santalum album . Sci. Rep. 2015, 5, 10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woodside, A. B. , Huang, Z. , Poulter, C. D. , Trisammonium geranyl diphosphate. Org. synth. 1988, 66, 211. [Google Scholar]
- 31. Bradford, M. M. , A rapid and sensitive method for the quantitation microgram quantities of protein utilizing the principle of protein‐dye binding. Anal. Biochem. 1976, 254, 248–254. [DOI] [PubMed] [Google Scholar]
- 32. Arnold, K. , Bordoli, L. , Kopp, J. , Schwede, T. , The SWISS‐MODEL workspace: a web‐based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [DOI] [PubMed] [Google Scholar]
- 33. Yang, J. , Yan, R. , Roy, A. , Xu, D. , et al., The I‐TASSER Suite: protein structure and function prediction. Nat. Methods 2014, 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ponnusamy, S. , Zinjarde, S. , Bhargava, S. , Kulkarni‐kale, U. , et al., Deciphering the inactivation of human pancreatic ‐amylase, an anti‐ diabetic target, by bisdemethoxycurcumin, a small molecule inhibitor, isolated from Curcuma longa . Nat. Prod. J. 2013, 3, 15–25. [Google Scholar]
- 35. Rosano, G. L. , Ceccarelli, E. A. , Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 2014, 5, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mótyán, J. A. , Tóth, F. , Tőzsér, J. , Research applications of proteolytic enzymes in molecular biology. Biomolecules 2013, 3, 923–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Young, C. L. , Britton, Z. T. , Robinson, A. S. , Recombinant protein expression and purification: a comprehensive review of affinity tags and microbial applications. Biotechnol. J. 2012, 7, 620–634. [DOI] [PubMed] [Google Scholar]
- 38. Zhang, Y.‐H. P. , Substrate channeling and enzyme complexes for biotechnological applications. Biotechnol. Adv. 2011, 29, 715–725. [DOI] [PubMed] [Google Scholar]
- 39. Rohdich, F. , Zepeck, F. , Adam, P. , Hecht, S. , et al., The deoxyxylulose phosphate pathway of isoprenoid biosynthesis: studies on the mechanisms of the reactions catalyzed by IspG and IspH protein. Proc. Natl. Acad. Sci. 2003, 100, 1586–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang, C. , Zada, B. , Wei, G. , Kim, S. W. , Metabolic engineering and synthetic biology approaches driving isoprenoid production in Escherichia coli . Bioresour. Technol. 2017, 241, 430–438. [DOI] [PubMed] [Google Scholar]
- 41. Banerjee, A. , Sharkey, T. D. , Methylerythritol 4‐phosphate (MEP) pathway metabolic regulation. Nat. Prod. Rep. 2014, 31, 1043–1055. [DOI] [PubMed] [Google Scholar]
- 42. Rozkov, A. , Avignone‐Rossa, C. A. , Ertl, P. F. , Jones, P. , et al., Characterization of the metabolic burden on Escherichia coli DH1 cells imposed by the presence of a plasmid containing a gene therapy sequence. Biotechnol. Bioeng. 2004, 88, 909–915. [DOI] [PubMed] [Google Scholar]
- 43. Tyo, K. E. J. , Ajikumar, P. K. , Stephanopoulos, G. , Stabilized gene duplication enables long‐term selection‐free heterologous pathway expression. Nat. Biotechnol. 2009, 27, 760–765. [DOI] [PubMed] [Google Scholar]
- 44. Jackson, B. E. , Hart‐Wells, E. A. , Matsuda, S. P. T. , Metabolic engineering to produce sesquiterpenes in yeast. Org. Lett. 2003, 5, 1629–1632. [DOI] [PubMed] [Google Scholar]
- 45. Chan, Y.‐T. , Ko, T.‐P. , Yao, S.‐H. , Chen, Y.‐W. , et al., Crystal structure and potential head‐to‐middle condensation function of a Z, Z ‐farnesyl diphosphate synthase. ACS Omega 2017, 2, 930–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Degenhardt, J. , Köllner, T. G. , Gershenzon, J. , Phytochemistry Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 2009, 70, 1621–1637. [DOI] [PubMed] [Google Scholar]
- 47. Shrivastava, P. L. , Characterization of the Genes Involved in Santalene Biosynthetic Pathway in Indian Sandalwood Santalum album Linn., (PhD. Thesis) Univerisity of Pune, 2014.
- 48. Mercke, P. , Crock, J. , Croteau, R. , Brodelius, P. E. , Cloning, expression, and characterization of epi‐cedrol synthase, a sesquiterpene cyclase from Artemisia annua L. Arch. Biochem. Biophys. 1999, 369, 213–222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information