

ABSTRACT
Macroautophagy/autophagy is an essential process for the maintenance of cellular homeostasis by recycling macromolecules under normal and stress conditions. ATG9 (autophagy related 9) is the only integral membrane protein in the autophagy core machinery and has a central role in mediating autophagosome formation. In cells, ATG9 exists on mobile vesicles that traffic to the growing phagophore, providing an essential membrane source for the formation of autophagosomes. Here we report the three-dimensional structure of ATG9 from Arabidopsis thaliana at 7.8 Å resolution, determined by single particle cryo-electron microscopy. ATG9 organizes into a homotrimer, with each protomer contributing at least six transmembrane α-helices. At the center of the trimer, the protomers interact via their membrane-embedded and C-terminal cytoplasmic regions. Combined with prediction of protein contacts using sequence co-evolutionary information, the structure provides molecular insights into the ATG9 architecture and testable hypotheses for the molecular mechanism of autophagy progression regulated by ATG9.
Abbreviations: 2D: 2-dimensional; 3D: 3-dimensional; AtATG9: Arabidopsis ATG9; Atg: autophagy-related; ATG9: autophagy-related protein 9; cryo-EM: cryo-electron microscopy; DDM: dodecyl maltoside; GraDeR: gradient-based detergent removal; LMNG: lauryl maltose-neopentyl glycol; PAS: phagophore assembly site; PtdIns3K: phosphatidylinositol 3-kinase.
KEYWORDS: ATG9, autophagosome, autophagy, cryo-electron microscopy, single particle
Introduction
Macroautophagy, henceforth called autophagy, is a major bulk degradation pathway responsible for the removal of unwanted or harmful materials from cells, including protein aggregates, damaged organelles and invading pathogens [1–3]. This process is critical to cell homeostasis and is highly conserved throughout the eukaryotes [4–6]. The dysregulation of autophagy is associated with the pathogenesis of cancers, neurodegenerative diseases and cardiometabolic diseases. In plants, autophagy plays important roles in development, nutrient recycling, environmental stress responses, and immune response [7,8]. Autophagy proceeds by the de novo formation of a cup-shaped membrane, the phagophore, that expands and engulfs cytoplasmic cargo. The expanding membrane eventually seals to generate a double-membrane compartment termed the autophagosome, which then fuses with the lysosome or the vacuole where the sequestered cargo is degraded and recycled [9].
A dedicated set of autophagy-related (Atg) proteins tightly regulates the process of autophagosome formation [10–12]. These core Atg proteins can be categorized into 6 functional groups based on identified protein-protein interactions: the Atg1 kinase complex, the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, Atg9, the Atg2-Atg18 complex, the Atg12 conjugation system, and the Atg8 conjugation system. Under nutrient starvation, the inhibition of MTORC1 (mechanistic target of rapamycin kinase complex 1) signalling induces the activation and localization of the Atg1 complex to the phagophore assembly site (PAS), leading to the initiation of autophagy [13]. Activated Atg1 complex then recruits and phosphorylates the class III PtdIns3K complex. During the nucleation step at which the phagophore forms and develops, the class III PtdIns3K complex produces phosphatidylinositol 3-phosphate to facilitate the subsequent recruitment of the Atg2-Atg18 complex. Atg9 is also recruited in this step through interacting with the Atg1 [14–16] and the Atg2-Atg18 complexes [17,18]. Finally, the Atg12 and the Atg8 conjugation systems cooperatively mediate autophagosome expansion and closure. Atg9 is the only integral membrane protein within the core Atg machinery. In the cytosol, Atg9 is incorporated into single-membrane vesicles with a diameter of 30 to 60 nm [19,20], and normally resides on trans-Golgi network and endosomal compartments before being recruited to the PAS upon autophagy induction [21,22], a process that requires membrane trafficking regulators [23–25]. Although the precise function of Atg9 is still not known, there is an increasing amount of evidence that Atg9-containing vesicles act as an organizing center and, at the same time, provide an essential source of lipids/membranes for phagophore nucleation [21,26–29]. Notably, the amount of Atg9 protein present in the cell is transcriptionally regulated by the master transcriptional repressor Pho23 and directly correlates with the frequency of autophagosome formation [30,31]. Consistent with its essential role in autophagy, Atg9 depletion inhibits autophagosome formation in both yeast and mammals [21,26], and atg9-knockout mice is embryonic lethal [32]. Interestingly, the Arabidopsis atg9 mutant exhibited abnormal tubular autophagosome structures with direct connection with the endoplasmic reticulum upon autophagy induction, indicating an essential and unique role of ATG9 vesicles and endoplasmic reticulum-origins of autophagosomes in plants [5,18].
Currently, the molecular events and mechanisms underlying phagophore nucleation remain poorly understood. In particular, unlike other core Atg proteins, no structural studies have yet been performed on Atg9 [12,33–35]. Using single-particle cryo-electron microscopy (cryo-EM), we determined the first 3-dimensional (3D) structure of ATG9 from Arabidopsis thaliana at subnanometer resolution and revealed its overall architecture and domain organization. Our structure allows transmembrane helix assignment on the basis of evolutionary covariance analysis and shows that ATG9 forms a trimer via intramolecular interfaces involving the soluble and the membrane-bound regions.
Results
Isolation and characterization of Arabidopsis ATG9
The full-length plant ATG9 was expressed in suspension HEK293F cell culture. After solubilizing ATG9 from the membrane fraction using dodecyl maltoside (DDM), the protein-detergent complex was exchanged into lauryl maltose-neopentyl glycol (LMNG) and purified using M2 affinity resin. The purified protein-detergent complex eluted as a single broad peak both from gel filtration column and in linear glycerol gradient (Figure 1A and Fig. S1). To remove excess detergent monomers and micelles for cryo-EM grid preparation, the previously established gradient-based detergent removal (GraDeR) approach was used as the final polishing step (Figure 1A and Fig. S1) [36]. The GraDeR approach was needed to increase particle density on holely carbon grids for diluted membrane protein concentration (final concentration at ~0.4 mg/ml). In addition, a low concentration of chemical crosslinker was included in the gradient to reduce ATG9 aggregation upon vitrification. After the gradient centrifugation, the major peak fractions consisting of purified ATG9 were pooled and concentrated for cryo-EM analysis (Figure 1B). Initial 2-dimensional (2D) classification of the data revealed the presence of monomers, dimers and trimers, consistent with previous findings suggesting that ATG9 self-interacts independent of other autophagy factors (Figure 1C; Fig. S2) [37,38]. ATG9 consists of a highly conserved membrane-bound core region consisting of six transmembrane helices (TM1-6) flanked by intrinsically disordered N- and C-terminal cytoplasmic regions (Fig. S3). This membrane-bound core region, surrounded by a detergent micelle, was readily identified in all class averages. Of the three morphologically distinct populations of particles, only the trimers appeared to form homogeneous complexes. Notably, blurry densities presumably representing the flexible cytoplasmic regions were found associating with the monomer and dimer populations, contributing to the observed particle heterogeneity. The disordered cytoplasmic regions appeared to mediate the association of the subunits within the dimers, possibly through transient interactions, with the core regions not coming into contact with one another. The lack of observed low-resolution densities in the trimers implies that these regions are folded into a more stable structure in these particles.
Figure 1.
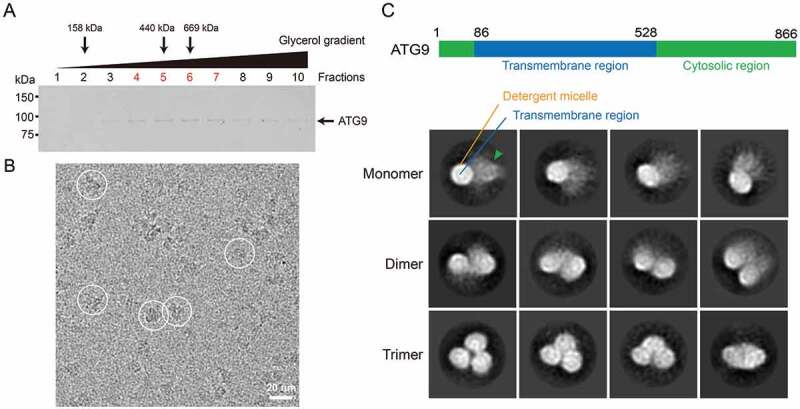
Purification and EM analysis of ATG9. (A) SDS-PAGE gel of the glycerol gradient fractions. Purified ATG9 was applied to GraDeR with 10–30% glycerol gradient. Fractions were collected from top to bottom and analyzed by SDS-PAGE. The sedimentation profile was pre-calibrated by using aldolase (158 kDa), apoferritin (440 kDa), thyroglobulin (669 kDa). Peak fractions of these protein standards were marked by arrow. ATG9 peaked at fractions 4–7 (labelled in red). (B) Representative cryo-EM micrograph with ATG9 particles circled in white. Scale bar: 20 nm. (C) Reference-free 2D class averages of particle images from cryo-EM micrographs, which were classified into 3 groups, monomer, dimer and trimer of ATG9. The blurry density representing the flexible cytosolic regions is indicated by arrowhead. The box size is 271 Å.
Cryo-EM reconstruction of Arabidopsis ATG9
Owing to the substantial amount of heterogeneity of the ATG9 monomers and dimers and the small size of the ordered membrane-bound region (~50 kDa), structural analysis was not pursued for these particles. Exhaustive image sorting was employed to enrich the homogeneous trimer particles and refinement of this subset attained a global resolution of 10 Å with C3 symmetry imposed, using the best 45,379 out of 73,531 trimer particles selected (Figure 2; Fig. S4). In an attempt to improve the map, a double signal subtraction protocol was performed to eliminate the strong signal of the LMNG detergent micelle density from the particle images [39], and subsequent focused refinement these signal subtracted particle images yielded an improved reconstruction at 7.8 Å resolution (Figure 2; Fig. S4 and S5). Further focused classification and refinement by masking out the single monomer did not yield an improved reconstruction. Figure 2A shows the overall map, filtered to the estimated local resolution. The majority of protein mass is buried in the lipid bilayer, as delineated by the detergent micelle in our reconstruction. The final attainable resolution of the map was likely limited by the presence of residual conformational variability in the trimer, the number of the trimer particles collected, and the lack of large membrane-extrinsic protein mass that precludes accurate image alignment. The protein complex measures 110 Å by 110 Å and has a height of 100 Å.
Figure 2.
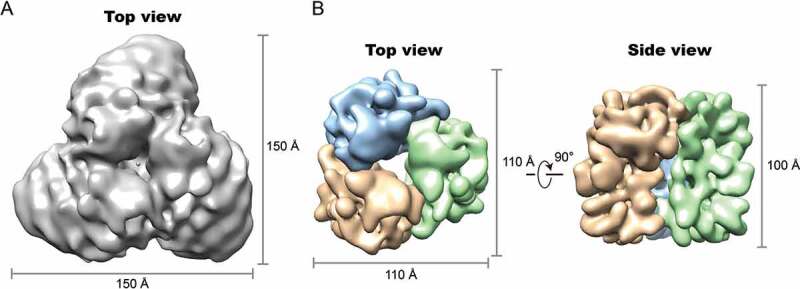
Cryo-EM map of the ATG9 trimer. (A) Surface view of the ATG9-detergent complex map with resolution at 10 Å. (B) Focused-refined map at 7.8 Å showing ATG9 protein density. Protomers are colored in orange, light blue and green. Dimensions of the maps are indicated.
The cytoplasmic region
Because no high-resolution model of any domain in ATG9 is available, we first used homology modeling to interpret the map. Using RaptorX [40], structural homologs were identified for a ordered segment of the N-terminal region (residues 58–81) as well as the large cytosol-facing loop of 140 residues in length between TM2 and TM3 (residues 180–319, hereafter referred to as the middle loop), which shares homology with the methyltransferases and myosin chain, respectively (Fig. S6). Both of these homology models could be reliably fitted into the cryo-EM map with minimal alterations (Figure 3). In yeast, the middle loop has been demonstrated to mediate the interaction of Atg9 with the adaptor protein Atg17 in the Atg1 complex [14]. The C-terminal region is much longer than the N-terminal region in Arabidopsis ATG9. Although being intrinsically disordered, the C-terminal region is predicted to contain some ordered segments and stretches of residues that undergo disorder-to-order transition upon protein binding (Fig. S3) [41]. It has been reported that the yeast Atg9 oligomerizes primarily through its C-terminal region, since deletion of the critical residues within this region compromised self-interactions [38]. Consequently, we ascribe the remaining density in our reconstruction to largely represent the structured domains of the C-terminal regions. The estimated volume of this region is ~20% of the density of the entire protomer, which approximately corresponds to the size of a 20 kDa polypeptide and is consistent with the calculated mass of the structured domain (16 kDa). The C-terminal region forms three distinct petal-shaped features around the three-fold axis where the middle loop is tucked behind.
Figure 3.
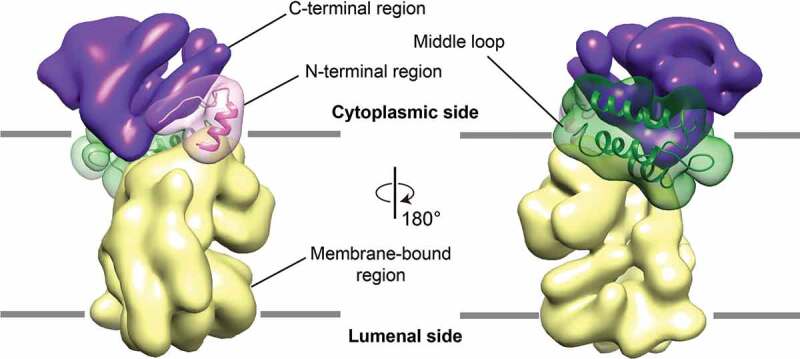
Segmentation of ATG9 and homology model fitting. Homology models of N-terminal region (pink) and middle loops regions (dark green) are docked into cytoplasmic regions of ATG9 protomers. The map density representing N-terminal region, C-terminal region, the middle loop and the membrane-bound region are colored in pink, purple, dark green, and pale yellow respectively.
The transmembrane helix organization
The map is of sufficient quality to resolve the rod-shaped, transmembrane α-helices in the membrane-embedded region. At this resolution, at least six transmembrane helices were identified, in agreement with secondary structure prediction (Figure 4A and Fig. S3). Two of these are bent and highly tilted helices. To assist the assignment of the transmembrane helices, we employed a machine learning method that utilizes an ultra-deep neural network to predict protein contacts from sequence conservation and co-evolutionary information [42]. Analysis of co-evolution of amino acids through multiple sequence alignment can identify direct spatial interactions between pairs of residues [43–46] and has been used to trace amino acid sequences through intermediate resolution cryo-EM maps [44,45]. Integration of this information into a deep learning model has been shown to greatly improve the accuracy of protein contact prediction for proteins with limited number of sequence homologs [42], as in the case for ATG9.
Figure 4.
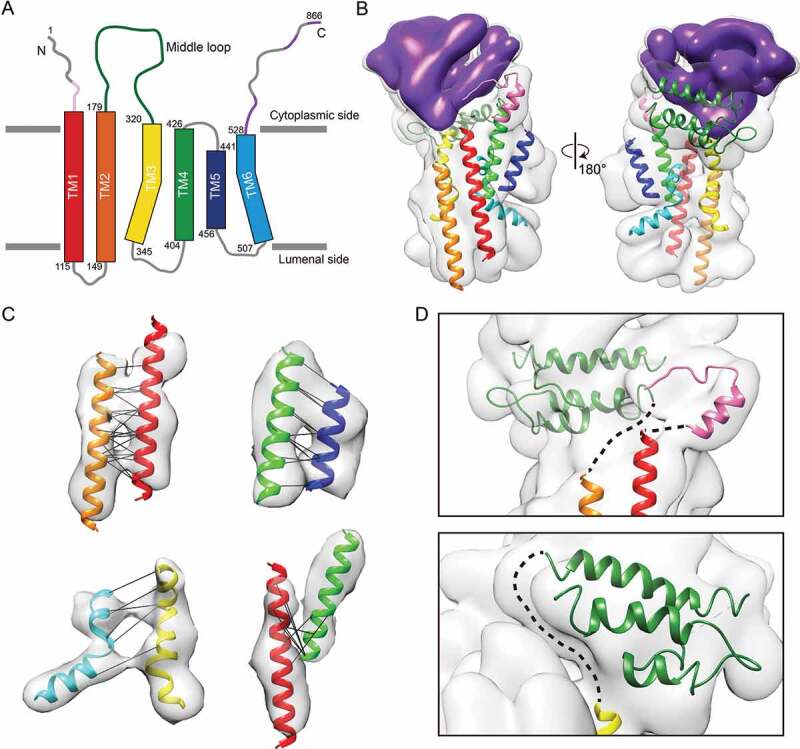
Transmembrane helix arrangement based on de novo prediction of protein contact by ultra-deep learning model. (A) Schematic drawing of ATG9 topology with transmembrane helix boundaries predicted with MEMSAT-SVM algorithm. Long loop (140 residues) between TM2 and TM3 is colored in dark green. Structured and disordered protein binding regions predicted to undergo disorder-to-order transition upon protein-protein interaction (shown in Figure S3) are colored in purple. Structured N-terminal region is colored in pink. (B) TMs and homology models-fitted density map. TMs are assigned to the map based on the distance constraints from deep-learning based protein contact prediction, length of TMs and loops connections. Same color code as shown in Figure 4A is used to represent TMs. (C) Evolutionary co-varying residue pairs between TMs are shown by black lines. Only pairs between TMs predicted with contact probability > 90% and Cα-Cα proximity < 15 Å in the model (43/46 pairs) are shown. A detailed schematic plot is displayed in Figure S7. (D) Enlarged view showing connections of TM1 to the N-terminal region, TM2 to middle loop (upper), and TM3 to the middle loop, which are indicated by dashed line.
The predicted contacts were used as distance constraints to build a model for the arrangement of the transmembrane segments by fitting the 6 predicted α-helices into the map (Figure 4B,C; Fig. S7). The final model places Cα of the pairwise residues are within 15 Å of each other in 43 out of the top 46 identified pairs, with an average distance of 10.2 Å. The 7% violated Cα- Cα distance constraints is in agreement with the observed false-positive rate in previous structure prediction studies of known protein structures using evolutionary covariance [47]. While TM1, TM2, TM4 and TM5 could be fitted as straight helices, TM3 and TM6 were flexibly fitted into map, as each harbors a single proline residue (P332 in TM3 and P521 in TM6) that introduces the apparent kinks in the transmembrane helices. Proline-induced distortions of transmembrane helices have been shown to facilitate inter-helical interactions [48]. The available distance constraints combined with the map density allowed unique assignment of all six transmembrane helices. In our model, the N termini of TM1 and TM3 are in proximity to the C termini of the N-terminal region and the middle loop, respectively (Figure 4D). This model is further reinforced by the continuous densities connecting these structural features. Of note, despite our transmembrane helix model is appealing and is built based on the map density and 93% of the distance constraints from co-evolving residue pairs, due to the limited resolution of the map, the current proposed model remains tentative.
After assigning the transmembrane helix densities and the density of the cytoplasmic region, there is remaining unoccupied density on the luminal border as well as in the membrane that can be accounted for by two extremely long stretches of residues connecting TM3 to TM4 (58 residues) and TM5 to TM6 (50 residues) (Figure 4A). This analysis also suggests that these residues not only constitute membrane-extrinsic loops but also additional membrane-embedded secondary structural elements. On the other hand, no observable density can be attributed to the residues connecting TM1 and TM2, suggesting that it forms a luminal loop and is highly flexible.
The trimer interface
Our map reveals extensive interactions between the cytoplasmic and membrane-embedded regions of adjacent protomers, forming the trimer interface. The N- and C-terminal regions, the middle loop as well as the membrane-embedded region all appear to contribute to the trimerization of the complex (Figure 5A,B). The contact between adjacent subunits in the membrane-embedded region, where the detergents are excluded in the map, creates a central cavity of ~20 Å in diameter. Considering the hydrophobic nature of the transmembrane helices lining the wall of this cavity and the presence of a central plug protruding from the membrane side (Fig. S8), this cavity is presumably filled with residual lipids, consistent with the observations seen in previous studies on F-/V-ATPase rotor rings [49–51]. TM2 and TM3 are positioned at the trimer interface, with TM2 from one subunit contacting TM3 of the other, mediating the self-interaction of ATG9 in the membrane (Figure 5B). We reason that changing the conformation of TM2 or TM3 could have an influence on the trimer stability. To alter the conformation of T3, we attempted to disrupt the TM3-TM6 interaction by introducing the proline to alanine mutations in the helices (Figure 5C). While mutating P332A on TM3 resulted in no expression of the protein, mutating P521A on TM6 resulted in an increase in interaction between protomers. Our experimental finding suggests that TM3-TM6 pair is involved at the trimer interface and we speculate that an impaired TM3-TM6 interaction favors the closer contact of TM3 with a neighboring TM, thus promoting trimer assembly.
Figure 5.
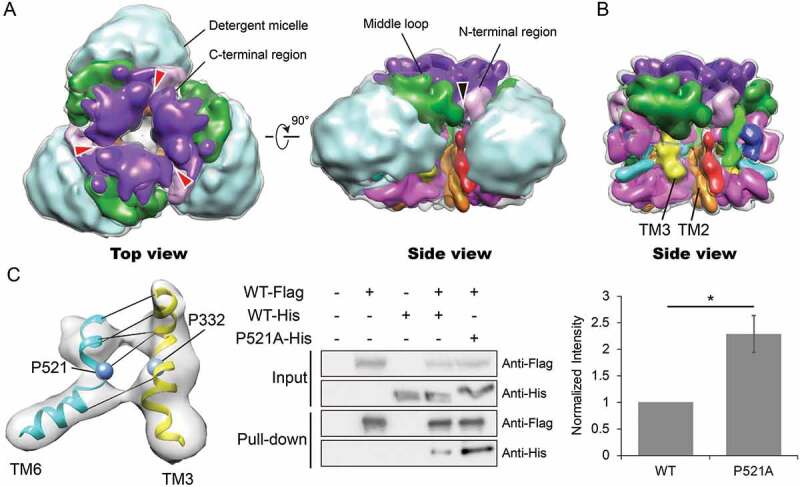
Interaction interface within membrane-bound regions and cytoplasmic regions facilitate ATG9 trimerization. (A) Segmented map delineating densities from different regions with the same color code as in Figures 3 and 4. The density corresponding to residues connecting TM3 to TM4 and TM5 to TM6 is colored in magenta. Detergent density is colored in light blue. Interactions between inter-subunit C-terminal regions, N-terminal regions and middle loops are indicated by red and black arrows, respectively. (B) Segmented map without detergent micelles showing close proximity of TM2 and TM3 within the membrane. (C) Presence of proline residues in TM3 and TM6 results in bending and interaction of the TM helices. Affinity-isolation assay testing the self-interaction ability of ATG9P521A mutant. Flag and His-tagged ATG9s were co-expressed in HEK293 cells. Cell lysates supplemented with DDM were incubated with anti-Flag M2 magnetic beads, followed by washing and elution with 3× Flag peptide. ATG9 proteins were detected by western blotting using anti-Flag and anti-His antibody (left panel). The quantification of the results are shown in right panel, n = 4. Asterisk indicates p-value < 0.05.
Discussion
Our structural analysis shows that monomers in the plant ATG9 self-interacts similar to its yeast and the human counterparts [37,38]. For yeast Atg9, a self-interaction motif has been identified in the C-terminal region of the protein, consisting of the amino acid residues 766–770, and deletion of this motif has been shown to be sufficient to impair its ability to form oligomers thus acutely blocking autophagy progression [38]. Blocking of autophagy was, in part, due to a partial defect in the anterograde transport of Atg9 to the PAS from peripheral compartments. This data suggests that oligomerization of Atg9 is crucial for the recruitment of other autophagy factors or adaptor proteins in order to facilitate its arrival to the PAS. According to our cryo-EM analyses, the intrinsically disordered nature of the C-terminal region folds into a more stable conformation upon trimer assembly, which in turn, could provide a scaffold for subsequent protein-protein interactions. Such interactions may promote budding of ATG9 from its membrane origin into transport vesicles, intracellular trafficking of ATG9 and/or its retention at the PAS. Interestingly, Atg1 was previously shown to phosphorylate multiple Ser/Tyr residues at the C-terminal region of Atg9 to regulate its interaction with the Atg2-Atg18 complex [16], which is required for phagophore expansion. As phosphorylation represents a major molecular mechanism for controlling the disorder-to-order transitions of intrinsically disordered proteins [52,53], it is tempting to speculate that phosphorylation of ATG9 near its C terminus might provide a means to regulate its interaction with downstream effectors, through modulating the conformation of the C-terminal region and therefore the efficiency of the trimer formation.
The study by Klionsky and coworkers demonstrated that overexpression of an adaptor protein could restore the trafficking of the Atg9Δ766-770 self-interaction mutant to the PAS but not the autophagy activity [38]. This observation indicates a second essential function for Atg9 oligomerization at the PAS. Moreover, the phagophore structures are fragmented with less total membrane present at the PAS in cells expressing the Atg9Δ766-770 mutant compared to the wild-type Atg9. Hence, the authors proposed a direct role of Atg9 in phagophore formation that involves a membrane-supplying and fusion mechanism through Atg9 oligomerization. In the current work, the identification of a new specific trimer interface involving the membrane-bound regions lends support to this hypothesis. It is conceivable that during phagophore nucleation, binding of an effector to the C-terminal regions of ATG9 localized on separate vesicles leads to the stabilization of the otherwise transient interactions between these regions. Then, the folding of the C-terminal regions brings the membrane-bound regions initially anchored in opposing membrane vesicles into close juxtaposition through trimerization, thereby promoting their specific interactions and resulting in the fusion of the membranes. Interestingly, coalescence of approximately three Atg9-vesicles has been proposed to be necessary for the formation of the phagophore [21,27], which is consistent with trimeric configuration of the ATG9 structure.
The high degree of sequence conservation between the membrane-bound regions of yeast, plants and humans suggests that these homologs exhibit similar structural properties for this region (Fig. S9 and Fig. S10). Furthermore, our study revealed that plant ATG9 self-interacts via multiple regions, unlike the yeast counterpart. In humans, since mutations introduced in the C-terminal region also failed to disrupt ATG9 self-interaction [37], it is likely that human ATG9A also trimerizes via multiple interaction interfaces.
We have provided structural insights into the mode of oligomerization of ATG9, identified the essential interaction interfaces, and tentatively assigned the transmembrane helices within the membrane-embedded region. Structures of ATG9 alone and in complex with other autophagy machinery at higher resolution will shed more light on the mechanisms by which ATG9 regulates autophagosome biogenesis.
Materials and methods
Expression and purification of Arabidopsis ATG9 (AtATG9)
The gene encoding the Arabidopsis full-length ATG9 was amplified from 221-UBQ-ATG9-GFP [18] and cloned into pEGFP-N1 vector (Clontech, 6085–1) by replacing the EGFP tag with a FLAG tag at the C terminus. The construct was verified by DNA sequencing. Expi293F cells (ThermoFisher Scientific, A14527) were cultured in Expi293 Expression Medium (ThermoFisher Scientific, A1435102) at 37°C supplemented with 8% CO2 at 125 rpm in an orbital incubator. Cells were transiently transfected with the ATG9-encoding plasmid using the cationic lipid-based ExpiFectamine Reagent (ThermoFisher Scientific, A14524) following the manufacturer’s protocol. Cells was harvested 48 h after transfection, and resuspended in Buffer A (50 mM Tris-HCl, 150 mM NaCl, 10% [v:v] glycerol, pH 7.4) supplemented with cOmplete, EDTA-free protease inhibitor cocktail (Roche, COEDTAF-RO). After disruption by sonication on ice, the membrane fraction was collected by ultracentrifugation at 150,000g for 1 h at 4°C. The isolated membranes were solubilized with 1% (w:v) DDM (Anatrace, D310) in Buffer A for 2 h at 4°C, followed by ultracentrifugation to remove insoluble materials. The supernatant was incubated with 3 mL anti-Flag M2 magnetic beads (Sigma-Aldrich, M8823) overnight at 4°C. To facilitate on-column detergent exchange to LMNG (Anatrace, NG310) and removal of contaminants, beads were incubated in Buffer A containing 1% (w:v) LMNG in the presence of 5 mM ATP (Sigma-Aldrich, A2383) and 20 mM MgCl2 followed by extensive washing. Proteins were eluted in Buffer A containing 0.05% (w:v) LMNG supplemented with 0.2–0.4 mg/ml 3× Flag peptide (Sigma-Aldrich, F4799) and concentrated.
The GraDeR and the GraFix methods were combined to remove free detergent micelles and stabilize the complex in one single step, as described previously [36,54], with a triple gradient of 10 to 30% (v:v) glycerol, 0.0015 to 0% (w:v) LMNG, and 0 to 0.2% (v:v) glutaraldehyde. Purified AtATG9 was applied at the top of the gradient and centrifuged at 50,000 × g for 18 h at 4°C in a swinging bucket rotor. The sample was fractionated into 10 fractions from top to bottom. To quench the crosslinking reaction, Tris-HCl, pH 7.4 was added immediately to a final concentration of 100 mM after the density gradient centrifugation. The desired fractions were pooled and concentrated. Purified proteins were flash-frozen in liquid nitrogen and stored at −80°C until use.
Sample preparation and cryo-EM data acquisition
Sample was dialyzed in a 50 mM Tris-HCl, 500 mM NaCl, pH 8.5 to remove glycerol prior to cryo-EM. Dialyzed sample at ~0.4 mg/ml (3 μL) was applied to a glow-discharged holey carbon grid (Quantifoil Micro Tools GmbH, R1.2/1.3; 400 mesh). Grids were blotted with a Vitrobot Mark IV (FEI Company) at 4°C and 100% relative humidity, and vitrified by plunging into liquid ethane cooled by liquid nitrogen. Images were collected with a ThermoFisher Scientific Titan Krios G3 electron microscope, operated at 300 kV and equipped with a FEI Falcon 3EC direct electron detector. A total of 2600 movies were recorded at a nominal magnification of 75000x, resulting in a calibrated pixel size of 1.06 Å. Movies were acquired in electron counting mode with an exposure rate of 0.8 electrons per pixel per second, a total exposure time of 60 s fractionated in 30 frames. The defocus ranged from −1.5 to −2.5 µm.
Image processing
The overall procedure for data processing is summarized in Fig. S3. Briefly, frame alignment and exposure weighting were performed with MotionCor2 [55] and contrast transfer function parameters were estimated with CTFFIND4 [56]. Movies with significant drift were discarded. All subsequent image processing steps were performed with RELION 2.1 [57]. A small set of particle images was selected manually and subjected to 2D reference-free classification. The resulting classes were used as templates for automatically selecting 770,775 particle images. After particle extraction, particle images were binned 2 × 2, and then subjected to three rounds of 2D classification to select the trimer particles for initial model building. The initial model, generated from 9929 best particle images, was further refined to 15 Å resolution. Since initial inspection of the 2D classification results clearly indicated the presence of monomers, dimers and trimers of ATG9, a 3D reference-based classification approach was used to separate a subset of 215,795 particle images according to their oligomeric state. The references for the dimer and monomer were obtained by deleting one and two subunits from the trimer map, respectively. All three references were low-pass filtered to 60 Å resolution. The resulting trimer particle population (73,531 particle images) was further classified into 12 classes in 3D, and a total of six good classes consisting of 45,379 particle images were selected and subjected to auto-refinement and post-processing to yield a map of 10 Å resolution. In the last step, focused refinement coupled with double signal subtraction approach specific to LMNG detergent was employed [58]. Signal corresponding to the LMNG micelle signal outside of the hydrophobic region was subtracted from each particle image based on the orientation determined in the previous round of auto-refinement. This procedure yielded a final reconstruction at 7.8 Å resolution after post-processing. All resolutions were determined Fourier shell correlation with the 0.143 criterion following a gold standard refinement. All 3D classification and auto-refinement were performed with C3 symmetry imposed, with the exception of the reference-based classification. For interpretation, the final map was sharpened and locally filtered according to the local-resolution estimation routine within RELION.
Map interpretation and structural modelling
Map segmentation was performed with UCSF Chimera [59]. Homology modeling of the N-terminal region (58–81 residues) and the middle loop (180–319 residues) was performed with the RaptorX webserver [40]. Fitting of the homology models were fitted first using the ‘Fit in map’ option in UCSF Chimera, followed by molecular dynamics flexible fitting [60]. The presumably flexible loop region (residues 248–276) in the middle loop domain was truncated during the fitting. De novo protein contact prediction was carried out to derive a model for the arrangement of the TMs in the hydrophobic region using RaptorX-Contact [42]. Of the 81 distance constraints with >90% probability of being in contact with each other, only the top 43 out of the 46 residue pairs between the TMs were included in our model building. The 7% false-positive rate (3 residue pairs) is considered acceptable, as described [44,45,47]. Six straight α-helices were created in UCSF Chimera based on the MEMSAT-SVM [61] predictions of transmembrane helix boundaries and then fitted in the map in orientations that satisfied the distance constraints. The fitting of the TM3 and TM6 into map was further optimized by molecular dynamics flexible fitting. In the final model, the Cα of the pairwise residues are within 15 Å of each other, with an average distance of 10.2 Å and a standard deviation of 2.4 Å. The current resolution of the map prohibited the modelling of the long stretches of residues between the TMs, ranging from 14–140 residues. For the same reason, the potentially ordered domain in the C-terminal region was also not modelled.
Affinity-isolation assay
Site-directed mutagenesis was carried out by overlapping PCR. The indicated plasmids were co-transfected into 30 ml of Expi293F cells and cultured for 48 h at 37°C supplemented with 8% CO2. After harvesting and lysing the cells, cell lysates were solubilized with 1% (w:v) DDM for 2 h at 4°C, followed by ultracentrifugation to remove insoluble materials. Supernatants were incubated with 100 μL of anti-Flag M2 magnetic beads at 4°C for 2 h. After extensive washing with Buffer A, the immobilized proteins were eluted with 3x Flag peptides and then subjected to SDS-PAGE. Eluted proteins were visualized by western blotting using the indicated antibodies.
Statistical analyses
Two-tailed Student t test was used to determine statistical significance.
Funding Statement
This work was supported by grants from the Research Grants Council of Hong Kong (14105517 to W.C.Y.L.) and (C4011-14R, C4012-16E, C4002-17G and AoE/M-05/12 to LJ.), National Natural Science Foundation of China (31670179 and 91854201 to L.J.), CUHK Faculty Strategic Development funding (to L.J.) and Research Committee of CUHK Direct Grant for Research (4053182 to W.C.Y.L.).
Acknowledgments
We thank Prof. John Rubinstein for providing access to the Titan Krios and critical reading of this manuscript. We thank Prof. Xiaohong Zhuang for providing the ATG9 DNA construct for cloning. Cryo-EM data were acquired at the Toronto High-Resolution High-Throughput cryo-EM facility, supported by the Canada Foundation for Innovation and Ontario Research Fund.
Data availability
Cryo-EM map for the AtATG9 has been deposited with the EMDB under accession code EMD-9681. Other data supporting the findings of this manuscript are available from the corresponding author upon reasonable request.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Mizushima N, Komatsu M.. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- [2].Shaid S, Brandts CH, Serve H, et al. Ubiquitination and selective autophagy. Cell Death Differ. 2013;20:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zaffagnini G, Martens S.. Mechanisms of selective autophagy. J Mol Biol. 2016;428:1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wen X, Klionsky DJ. An overview of macroautophagy in yeast. J Mol Biol. 2016;428:1681–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhuang X, Chung KP, Luo M, et al. Autophagosome biogenesis and the endoplasmic reticulum: a plant perspective. Trends Plant Sci. 2018;23:677–692. [DOI] [PubMed] [Google Scholar]
- [6].Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016;85:685–713. [DOI] [PubMed] [Google Scholar]
- [7].Wang P, Mugume Y, Bassham DC. New advances in autophagy in plants: regulation, selectivity and function. Semin Cell Dev Biol. 2018;80:113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang X, Bassham DC. New insight into the mechanism and function of autophagy in plant cells. Int Rev Cell Mol Biol. 2015;320:1–40. [DOI] [PubMed] [Google Scholar]
- [9].Noda NN, Inagaki F. Mechanisms of autophagy. Annu Rev Biophys. 2015;44:101–122. [DOI] [PubMed] [Google Scholar]
- [10].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. [DOI] [PubMed] [Google Scholar]
- [11].Hurley JH, Young LN. Mechanisms of autophagy initiation. Annu Rev Biochem. 2017;86:225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24:24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014;24:400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rao Y, Perna MG, Hofmann B, et al. The Atg1-kinase complex tethers Atg9-vesicles to initiate autophagy. Nat Commun. 2016;7:10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Suzuki SW, Yamamoto H, Oikawa Y, et al. Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc Natl Acad Sci U S A. 2015;112:3350–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Papinski D, Schuschnig M, Reiter W, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014;53:471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gomez-Sanchez R, Rose J, Guimaraes R, et al. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J Cell Biol. 2018;217:2743–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhuang X, Chung KP, Cui Y, et al. ATG9 regulates autophagosome progression from the endoplasmic reticulum in Arabidopsis. Proc Natl Acad Sci U S A. 2017;114:E426–E435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Karanasios E, Walker SA, Okkenhaug H, et al. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun. 2016;7:12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mari M, Griffith J, Rieter E, et al. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol. 2010;190:1005–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yamamoto H, Kakuta S, Watanabe TM, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012;198:219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Orsi A, Razi M, Dooley HC, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell. 2012;23:1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Noda T. Autophagy in the context of the cellular membrane-trafficking system: the enigma of Atg9 vesicles. Biochem Soc Trans. 2017;45:1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Davies AK, Itzhak DN, Edgar JR, et al. AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A. Nat Commun. 2018;9:3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mattera R, Park SY, De Pace R, et al. AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc Natl Acad Sci U S A. 2017;114:E10697–E10706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–774. [DOI] [PubMed] [Google Scholar]
- [27].Bahrami AH, Lin MG, Ren X, et al. Scaffolding the cup-shaped double membrane in autophagy. PLoS Comput Biol. 2017;13:e1005817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ge L, Zhang M, Kenny SJ, et al. Remodeling of ER-exit sites initiates a membrane supply pathway for autophagosome biogenesis. Embo Rep. 2017;18:1586–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mari M, Reggiori F. Atg9 reservoirs, a new organelle of the yeast endomembrane system? Autophagy. 2010;6:1221–1223. [DOI] [PubMed] [Google Scholar]
- [30].Jin M, Klionsky DJ. Transcriptional regulation of ATG9 by the Pho23-Rpd3 complex modulates the frequency of autophagosome formation. Autophagy. 2014;10:1681–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jin MY, He D, Backues SK, et al. Transcriptional regulation by Pho23 modulates the frequency of autophagosome formation. Curr Biol. 2014;24:1314–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Saitoh T, Fujita N, Hayashi T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106:20842–20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Suzuki H, Osawa T, Fujioka Y, et al. Structural biology of the core autophagy machinery. Curr Opin Struct Biol. 2017;43:10–17. [DOI] [PubMed] [Google Scholar]
- [34].Hurley JH, Schulman BA. Atomistic autophagy: the structures of cellular self-digestion. Cell. 2014;157:300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chew LH, Yip CK. Structural biology of the macroautophagy machinery. Front Biol (Beijing). 2014;9:18–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hauer F, Gerle C, Fischer N, et al. GraDeR: membrane protein complex preparation for single-particle cryo-EM. Structure. 2015;23:1769–1775. [DOI] [PubMed] [Google Scholar]
- [37].Staudt C, Gilis F, Tevel V, et al. A conserved glycine residue in the C-terminal region of human ATG9A is required for its transport from the endoplasmic reticulum to the Golgi apparatus. Biochem Biophys Res Commun. 2016;479:404–409. [DOI] [PubMed] [Google Scholar]
- [38].He C, Baba M, Cao Y, et al. Self-interaction is critical for Atg9 transport and function at the phagophore assembly site during autophagy. Mol Biol Cell. 2008;19:5506–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Garcia-Nafria J, Lee Y, Bai X, et al. Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. Elife. 2018;7:e35946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kallberg M, Margaryan G, Wang S, et al. RaptorX server: a resource for template-based protein structure modeling. Methods Mol Biol. 2014;1137:17–27. [DOI] [PubMed] [Google Scholar]
- [41].Jones DT, Cozzetto D. DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics. 2015;31:857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang S, Sun S, Li Z, et al. Accurate De Novo prediction of protein contact map by ultra-deep learning model. PLoS Comput Biol. 2017;13:e1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hopf TA, Colwell LJ, Sheridan R, et al. Three-dimensional structures of membrane proteins from genomic sequencing. Cell. 2012;149:1607–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhou A, Rohou A, Schep DG, et al. Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. Elife. 2015;4:e10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schep DG, Zhao J, Rubinstein JL. Models for the a subunits of the Thermus thermophilus V/A-ATPase and Saccharomyces cerevisiae V-ATPase enzymes by cryo-EM and evolutionary covariance. Proc Natl Acad Sci U S A. 2016;113:3245–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sjodt M, Brock K, Dobihal G, et al. Structure of the peptidoglycan polymerase RodA resolved by evolutionary coupling analysis. Nature. 2018;556:118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Marks DS, Colwell LJ, Sheridan R, et al. Protein 3D structure computed from evolutionary sequence variation. Plos One. 2011;6:e28766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schmidt T, Situ AJ, Ulmer TS. Structural and thermodynamic basis of proline-induced transmembrane complex stabilization. Sci Rep-Uk. 2016;6:29809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lau WCY, Rubinstein JL. Structure of intact Thermus thermophilus V-ATPase by cryo-EM reveals organization of the membrane-bound V-O motor. Proc Natl Acad Sci U S A. 2010;107:1367–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Meier T, Polzer P, Diederichs K, et al. Structure of the rotor ring of F-Type Na+-ATPase from Ilyobacter tartaricus. Science. 2005;308:659–662. [DOI] [PubMed] [Google Scholar]
- [51].Meier T, Matthey U, Henzen F, et al. The central plug in the reconstituted undecameric c cylinder of a bacterial ATP synthase consists of phospholipids. FEBS Lett. 2001;505:353–356. [DOI] [PubMed] [Google Scholar]
- [52].Bah A, Vernon RM, Siddiqui Z, et al. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2015;519:106–109. [DOI] [PubMed] [Google Scholar]
- [53].Kjaergaard M, Kragelund BB. Functions of intrinsic disorder in transmembrane proteins. Cell Mol Life Sci. 2017;74:3205–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kastner B, Fischer N, Golas MM, et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat Methods. 2008;5:53–55. [DOI] [PubMed] [Google Scholar]
- [55].Zheng SQ, Palovcak E, Armache JP, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods. 2017;14:331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rohou A, Grigorieff N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bai XC, Rajendra E, Yang GH, et al. Sampling the conformational space of the catalytic subunit of human gamma-secretase. Elife. 2015;4:e11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- [60].Trabuco LG, Villa E, Mitra K, et al. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nugent T, Jones DT. Transmembrane protein topology prediction using support vector machines. Bmc Bioinformatics. 2009;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM map for the AtATG9 has been deposited with the EMDB under accession code EMD-9681. Other data supporting the findings of this manuscript are available from the corresponding author upon reasonable request.