

ABSTRACT
DDX58/RIG-I, is a critical pattern recognition receptor for viral RNA, which plays an essential role in antiviral immunity. Its posttranslational modifications and stability are tightly regulated to mediate the moderate production of type I IFN to maintain the immune homeostasis. Recently, we reported that macroautophagy/autophagy balances type I IFN signaling through selective degradation of ISG15-associated DDX58 via LRRC25. However, the regulatory mechanism about the autophagic degradation of DDX58 remains largely undefined. Here, we identified LRRC59 as a vital positive regulator of DDX58-mediated type I IFN signaling. Upon virus infection, LRRC59 specifically interacted with ISG15-associated DDX58 and blocked its association with LRRC25, the secondary receptor to deliver DDX58 to autophagosomes for SQSTM1/p62-dependent degradation, leading to the stronger antiviral immune responses. Thus, our study reveals a novel regulatory role of selective autophagy in innate antiviral responses mediated by the cross-regulation of LRRC family members. These data further provide insights into the crosstalk between autophagy and innate immune responses.
Abbreviations: ATG: Autophagy-related; Baf A1: Bafilomycin A1; DDX58/RIG-I: DEAD [Asp-Glu-Ala-Asp] box polypeptide 58; EV: Empty vector; IC poly[I:C]: Intracellular polyriboinosinic polyribocytidylic acid; IFIH1/MDA5: Interferon induced with helicase C domain 1; IFN: Interferon; ISG15: ISG15 ubiquitin like modifier; IKBKE: Inhibitor of nuclear factor kappa B kinase subunit epsilon; IRF3: Interferon regulatory factor 3; KO: Knockout; LRRC: Leucine rich repeat containing; MAVS: Mitochondrial antiviral signaling protein; CGAS/MB21D1: Cyclic GMP-AMP synthase; SeV: Sendai virus; siRNA: small interfering RNA; SQSTM1/p62: Sequestosome 1; TBK1: TANK binding kinase 1; TLR: Toll like receptor; TMEM173/STING: Transmembrane protein 173; VSV: Vesicular stomatitis virus; WT: Wild type
KEYWORDS: Autophagic degradation, cargo receptor, DDX58/RIG-I, LRRC59, type I interferon
Introduction
Innate immune responses triggered by pathogen-associated molecular patterns (PAMPs) provide the first line of host’s immune defenses, which are characterized by the production of type I interferons (IFNs), type III IFNs, proinflammatory cytokines and chemokines [1]. Upon viral infection, pattern recognition receptors (PPRs), such as RIG-I-like receptors (RLRs) and DNA sensor CGAS/MB21D1 (cyclic GMP-AMP synthase), are employed to detect viral nucleic acids, hence initiating the transcription of type I IFN and provoking the comprehensive host defenses [2,3]. DDX58/RIG-I is one of the crucial members of RLRs family, which recognizes short double-strand viral RNA (dsRNA) and signals through MAVS (mitochondrial antiviral signaling protein) to activate IRF3 (interferon regulatory factor 3), leading to the translocation of IRF3 and the production of type I IFN [4–6]. Secreted IFNs induce the transcription of hundreds of IFN-stimulated genes (ISGs), which amplify and broaden innate immune responses to effectively control pathogens invasion [7].
Given that DDX58 is a critical mediator of antiviral immunity and inflammation, multiple posttranscriptional mechanisms, including the ligand recognition, conformation change, dephosphorylation, ubiquitination and tetramerization, are employed to adjust the activity of DDX58 [5,8]. In addition, the stability of DDX58 is also subtly modulated to perform the function of DDX58. Several reports indicated that DDX58 can be degraded through ubiquitin-proteasome pathway by SIGLEC10 (sialic acid binding Ig like lectin 10), RNF125 (ring finger protein 125), and IFI35 (interferon induced protein 35) [9–11]. However, whether DDX58 can be degraded through other degradation pathway remains poorly understood.
Autophagy is a powerful bio-progress which is used to defend against invading pathogens in host cells [12]. It is orchestrated by a series of autophagy-related (ATG) proteins and forms double-membrane structures called phagophores that sequester cytoplasmic components. The phagophores mature into autophagosomes, which deliver the cargos directly into the lysosomes for acidic hydrolases-mediated degradation [13]. Our recent study reported that autophagy plays a crucial role in the degradation of DDX58 for the first time. We identified LRRC25 functions as a secondary receptor to promote the recognition of ISG15-associated DDX58 by SQSTM1/p62, which mediates the degradation of DDX58 through selective autophagy [14]. However, whether positive regulators are involved in the autophagic regulation of DDX58 activity remain unclear.
Leucine-rich repeat (LRR) domains are frequently found in proteins involved in innate immunity and other multifarious cellular processes, such as apoptosis, autophagy and nuclear mRNA transport [15,16]. In mammals, the functional LRR-containing proteins, including NOD-like receptors and Toll-like receptors (TLRs) have been well characterized in innate immunity [15]. Besides NOD-like receptors and TLRs, the roles of other LRR-containing proteins in innate immune responses remain unclarified, which need to be further studied. In this study, we identified LRRC59 as a positive regulator of type I IFN signaling induced by DDX58, but not IFIH1/MDA5 or CGAS. Interestingly, we found LRRC59 targeted ISG15-associated DDX58 to inhibit the association between LRRC25 and DDX58, thus disrupting the recognition of DDX58 by SQSTM1. Collectively, the current study reveals that LRRC25 and LRRC59 function as secondary cargo receptor and inhibitor to modulate type I IFN signaling through manipulating DDX58 stability, respectively, and provides insights of the cross-regulation of LRRC family members in the crosstalk between innate immunity and selective autophagy.
Results
LRRC59 potentiates DDX58-induced antiviral immune responses
We performed a screening on the roles of LRRC family proteins in innate immunity and found that LRRC59 significantly promoted the activation of type I IFN signaling upon viral infection (Fig. S1A). LRRC59 was previously found to be important for nuclear transportation and TLRs translocation upon viral infection [16,17]. However, detailed information of LRRC59 in antiviral innate immune responses still needs to be explored. Real-time PCR and immunoblot analysis results showed that LRRC59 was ubiquitously expressed in various cells (Fig. S1B and S1C). In addition, the protein abundances of LRRC59 kept the equal level under Sendai virus (SeV) infection or intracellular (IC) poly[I:C] stimulation (Fig. S1D and S1E). To confirm the function of LRRC59 in type I IFN signaling, we transfected IFNB1/IFN-β luciferase reporter and the internal control renilla luciferase into 293T cells, together with increasing amount of LRRC59. Upon SeV or vesicular stomatitis virus (VSV) stimulation, we found that LRRC59 potently promoted the activation of type I IFN signaling in a dose-dependent manner (Figure 1(a)).
Figure 1.
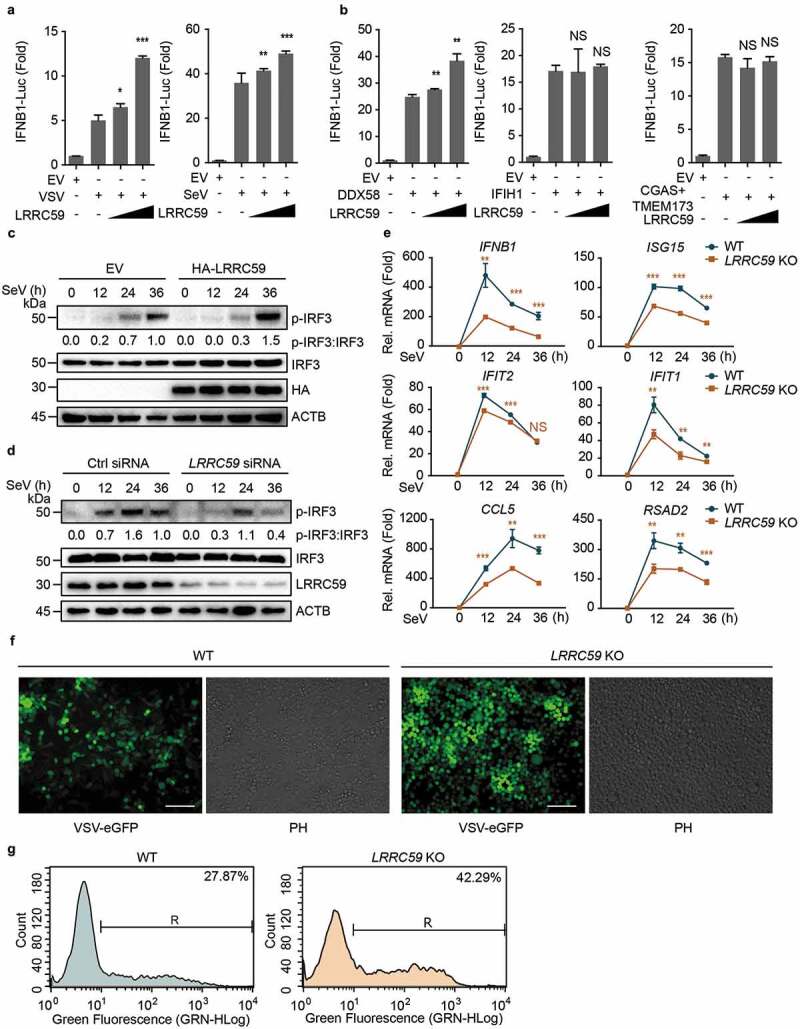
LRRC59 potentiates DDX58-mediated type I IFN antiviral immune response. (a,b) Luciferase activity in 293T cells transfected with an IFNB1 luciferase reporter (IFNB1-Luc), together with an empty vector (EV) or increasing amounts (100 ng and 300 ng per well, the same with following experiments) of plasmid encoding LRRC59, followed by infection with or without VSV or SeV (A) or activated by DDX58 (with IC poly[I:C] treatment; 5 μg/ml for 12 h in all IC poly[I:C] stimulation if no additional annotation), IFIH1 or co-expression of CGAS and TMEM173 together (CGAS+TMEM173) (b). (c) Immunoblot analysis of extracts of 293T cells transfected with EV or HA-LRRC59 and infected with SeV for the indicated time points. (d) Immunoblot analysis of extracts of A549 cells transfected with control siRNA or LRRC59-specific siRNA, followed by the infection with SeV for indicated time points. (e) Real-time PCR analysis of IFNB1 and other ISGs in A549 wild type (WT) cells or LRRC59 knockout (KO) A549 cells after SeV infection. (f,g) Phase-contrast (PH) and fluorescence microscopy analyses (f) or flow cytometric analyses (g) of WT and LRRC59 KO A549 cells infected with VSV-eGFP for 18 h. Scale bars, 200 μm. Data in (a, b, and e) are means ± SD of 3 independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001. Data in (c, d, f, and g) are representative of 3 independent experiments.
To determine whether LRRC59 is involved in the regulation of RNA or DNA virus-induced type I IFN signaling, we performed IFNB1 luciferase reporter assay with the expression of several key viral nucleotide receptors and signaling proteins, including DDX58 (in the presence of IC poly[I:C] to activate it), IFIH1 or co-expression of CGAS and TMEM173 (transmembrane protein 173) together. We observed that LRRC59 could only positively regulate DDX58-mediated, but not IFIH1 or CGAS-mediated type I IFN signaling (Figure 1(b)), suggesting that LRRC59 functions as a positive regulator of RNA virus-induced type I IFN signaling through DDX58. To further confirm the function of LRRC59 in RNA virus infection, we overexpressed or silenced LRRC59 in 293T cells or A549 cells and examined the phosphorylation levels of IRF3, the key transcriptional factor of type I IFN signaling under SeV or VSV infection. We found that overexpression of LRRC59 promoted the phosphorylation levels of IRF3, while LRRC59 deficiency decreased the phosphorylation levels of IRF3 (Figure 1(c,d) and S1F). Together, these results suggest that LRRC59 functions as a positive regulator of DDX58-mediated type I IFN signaling.
Newly synthesized IFN is secreted and activates JAK-STAT signaling components to induce the expression of ISGs, which are required to restrain viral infection [7]. We generated LRRC59 knockout (KO) A549 cells (Fig. S1G) and found that the expression of ISG messenger RNAs (mRNAs) was decreased in LRRC59 KO cells (Figure 1(e)). This result concurred with the attenuated IRF3 phosphorylation in LRRC59 depletion cells. To further assess the antiviral capability of LRRC59, we infected wild type (WT) and LRRC59 KO A549 cells with VSV-eGFP for 18 h. Fluorescence microscopy and flow cytometry analysis showed the markedly enhanced viral load in LRRC59 KO A549 cells (Figure 1(f,g)). Taken together, these data indicate that LRRC59 potentiates DDX58-mediated antiviral immune responses.
LRRC59 mediates type I IFN signaling at DDX58 level
To determine the molecular mechanism underpinning the activation of type I IFN signaling by LRRC59, we overexpressed the increasing amount of LRRC59 and DDX58 amino terminus (DDX58-N, an active deletion of DDX58), MAVS, TBK1 (TANK binding kinase 1) or IRF35D (a persistent active form of IRF3) [18], together with IFNB1 luciferase reporter in 293T cells and observed that LRRC59 promoted type I IFN signaling induced by DDX58-N, but not by MAVS, TBK1 or IRF35D (Figure 2(a,b)). Consistently, we only observed the prominent inhibition of DDX58-N-mediated type I IFN signaling in LRRC59 depletion cells (Figure 2(c)). To further confirm that LRRC59 specifically affects type I IFN signaling via DDX58, we generated DDX58 KO 293T cells (Fig. S2) and found that knockdown of LRRC59 couldn’t influence IFNB1 activation mediated by IFIH1 or CGAS (co-expressed with TMEM173) in the absence of DDX58 (Figure 2(d)). Thus, these data indicate that LRRC59 regulates type I IFN signaling at DDX58 level. A previous study reported that LRRC59 regulates TLR3 trafficking via association with UNC93B1 [17]. We further detected the function of LRRC59 in 293T cells (with no TLR3 expression) transfected with control or UNC93B1-specific siRNA. Positive regulatory role of LRRC59 in type I IFN signaling could still be observed in the absence of UNC93B1 (Figure 2(e)), suggesting that LRRC59 can promote type I IFN signaling in a TLR3 independent manner.
Figure 2.
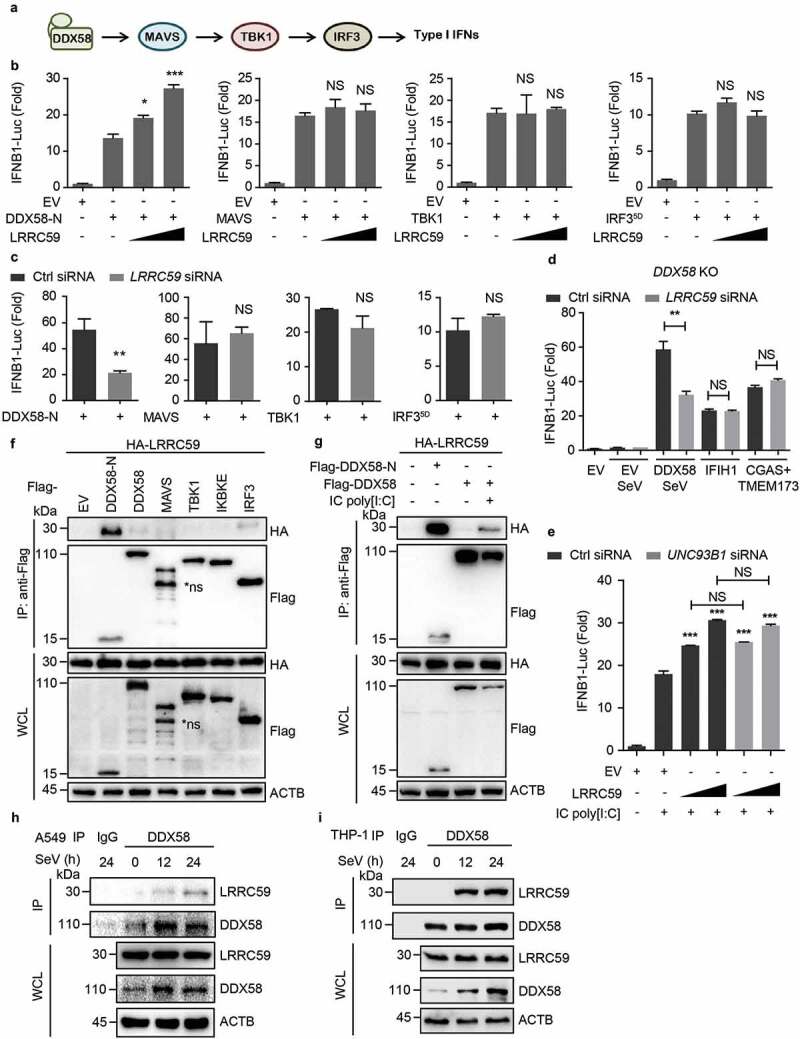
LRRC59 positively regulates type I IFN signaling by targeting activated DDX58. (a) Schematic of DDX58-mediated type I IFN signaling pathway. (b,c) Luciferase activity in 293T cells transfected with an IFNB1 luciferase reporter, together with an EV or increasing amount of plasmid expressing LRRC59 (b) or with control siRNA or LRRC59-specific siRNA (c), activated by DDX58-N, MAVS, TBK1 or IRF35D. (d) Luciferase activity in DDX58 KO 293T cells transfected with control siRNA or LRRC59-specific siRNA, subsequently transfected with an IFNB1 luciferase reporter (IFNB1-Luc), together with an EV, DDX58, IFIH1 or co-expression of CGAS and TMEM173 together (CGAS+TMEM173), followed with or without SeV infection. (e) Luciferase activity of 293T cells transfected with control siRNA or UNC93B1-specific siRNA, subsequently transfected with an IFNB1 luciferase reporter, together with plasmids expressing EV or HA-LRRC59, followed with or without IC poly[I:C] stimulation. (f) Co-immunoprecipitation and immunoblot analysis of extracts of 293T cells transfected with HA-LRRC59 and EV, Flag-tagged DDX58-N, DDX58, MAVS, TBK1, IKBKE or IRF3. (g) Co-immunoprecipitation and immunoblot analysis of extracts of 293T cells transfected with plasmids encoding HA-LRRC59 and Flag-tagged DDX58-N or DDX58, followed by the treatment of IC poly[I:C]. (h,i) Co-immunoprecipitation and immunoblot analysis of extracts of A549 cells (h) or THP-1 cells (i) infected with SeV for indicated time points. Data in (B to E) are means ± SD of 3 independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001. Data in (f–i) are representative of 3 independent experiments.
LRRC59 interacts with DDX58 after viral infection
Recently, we found that the LRR domain of LRRC25 interacts with DDX58 N-terminus [14]. Thus, we wondered whether LRRC59 can compete with LRRC25 for DDX58 binding. Co-immunoprecipitation and immunoblot analysis revealed that LRRC59 strongly interacted with DDX58-N, but not the full-length form of DDX58, MAVS, TBK1, IKBKE or IRF3 (Figure 2(f)). As the functional domain, N-terminus of DDX58 is shielded by its regulatory domain in physiological condition and is exposed after ligand stimulation [4]. We found that the full-length DDX58 could interact with LRRC59 after IC poly[I:C] treatment, suggesting that LRRC59 specifically binds to the N-terminus of activated DDX58 (Figure 2(g)). To rigorously observe the physiological relevance of these findings, we infected A549 cells and THP-1 cells with SeV and found that endogenous DDX58 associated together with LRRC59 after RNA virus infection (Figure 2(h,i)). Collectively, our results indicate that LRRC59 promotes type I IFN signaling by targeting DDX58-N.
LRRC59 promotes the stabilization of DDX58 upon viral infection
We next sought to determine the precise mechanism underlying LRRC59-promoted type I IFN signaling by targeting DDX58. We co-expressed the plasmids encoding LRRC59 and DDX58 and found that the concentrations of DDX58 protein increased considerably with the presence of LRRC59 after stimulation (Figure 3(a)). However, protein levels of MAVS, TBK1, IKBKE and IRF3 were not influenced by LRRC59 overexpression (Fig. S3). To further verify this result, we detected the endogenous protein levels of DDX58 in WT and LRRC59 KO A549 cells after SeV infection, and found that LRRC59 deletion accelerated the degradation of endogenous DDX58 protein (Figure 3(b)). Taken together, these data suggest that LRRC59 stabilizes DDX58 protein upon viral infection.
Figure 3.
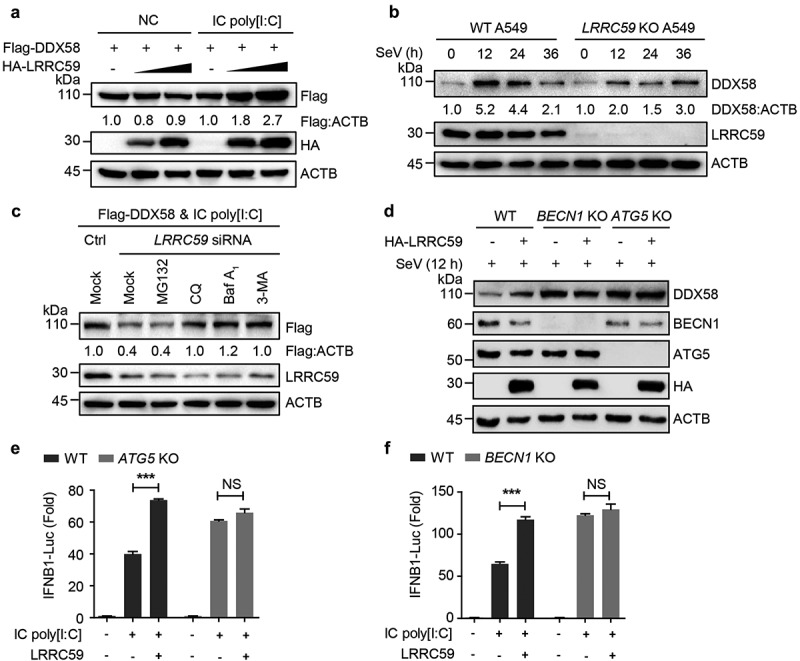
LRRC59 stabilizes activated DDX58 by restraining its autophagic degradation. (a) Immunoblot analysis of extracts of 293T cells transfected with plasmids expressing Flag-tagged DDX58 together with EV or HA-LRRC59, with or without IC poly[I:C] treatment. (b) Immunoblot analysis of extracts of wild type (WT) and LRRC59 KO A549 cells infected with SeV for indicated time points. (c) Immunoblot analysis of extracts of 293T cells transfected with Flag-DDX58 vector and poly[I:C], together with control siRNA or LRRC59-specific siRNA, followed by the treatment of mock, MG132 (10 μM), chloroquine (CQ; 50 μM), Baf A1 (0.2 μM) or 3-methyladenine (3-MA) (10 mM). (d) Immunoblot analysis of extracts of WT and ATG5 KO, BECN1 KO 293T cells transfected with EV or plasmid expressing HA-LRRC59 followed by SeV infection for indicated time points. (e,f) Luciferase activity in WT or ATG5 KO (D), BECN1 KO (e) 293T cells transfected with an IFNB1 luciferase reporter (IFNB1-Luc), together with an EV or plasmid encoding LRRC59 activated by IC poly[I:C]. Data in (A to D) are representative of 3 independent experiments. Data in (E and F) are means ± SD of 3 independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001.
LRRC59 restrains autophagic degradation of DDX58
Eukaryotic cells use three major systems to orchestrate protein degradation: the proteasome, lysosome and autolysosome pathway [19]. We next detected which degradation system contributed to LRRC59-prevented DDX58 degradation. We observed that the down-regulation of DDX58 protein abundances with LRRC59 deficiency was rescued by treatment with the autophagy inhibitor 3-methyladenine and autolysosome inhibitor chloroquine, bafilomycin A1 (Baf A1), but not the proteasome inhibitor MG132 (Figure 3(c)), suggesting that LRRC59 inhibits the autophagic degradation of DDX58. We further assessed the function of LRRC59 in BECN1 KO or ATG5 KO 293T cells, in which the autophagy is impaired. We found that expression levels of DDX58 in BECN1 KO or ATG5 KO cells were stable in the absence of LRRC59 (Figure 3(d)). Additionally, LRRC59-promoting IFN signaling was also totally abolished in autophagy-deficient cells (Figure 3(e,f)). Together, these results demonstrate that LRRC59 blocks the autophagic degradation of DDX58, thus potentiating the DDX58-mediated type I IFN signaling.
LRRC59 competes with LRRC25 for DDX58 N-terminus binding to block SQSTM1-DDX58 interaction
Previously we reported that LRRC25 is a critical regulator to mediate autophagic degradation of DDX58 [14]. We wondered whether LRRC59 perturbs LRRC25-DDX58 association, thus suppressing SQSTM1-mediated autophagic degradation of DDX58. By silencing the endogenous expression of LRRC25, we found that LRRC59 no longer elevated IFNB1 luciferase activity (Figure 4(a)). Moreover, the increased transcription of IFNB1 and ISGs and production of IFNB1 with LRRC59 overexpression were also impaired in LRRC25-deficient cells (Figure 4(b,c)). Collectively, these data reveal that LRRC59 serves as a positive regulator in type I IFN signaling in an LRRC25-dependent manner.
Figure 4.
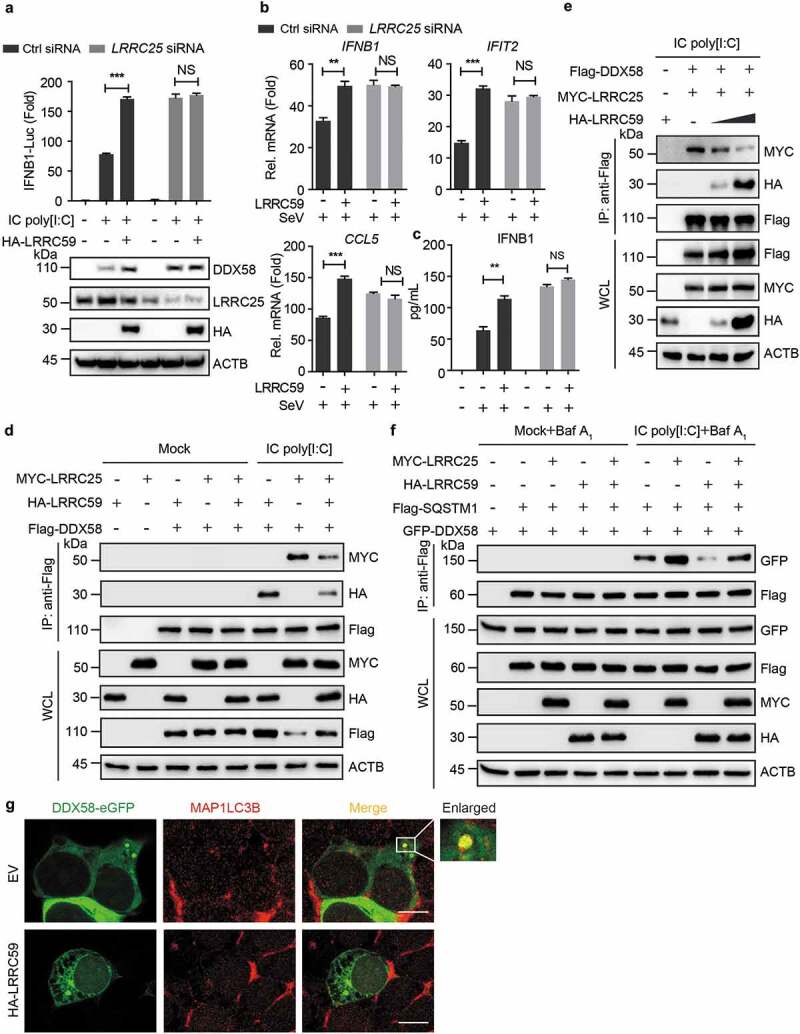
LRRC59 competes with LRRC25 for DDX58 N-terminus binding. (a) Luciferase activity of 293T cells transfected with control siRNA or LRRC25-specific siRNA, subsequently transfected with an IFNB1 luciferase reporter, together with plasmids expressing EV or HA-LRRC59, followed with or without IC poly[I:C] stimulation (Upper). Immunoblot analysis of similar setting without luciferase reporter plasmids (Below). (b,c) Real-time PCR analysis (B) or enzyme-linked immunosorbent assay analysis (ELISA) (C) of 293T cells (B) or supernatants of 293T cells (C) transfected with control siRNA or LRRC25-specific siRNA, subsequently transfected with EV or HA-LRRC59, followed with or without SeV infection. (d,e) Co-immunoprecipitation and immunoblot analysis of extracts of 293T cells transfected with vectors of Flag-DDX58, MYC-LRRC25 and HA-LRRC59 (d) or increasing dose of HA-LRRC59 (e), followed by the indicated treatment of mock or IC poly[I:C]. (f) Co-immunoprecipitation and immunoblot analysis of extracts of 293T cells transfected with plasmids encoding GFP-DDX58, Flag-SQSTM1 and MYC-LRRC25 or HA-LRRC59, followed by the treatment of mock or IC poly[I:C] together with Baf A1. (g) HEK 293T cells transfected with EV or HA-LRRC59 were subjected to immunofluorescence analysis using MAP1LC3B-specific antibody after stimulating with IC poly[I:C] for 8 h. Scale bars, 10 μm. Data in (A to C) are means ± SD of 3 independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001. Data in (D to G) are representative of 3 independent experiments.
Since both LRRC59 and LRRC25 interact with the N-terminus of DDX58 (Figure 2(f) and S4), we wondered if LRRC59 competes with LRRC25 for DDX58 N-terminus binding. Co-immunoprecipitation results showed that LRRC59 reduced the association between DDX58 and LRRC25 in a dose-dependent manner (Figure 4(d,e)). SQSTM1 is a cargo receptor required for the autophagic degradation of DDX58. Thus, we wondered whether LRRC59 can restrain SQSTM1-DDX58 association. LRRC59 was found to significantly suppress the interaction between SQSTM1 and DDX58 (Figure 4(f)). Immunofluorescence analysis further confirmed that LRRC59 restricted the co-localization between DDX58 and MAP1LC3B (Figure 4(g)). Collectively, these data demonstrate that LRRC59 stabilizes DDX58 protein by impairing the DDX58-SQSTM1 and DDX58-LRRC25 interactions.
LRRC59 specifically binds to ISG15-associated DDX58
ISG15 is widely known for its function in ISGylation, which may bind with over 150 kinds of proteins involved in immune response signaling and diverse cellular processes [20,21]. Recently, the conjugation of ISG15 to DDX58 has been shown to suppress type I IFN signaling by promoting the proteasomal and autophagic degradation of DDX58 [14,21–23]. To determine whether downstream signaling of type I IFN is necessary for the interaction with between LRRC59 and DDX58, we studied the role of LRRC59 in IRF3 KO cells. We observed that LRRC59 failed to maintain DDX58 protein abundance in IRF3 KO 293T cells, while this phenomenon could be rescued by the reintroduction of ISG15 (Fig. S5A and S5B). To illuminate the direct role of ISG15 in LRRC59-mediated type I IFN signaling, we generated ISG15 KO 293T cells (Figure 5(a)). Immunoblot analysis revealed that LRRC59 lost its ability to up-regulate the protein abundance of DDX58 and the phosphorylation of TBK1 upon IC poly[I:C] stimulation in ISG15 KO cells (Figure 5(a)). Furthermore, we performed luciferase reporter assay and found that the activation of type I IFN signaling induced by LRRC59 was totally abrogated with ISG15 deficiency (Figure 5(b)). Altogether, these results indicate that LRRC59 prevents the degradation of ISG15-associated DDX58, hence promoting the DDX58-mediated type I IFN signaling.
Figure 5.
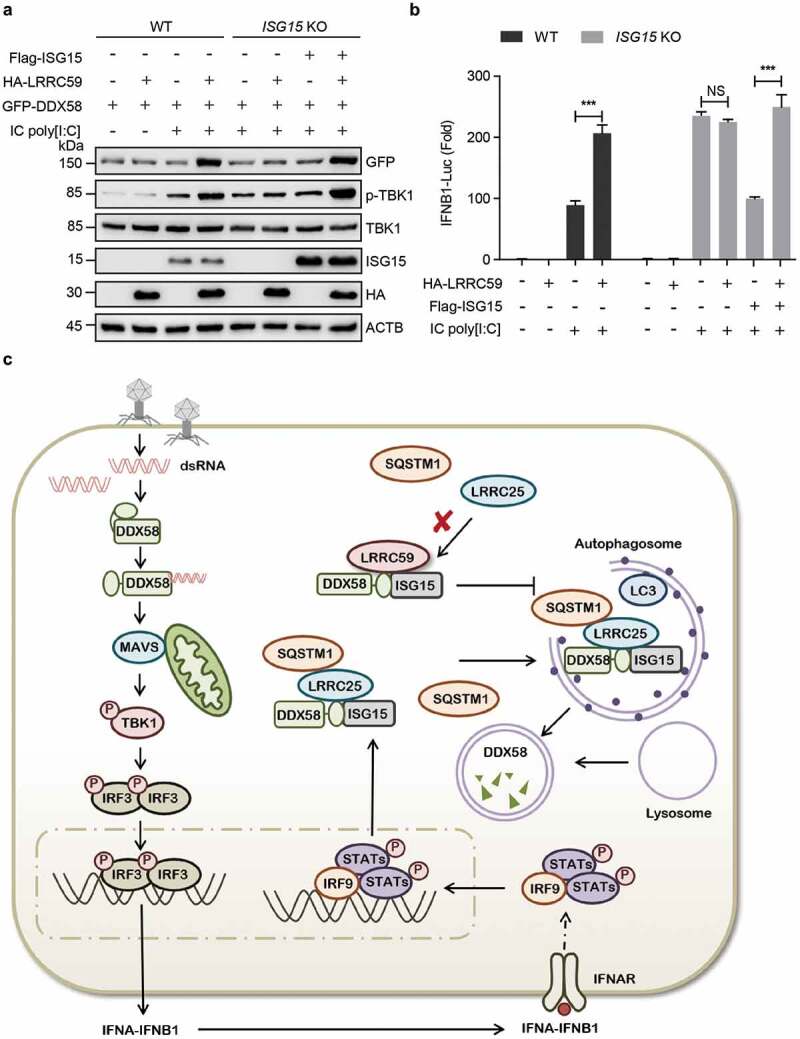
ISG15 is requisite for DDX58-LRRC59 interaction. (a) Immunoblot analysis of WT or ISG15 KO 293T cells transfected with EV or HA-LRRC59, together with plasmid encoding GFP-DDX58 and stimulated by IC poly[I:C]. (b) Luciferase activity of WT or ISG15 KO 293T cells transfected with EV, HA-LRRC59 or Flag-ISG15, followed with or without IC poly[I:C] stimulation. (c) The working model of the positive regulation of type I IFN signaling pathway by LRRC59. Data in (A) is representative of 3 independent experiments. Data in (B) are means ± SD of 3 independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001.
Discussion
Accumulating evidence demonstrate that autophagy plays a vital role in the regulation of type I IFN signaling as well as antiviral immunity [24–26]. Several key regulators of autophagy, including ATG12–ATG5, ATG9 and ULK1, are reported to play essential roles in antiviral immunity through an autophagic degradation-independent manner [27–29]. Our previous studies also demonstrated that BECN1 functions as an inhibitor in type I IFN signaling in an autophagy-independent manner by hampering the DDX58-MAVS association [19]. Moreover, autophagy can influence antiviral immune responses by down-regulating various immune factors through selective autophagic degradation. Type I IFN-induced BST2/Tetherin (bone marrow stromal cell antigen 2) promotes CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2)-dependent autophagic degradation of MAVS, serving as a suppressor to prevent the sustained activation of DDX58-mediated type I IFN signaling via a negative feedback loop [19]. TRIM14 (tripartite motif containing 14) recruits USP14 (ubiquitin specific peptidase 14) to cleave the K48-linked ubiquitination of CGAS, thereby inhibiting SQSTM1-mediated autophagic degradation of CGAS to enhance the activation of type I IFN signaling [30].
LRR domain is conserved in a wide variety of eukaryotic proteins, which plays an important role in innate immune responses [14,15]. Our recent study showed that LRRC25 promotes the autophagic degradation of DDX58 [14]. Moreover, LRRC25 also interacts with RELA/p65 (RELA proto-oncogene, NF-kB subunit) and enhances the association between RELA and SQSTM1, thus facilitating the degradation of RELA through selective autophagy [31]. These studies suggest that leucine-rich repeat containing protein family members can serve as secondary receptors to deliver the substrates to autophagosomes for degradation.
Here, we identified LRRC59 as a new regulator in DDX58-mediated type I IFN signaling by targeting the active form of DDX58. Through disrupting the interaction between ISG15-associated DDX58 and LRRC25, LRRC59 suppresses the SQSTM1-directed autophagic degradation of DDX58 in an LRRC25-dependent manner, hence stabilizing DDX58 protein to positively regulate DDX58-mediated type I IFN signaling (Figure 5(c)). The antagonism of LRRC59 in LRRC25-SQSTM1 axis-directed autophagic degradation of DDX58 implies that the leucine-rich repeat containing protein family members generate a cross-regulation network in selective autophagy, thus keeping a precise control of the crosstalk between autophagy and innate immune responses. In view of the indispensable role of DDX58 in antiviral immune response and antitumor therapy, our findings enrich the overall comprehending of DDX58 regulation, and might provide an opportunity to develop the optimal therapeutic strategies based on selective autophagy.
Materials and methods
Cell culture and transfection
All the experiments were performed in human cell lines. HEK293T (human embryonic kidney 293T), HeLa and A549 cells were cultivated in Dulbecco’s modified Eagle’s medium (CORNING, 10–013-CVR), and THP-1 cells were maintained in 1640 medium (Gibco, C22400500BT) supplemented with 10% fetal bovine serum (GenStar, C511-10) and 1% L-glutamine (Gibco, 35,050,061) at 37°C in 5% CO2. Overexpression plasmids were transfected using Lipofectamine 2000 reagent (Invitrogen, 11,668–019) according to the manufacturer’s instructions. Poly[I:C]-LMW was purchased from Invivogen (tlrl-picw).
Antibodies and reagents
Monoclonal anti-Flag M2-peroxidase (A8592), monoclonal anti-β-actin antibody produced in mouse AC-74 (A2228), and anti-Flag M2 affinity gel (A2220) were purchased from Sigma. Anti-MYC-horseradish peroxidase (11,814,150,001) and anti-HA-peroxidase (high affinity from rat immunoglobulin G1) (12,013,819,001) were purchased from Roche. DDX58/RIG-I (D14G6) rabbit monoclonal antibody (mAb) (3743S), phospho-IRF3 (Ser396, 4D4G) rabbit mAb (4947S), and MAP1LC3B (3868) were purchased from Cell Signaling Technology. IRF3 (FL-425; sc-9082) was purchased from Santa Cruz Biotechnology. Polyclonal anti-LRRC59 rabbit mAb (ab127912) was from Abcam.
Luciferase reporter assays
Cells were plated in 24-well plates and transfected with plasmids encoding the ISRE/IFNB1 luciferase reporter (firefly luciferase; 20 ng) and pRL-TK (Renilla luciferase plasmid; 8 ng), which were kindly provided by Dr. Rong-Fu Wang (Houston Methodist Research Institute), together with different plasmids (100 ng). Cells were harvested after IC poly[I:C] stimulation for the indicated times in passive lysis buffer (Promega, E1941). Enzyme activity was normalized by the efficiency of transfection on the basis of Renilla luciferase activity levels. Fold induction relative to the basal level was measured in cells. The values were means ± SD of 3 independent transfections performed in parallel.
Immunoprecipitation and immunoblot analysis
Cells were extracted in ice-cold low-salt lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1.5 mM MgCl2, 10% glycerol, 1% Triton X-100 [Sigma, T9284]) supplemented with a protease inhibitor cocktail (5 mg/mL; Roche, 04906837001). A 20 μL aliquot of each sample was subjected to SDS polyacrylamide gel electrophoresis. For immunoprecipitation experiments, whole-cell extracts were incubated with anti-Flag agarose gels (Sigma, A2220) overnight. The beads were washed 3 times with low-salt lysis buffer. The immunoprecipitates were resuspended in 3× SDS loading buffer (FD Biotechnology, PD006) and boiled for 5 min. The released proteins were electrophoresed on 8 to 12% SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes, with subsequent blocking using 5% skim milk. The membranes were incubated with the indicated antibodies and detected using enhanced chemiluminescence (Millipore).
Fluorescence microscopy
Cells were cultured on Glass Bottom culture dishes (Nest Scientific, 801,002) and directly observed as previously described [19]. For examination by immunofluorescence microscopy, cells were fixed with 4% paraformaldehyde for 15 min, and then permeabilized in methyl alcohol for 10 min at −20°C. After washing with PBS for 3 times, cells were blocked in 5% fetal goat serum (Boster Biological, AR1009) for 1 h, and then incubated with primary antibodies diluted in 10% bull serum albumin (Sigma, A1933) overnight. The cells were washed, and followed by a fluorescently labeled secondary antibody CF568 goat anti-rabbit IgG (H + L), highly cross-adsorbed (Biotium, 20,103–1). Confocal images were examined using a microscope (LSM710; Carl Zeiss) equipped with 100 × 1.40 NA oil objectives, with Immersol 518F (Carl Zeiss) as imaging medium and a camera (AxioCam HRc; Carl Zeiss) under the control of Zen 2008 software (Carl Zeiss). The images were processed for gamma adjustments using LSM Zen 2008 or ImageJ software (National Institutes of Health).
Knockout of LRRC59 by the CRISPR cas9 system
We analyzed guide RNA (gRNA) in the website http://crispr.mit.edu/ and chose the gRNA sequence with the highest score to design primers:
LRRC59 guide: 5′-GACCAGACGGCCAAAGTCTGC-3′;
DDX58 guide: 5′-AGATCAGAAATGATATCGGT-3′.
Annealing products were annealed and then linked to the pCRISPR-V2 vector.
RNA extraction and real-time PCR analysis
After total cellular RNA was isolated by TRIzol reagent (Invitrogen, 15,596,018), reverse transcription was performed using a reverse transcription kit (Vazyme, R223-01). Real-time PCR was performed with the SYBR Green qPCR Mix (GenStar, A311). Data were normalized to the GAPDH gene, and the relative abundance of transcripts was calculated by the Ct models. The primers used for real-time PCR are listed in Table S1.
RNA interference
Small interfering RNA (siRNA) duplexes (LRRC59, s30849 [siLRRC59]; negative control, AM4635) were obtained from Applied Biosystems (Ambion). Other small interfering RNA sequences are as follows:
siLRRC25 sense: 5′-GCACCAGUGGGAUGAACAA-3′;
siLRRC25 anti-sense: 5′-UUGUUCAUCCCACUGGUGC-3′;
siUNC93B1 sense: 5′-CACUACCUGUAUGACCUGA-3′;
siUNC93B1 anti-sense: 5′-UCAGGUCAUACAGGUAGUG-3′.
Statistical analysis
Student’s t-test was used for functional luciferase statistical analyses with GraphPad Prism 5.0 software. * P < 0.05, ** P < 0.01, *** P < 0.001.
Funding Statement
This work was supported by National Natural Science Foundation of China (31870862, 31700760, 31770978 and 91742109), Science and Technology Planning Project of Guangzhou, China (201804010385), and the Fundamental Research Funds for the Central Universities (18lgpy49).
Acknowledgments
We would like to thank Yang Du (Zhongshan School of Medicine, Sun Yat-sen University) for providing us with Flag-ISG15 vector and pCRISPR-V2 vector of sgISG15. We are grateful to Weihuang He (Zhongshan School of Medicine, Sun Yat-sen University) for the kind gift of HA-LRRC59 vector.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary data
Supplementary data can be accessed here.
References
- [1].Patel JR, Garcia-Sastre A.. Activation and regulation of pathogen sensor RIG-I. Cytokine Growth Factor Rev. 2014. October;25(5):513–523. .PubMed PMID: 25212896 [DOI] [PubMed] [Google Scholar]
- [2].Sparrer KM, Gack MU.. Intracellular detection of viral nucleic acids. Curr Opin Microbiol. 2015. August;26:1–9. PubMed PMID: 25795286; PubMed Central PMCID: PMC5084527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Goubau D, Deddouche S, Reis E Sousa C. Cytosolic sensing of viruses. Immunity. 2013. May 23;38(5):855–869. . PubMed PMID: 23706667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nakhaei P, Genin P, Civas A, et al. RIG-I-like receptors: sensing and responding to RNA virus infection. Semin Immunol. 2009. August;21(4):215–222. .PubMed PMID: 19539500 [DOI] [PubMed] [Google Scholar]
- [5].Eisenacher K, Krug A. Regulation of RLR-mediated innate immune signaling–it is all about keeping the balance. Eur J Cell Biol. 2012. January;91(1):36–47. .PubMed PMID: 21481967 [DOI] [PubMed] [Google Scholar]
- [6].Chiang JJ, Davis ME, Gack MU. Regulation of RIG-I-like receptor signaling by host and viral proteins. Cytokine Growth Factor Rev. 2014. October;25(5):491–505. .PubMed PMID: 25023063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. .PubMed PMID: 24555472; PubMed Central PMCID: PMCPMC4313732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Peisley A, Wu B, Xu H, et al. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature. 2014. May 1;509(7498):110–114. . PubMed PMID: 24590070; PubMed Central PMCID: PMCPMC6136653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen W, Han C, Xie B, et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell. 2013. January 31;152(3):467–478. PubMed PMID: 23374343. [DOI] [PubMed] [Google Scholar]
- [10].Arimoto K, Takahashi H, Hishiki T, et al. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci U S A. 2007. May 01;104(18):7500–7505. . PubMed PMID: 17460044; PubMed Central PMCID: PMC1863485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Das A, Dinh PX, Panda D, et al. Interferon-inducible protein IFI35 negatively regulates RIG-I antiviral signaling and supports vesicular stomatitis virus replication. J Virol. 2014. March;88(6):3103–3113. .PubMed PMID: 24371060; PubMed Central PMCID: PMC3957919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol. 2018. June;16(6):341–354. .PubMed PMID: 29556036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hansen M, Rubinsztein DC, Walker DW. Publisher Correction: autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018. September;19(9):611. .PubMed PMID: 30046055 [DOI] [PubMed] [Google Scholar]
- [14].Du Y, Duan T, Feng Y, et al. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. Embo J. 2018. February 1;37(3):351–366. . PubMed PMID: 29288164; PubMed Central PMCID: PMCPMC5793803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ng AC, Eisenberg JM, Heath RJ, et al. Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc Natl Acad Sci U S A. 2011. March 15;108(Suppl 1):4631–4638. . PubMed PMID: 20616063; PubMed Central PMCID: PMC3063585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhen Y, Sorensen V, Skjerpen CS, et al. Nuclear import of exogenous FGF1 requires the ER-protein LRRC59 and the importins Kpnalpha1 and Kpnbeta1. Traffic. 2012. May;13(5):650–664. .PubMed PMID: 22321063 [DOI] [PubMed] [Google Scholar]
- [17].Tatematsu M, Funami K, Ishii N, et al. LRRC59 regulates trafficking of nucleic acid-sensing TLRs from the endoplasmic reticulum via association with UNC93B1. J Immunol. 2015. November 15;195(10):4933–4942. . PubMed PMID: 26466955. [DOI] [PubMed] [Google Scholar]
- [18].Lin R, Mamane Y, Hiscott J. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol. 1999. April;19(4):2465–2474. PubMed PMID: 10082512; PubMed Central PMCID: PMC84039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jin S, Tian S, Luo M, et al. Tetherin suppresses Type I interferon signaling by targeting MAVS for NDP52-Mediated selective autophagic degradation in human cells. Mol Cell. 2017. October 19;68(2):308–322 e4. . PubMed PMID: 28965816. [DOI] [PubMed] [Google Scholar]
- [20].Dos Santos PF, Mansur DS. Beyond ISGlylation: functions of free intracellular and extracellular ISG15. J Interferon Cytokine Res. 2017. June;37(6):246–253. .PubMed PMID: 28467275 [DOI] [PubMed] [Google Scholar]
- [21].Zhao C, Denison C, Huibregtse JM, et al. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc Natl Acad Sci U S A. 2005. July 19;102(29):10200–10205. . PubMed PMID: 16009940; PubMed Central PMCID: PMC1177427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Arimoto K, Konishi H, Shimotohno K. UbcH8 regulates ubiquitin and ISG15 conjugation to RIG-I. Mol Immunol. 2008. February;45(4):1078–1084. .PubMed PMID: 17719635 [DOI] [PubMed] [Google Scholar]
- [23].Kim MJ, Hwang SY, Imaizumi T, et al. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J Virol. 2008. February;82(3):1474–1483. .PubMed PMID: 18057259; PubMed Central PMCID: PMCPMC2224411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].O’Connell D, Liang CY. Autophagy interaction with herpes simplex virus type-1 infection [Review]. Autophagy. 2016;12(3):451–459. .PubMed PMID: WOS:000373983300002; English [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liang Q, Seo GJ, Choi YJ, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. 2014. February 12;15(2):228–238. PubMed PMID: 24528868; PubMed Central PMCID: PMC3950946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jin S, Tian S, Chen Y, et al. USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. Embo J. 2016. April 15;35(8):866–880. . PubMed PMID: 26988033; PubMed Central PMCID: PMC4972138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013. October;13(10):722–737. .PubMed PMID: 24064518; PubMed Central PMCID: PMCPMC5340150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013. October 24;155(3):688–698. . PubMed PMID: 24119841; PubMed Central PMCID: PMCPMC3881181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Saitoh T, Fujita N, Hayashi T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009. December 8;106(49):20842–20846. PubMed PMID: 19926846; PubMed Central PMCID: PMCPMC2791563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen M, Meng Q, Qin Y, et al. TRIM14 Inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol Cell. 2016. October 6;64(1):105–119. PubMed PMID: 27666593. [DOI] [PubMed] [Google Scholar]
- [31].Feng YC, Duan TH, Du Y, et al. LRRC25 functions as an inhibitor of NF-κB signaling pathway by promoting p65/RelA for autophagic degradation [Article]. Sci Rep. 2017. October;7:12. PubMed PMID: WOS:000413188400029; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.