

ABSTRACT
Mutations in the macroautophagy/autophagy gene WDR45 cause β-propeller protein-associated neurodegeneration (BPAN); however the molecular and cellular mechanism of the disease process is largely unknown. Here we generated constitutive wdr45 knockout (KO) mice that displayed cognitive impairments, abnormal synaptic transmission and lesions in several brain regions. Immunohistochemistry analysis showed loss of neurons in prefrontal cortex and basal ganglion in aged mice, and increased apoptosis in prefrontal cortex, recapitulating a hallmark of neurodegeneration. Quantitative proteomic analysis showed accumulation of endoplasmic reticulum (ER) proteins in KO mouse. At the cellular level, accumulation of ER proteins due to WDR45 deficiency resulted in increased ER stress and impaired ER quality control. The unfolded protein response (UPR) was elevated through ERN1/IRE1 or EIF2AK3/PERK pathway, and eventually led to neuronal apoptosis. Suppression of ER stress or activation of autophagy through MTOR inhibition alleviated cell death. Thus, the loss of WDR45 cripples macroautophagy machinery in neurons and leads to impairment in organelle autophagy, which provides a mechanistic understanding of cause of BPAN and a potential therapeutic strategy to treat this genetic disorder.
Abbreviations: 7-ADD: 7-aminoactinomycin D; ASD: autistic spectrum disorder; ATF6: activating transcription factor 6; ATG: autophagy-related; BafA1: bafilomycin A1; BCAP31: B cell receptor associated protein 31; BPAN: β-propeller protein-associated neurodegeneration; CCCP: carbonyl cyanide m-chlorophenylhydrazone; CDIPT: CDP-diacylglycerol–inositol 3-phosphatidyltransferase (phosphatidylinositol synthase); DDIT3/CHOP: DNA-damage inducible transcript 3; EIF2A: eukaryotic translation initiation factor 2A; EIF2AK3/PERK: eukaryotic translation initiation factor 2 alpha kinase 3; ER: endoplasmic reticulum; ERN1/IRE1: endoplasmic reticulum to nucleus signaling 1; GFP: green fluorescent protein; HIP: hippocampus; HSPA5/GRP78: heat shock protein family A (HSP70) member 5; KO: knockout; LAMP1: lysosomal-associated membrane 1; mEPSCs: miniature excitatory postsynaptic currents; MG132: N-benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal; MIB: mid-brain; MTOR: mechanistic target of rapamycin kinase; PCR: polymerase chain reaction; PFA: paraformaldehyde; PFC: prefrontal cortex; PRM: parallel reaction monitoring; RBFOX3/NEUN: RNA binding protein, fox-1 homolog [C. elegans] 3; RTN3: reticulon 3; SEC22B: SEC22 homolog B, vesicle trafficking protein; SEC61B: SEC61 translocon beta subunit; SEM: standard error of the mean; SNR: substantia nigra; SQSTM1/p62: sequestosome 1; TH: tyrosine hydroxylase; Tm: tunicamycin; TMT: tandem mass tag; TUDCA: tauroursodeoxycholic acid; TUNEL: terminal deoxynucleotidyl transferase dUTP nick-end labeling; UPR: unfolded protein response; WDR45: WD repeat domain 45; WT: wild type; XBP1: X-box binding protein 1.
KEYWORDS: ER stress, neuronal apoptosis, quantitative proteomics, UPR, WDR45
Introduction
Macroautophagy (herein referred to as autophagy) is the major cellular catabolic process to degrade damaged organelles as well as protein aggregates. The process involves the engulfment of cellular compartments by a double-membrane structure and the fusion with lysosomes for degradation [1]. The importance of autophagy in preventing neurodegeneration has been greatly appreciated. In mice, deletion of the essential autophagy gene Atg5 or Atg7 in the central nervous system results in accumulation of intracellular inclusion bodies and neurodegeneration [2,3]. In humans, mutations in various genes functioning in multiple steps of autophagy cause a wide range of neurodegenerative diseases [4–6].
β-Propeller protein-associated neurodegeneration (BPAN) is a recently identified subtype of neurodegeneration with brain iron accumulation (NBIA) [7]. Since its first description in 2012, 68 cases of BPAN patients have been reported [8]. Among them only 9 cases are male and they generally show more severe endophenotype, probably because of somatic mosaicism in males and skewed X-chromosome inactivation (XCI) in females [8]. BPAN is characterized by global developmental delay in motor and cognitive function during early childhood and the intellectual disability is carried into adulthood. Various seizure types have been observed in these patients during childhood [9]. During early adulthood the patients develop sudden-onset dystonia, parkinsonism, and dementia [10–12]. To date, most studies on BPAN have focused on case reports and genetic analyses, confirming BPAN as a distinct class of neurodegenerative disease. Recently, de novo mutations in WDR45 (WD repeat domain 45) gene were identified in BPAN patients by exon sequencing [7,13,14]. Further studies on patient-derived lymphoblastoid cells show lower autophagic activity and accumulation of early autophagic structures [13], linking abnormal autophagy to the disease.
As a β-propeller-shaped scaffold protein, WDR45/WIPI4 is one of the 4 mammalian homologs of yeast Atg18, which plays important roles in autophagy [15]. These roles include recruitment of lipids to membranes via its conserved residues, specific binding to phosphatidylinositol-3-phosphate (PtdIns3P), and regulation of autophagosome formation via interacting with ATG2 and AMP-activated protein kinase (AMPK)-unc-51 like kinase 1 (ULK1) complexes [15–17]. Unlike other essential autophagy genes such as Atg1, Atg5, and Atg7, whose germ line deletion is lethal; germ line deletion of epg-6 (ectopic p granules 6), the worm homolog of WDR45, in Caenorhabditis elegans only causes abnormal autophagic structures [18]. Neuronal-specific knockout of Wdr45 in mouse results in dysfunction in autophagy and axonal degeneration, together with poor motor coordination and impaired learning and memory [19]. Thus, it appears that the major function of WDR45 is the regulation of autophagosome formation and its loss-of-function contributes to behavioral abnormalities in mice.
In spite of these studies, it remains a mystery how loss of WDR45 leads to neurodegeneration in BPAN, and whether dysfunction in autophagy is the major cellular underpinning of the disease. To address these questions, we generated constitutive wdr45 knockout (KO) mouse to model the complex processes of BPAN. Multiple levels of analyses showed that the KO mouse displays cognitive impairments, lesions in prefrontal cortex and basal ganglia. Comparative proteomic quantitation of brain regions showed a surprisingly large number of accumulated endoplasmic reticulum (ER) proteins in KO mouse. Mechanistically, our data indicated that loss of WDR45 resulted in elevated ER stress and impaired ER turnover. In KO cells, the unfolded protein response (UPR) was augmented under basal condition and was dramatically aggravated after inducing ER stress, which led to the induction of apoptosis. These abnormalities were alleviated by an MTOR inhibitor as well as by an inhibitor of ER stress. Thus, our study revealed that a dysfunction in the macroautophagy machinery leads to abnormal organelle autophagy, which results in BPAN-like pathology.
Results
Wdr45 knockout mouse displayed cognitive impairments
We generated wdr45 KO mouse using clusters of regularly interspaced short palindromic repeats-CRISPR associated protein 9 (CRISPR-Cas9) technique targeting the mutated sites found in BPAN patient (Formerly known as SENDA, subject 1) [13] (Figure 1a). The genomic DNA was extracted for polymerase chain reaction (PCR) using primers flanking the target site. DNA sequencing showed that in the mutant mouse two base pairs at the end of exon 6 were removed, resulting in frame-shift mutation and the introduction of a premature stop codon (Figure 1b). Moreover, the same Wdr45 mutation was present in the F1 mice, suggesting that this mutation is heritable. Real-time PCR confirmed that the mRNA of Wdr45 gene was significantly reduced in mutant mice (referred to as KO thereafter) in the brain (Figure 1c). Because commercially available antibodies were not able to detect endogenous WDR45 in various tissues in our hands, we used parallel reaction monitoring (PRM), a targeted mass spectrometry approach, to detect two specific WDR45 peptides (peptide1: 335-YVFTPDGNCNR-345, peptide2: 60-SNLLALVGGGSSPK-73). We were able to detect both WDR45 peptides in WT but not in KO mice (Figure 1d and Figure S1b-d).
Figure 1.
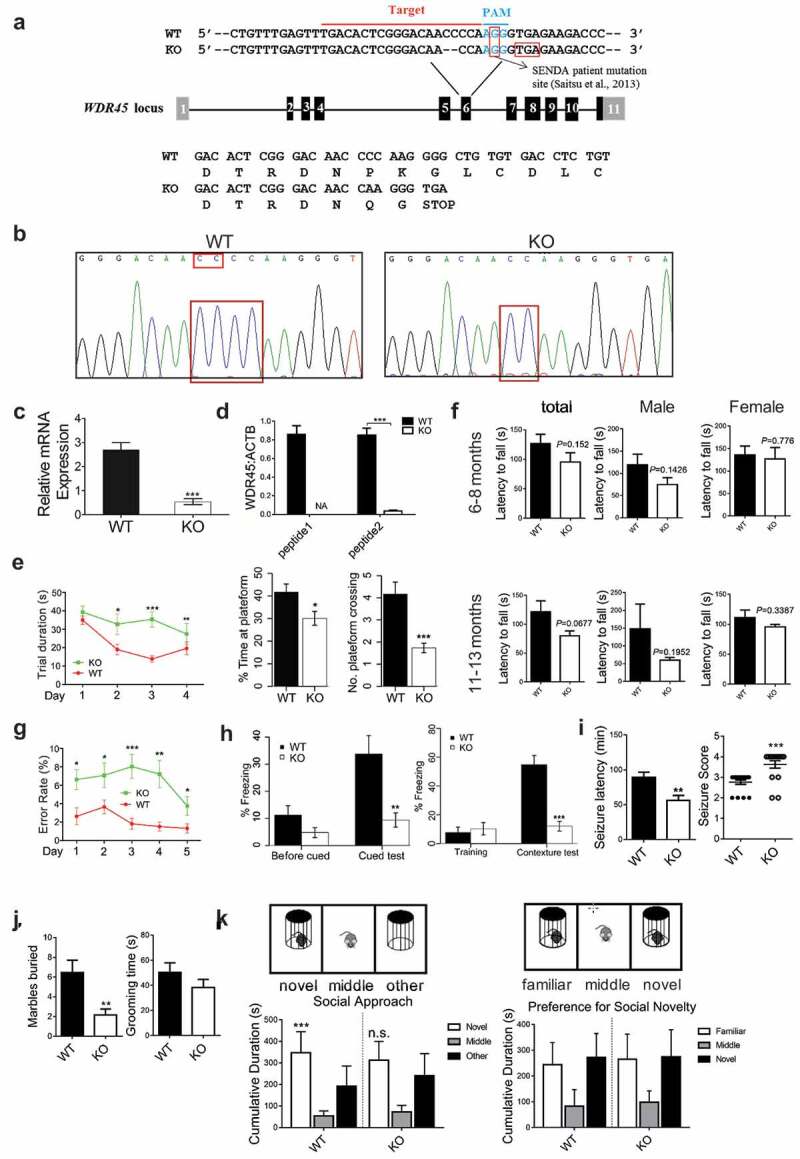
wdr45 knockout (KO) mice show cognitive impairments. (a) Generation of wdr45 KO mouse by deleting a CC dinucleotide at the exon 6 of Wdr45 gene. (b) DNA sequencing validation of the targeted sequence in WT and KO mouse. (c) Real-time PCR quantification of mRNA expression in WT (n = 3) and KO (n = 3) mice. (d) Protein expression quantified by parallel reaction monitoring (PRM) mass spectrometry of WDR45 peptides (peptide1:YVFTPDGNCNR, peptide2:SNLLALVGGGSSPK). NA, not available. (e) Morris water maze test of the trial period (left, P < 0.01, P = 0.006 and P < 0.001 on day 2, 3 and 4, respectively. WT: n = 16, KO; n = 16. All males), time spent at the platform and times of platform crossing (right bar graphs, P = 0.017 and P < 0.001 respectively). (f) Rotarod test showing the latency to fall at 6–8 and 11–13 month of age (WT: n = 10, 5 males, 5 females; KO: n = 10, 6 males, 4 females). (g) An 8-arm maze showing the error rate (*P < 0.05, **P < 0.001, ***P < 0.001. WT: n = 15, KO: n = 17, all males). (h) Freezing response to the shock in the cued test (left, P = 0.0023) and the contexture test (right, P < 0.001), compared to WT mice (n = 16 for both WT and KO, all males). (i) Seizure latency and severity in mice of 2 month old after pilocarpine administration (30 mg/kg) are shown (WT: n = 17, 10 males, 7 females; KO: n = 16, 8 males, 8 females). (j) Repetitive self-grooming and marble burying behavior were measured in mice of 2 month old (WT: n = 13, 8 males, 5 females; KO: n = 14, 7 males, 7 females). (k) Social approach test, WT mice spent significantly more time with novel conspecific compared to an empty test cage and no preference for novel conspecific was found for KO mice (WT: n = 13, 8 males, 5 females; KO: n = 14, 7 males, 7 females). Data shown are Mean ± SEM, P values: *P < 0.05, **P < 0.01, ***P < 0.001.
We then performed behavioral experiments using mice at 6 months of age. In the morris water maze test, we noted significant difference in trial duration between KO and WT mice during the training period. On day 2, 3 and 4 during the training, KO mice spent much more time to find the platform compared to WT mice (Figure 1e, left, P < 0.01, P = 0.006 and P < 0.001, respectively). During the test, KO mice stayed or crossed the target area with significantly less time (Figure 1e, right, P = 0.017 and P < 0.001, respectively). In the 8-arm maze test, the error rate in KO mice was significantly higher than that in WT mice on day 3 (P < 0.0001), and the error rate significantly declined over time for both KO and WT mice on day 5 (P = 0.026) (Figure 1g). The rotarod test did not show statistically significant difference (Figure 1f). We further analyzed the rotarod test results separately by gender. In general KO male mice are more likely to fall from the rotarod (Male WT:119.2 ± 24.05s vs male KO:74.83 ± 15.43s; Female WT: 136 ± 19.44s vs female KO: 126.8 ± 25.38s at 6–8 month), although the P value was not significant (Male:P = 0.1426 vs Female: P = 0.776). In the fear condition and cued tests, KO mice showed significantly reduced freezing response to fear in both the contextual and cued stimulation (Figure 1h). These data suggest that at the tested age wdr45 KO mice showed no sign of significant impairment in motor coordination. However, they suffer from deficits in spatial learning and conditioning memory.
Because a subset of BPAN patients displays epilepsy and autistic spectrum disorder (ASD), we tested whether wdr45 KO mice suffered from similar symptoms. We induced seizure with pilocarpine, and assessed seizure severity and latency. At 2 months of age, KO mice showed a significantly shorter latency and increased seizure severity compared to WT mice (Figure 1i). We also assessed characters of ASD. In the marble burying test, KO mice buried significantly less marbles than did WT mice, while the self-grooming test did not reveal statistically significant difference (Figure 1j). In the three-chamber social approach test, WT mice spent significantly more time in the novel proximity zone than the empty proximity zone, while KO mice showed no preference. When a second novel mouse was introduced to the remaining side chamber, both WT and KO mice show no preference of novel over familiar mouse (Figure 1k). Overall, younger KO mice suffered from epileptic seizures and displayed some ASD-related phenotypes.
Wdr45 knockout mouse showed neuronal loss in the brain
Since BPAN patients display neurodegeneration and parkinsonism in adulthood, we examined whether aged mice showed signs of neurodegeneration. We performed immunostaining in mice at 16 months of age using an antibody against TH (tyrosine hydroxylase) in substantia nigra (SNR) and RBFOX3/NeuN in prefrontal cortex (PFC), respectively. wdr45 KO mice displayed nearly 50% reduction of TH+ neurons in SNR regions and a 34% reduction of RBFOX3+ neurons in PFC regions (Figure 2a-c). These defects appeared to be age-related since they were not observed in 6-month-old KO mice (Figure S2a-2c). Terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) assay on brain regions showed that there were elevated TUNEL labeling in PFC of KO mice (Figure 2d and e). We also examined whether there were differences in brain iron accumulation, another neuropathological feature found in a subset of BPAN patients. We found essentially no change in Pearls’ Prussian blue stain of iron in KO mice at six month of age. However, at sixteen month of age there was statistically significant difference (Figure S1e and 1f). Thus, the KO mouse we generated shows neurodegeneration at older age, and mimics at least some pathological features of BPAN patients.
Figure 2.
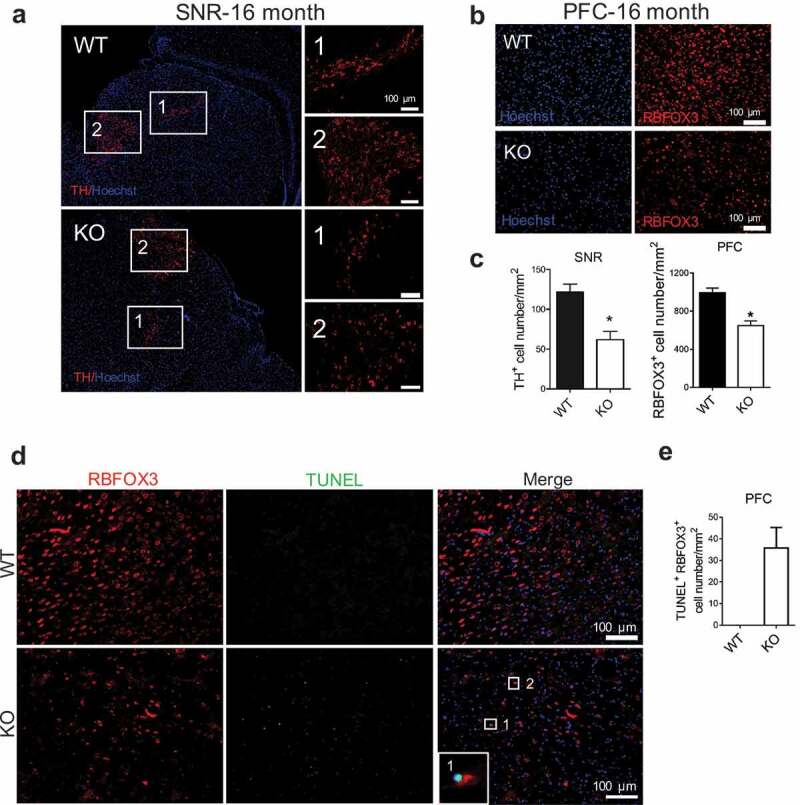
wdr45 KO mice show neuron loss in SNR and PFC at old age. (a and b) Immunofluorescence staining of brain sections from WT and KO mouse at 16 months of age. Scale bar: 100 μm. (c) Statistical analysis of TH-positive cells in SNR (a) and RBFOX3 positive cells in PFC (b), respectively. (d) TUNEL staining of brain sections from WT and KO mouse PFC at 16 months of age. (e) Statistical analysis of TUNEL positive- RBFOX3 positive cell number in PFC (d). Data were expressed as mean ± SEM (n = 3) and analyzed by two-tailed unpaired t-test. P values: * P < 0.05. PFC: prefrontal cortex, SNR: substantia nigra. Three mice brains per group and 3 sections per brain were used for quantification.
We then used whole-cell patch-clamp recordings to examine the effect of Wdr45 deletion on basal synaptic transmission of hippocampal CA1 neurons from 6-month-old mice. We found no significant difference in the frequency of miniature excitatory postsynaptic currents (mEPSCs) between WT and KO mice (Figure S2d). However, the amplitude of mEPSCs in the CA1 pyramidal neurons was significantly decreased in KO compared to the WT mice, indicating a deficit in post-synaptic function of the hippocampal synapses. In contrast, presynaptic function as assessed by paired-pulse facilitation (PPF) was unaffected (Figure S2e). Strikingly, the KO mouse shows axonal swelling at 7 month of age (Figure S2f and 2g), similar to the axonal pathology characterized by axonal imbalance and neural degeneration [20] found in a previous study [19] but in an earlier age.
Quantitative proteomic analysis of brain regions revealed ER protein accumulation in KO mouse
We dissected five pairs of WT and KO mice brains into pre-frontal cortex (PFC), hippocampus (HIP) and mid-brain (MIB), and performed quantitative tandem mass spectrometry experiments using 10-plex tandem mass tag (10xTMT) labeling (Figure S3a). In total we quantified nearly 4,000 proteins overlapped in three brain regions, and 67 proteins in PFC, 12 proteins in HIP, and 185 proteins in MIB were significantly changed (P < 0.05 after Benjamini-Hochberg correction, fold change ≥ 1.5), with PFC and MIB shared 30, and HIP and MIB shared 4 significantly changed proteins (Figure 3a and b, Table S1). The proteins quantified in each individual brain region are listed in Tables S2–4. Principal component analysis of the quantitative results shows that the protein expression profiles are well separated between the two genotypes in each brain region (Figure 3c), indicating that our quantitative mass spectrometry method is able to differentiate mouse brain proteome by the loss of WDR45. Volcano plots show quantified protein ratios between KO and WT and their P values in the three brain regions. Proteins significantly upregulated were labeled red and significantly down regulated were labeled green, and we observed more upregulated than down regulated proteins (Figure 3d). Surprisingly, regardless of well-established role of WDR45 in autophagy, among the 40 quantified proteins intimately participating the autophagy pathway, none of them showed statistically significant change except WDR45 (Figure S3b). To test whether changes in protein level were the results of changes in gene expression, we performed quantitative PCR experiments on 16 significantly changed proteins in the three brain regions and found that with very few exceptions, the mRNA of the majority of the proteins were unaltered (Figure S3c), ruling out the regulation of the protein abundance at the transcription level.
Figure 3.
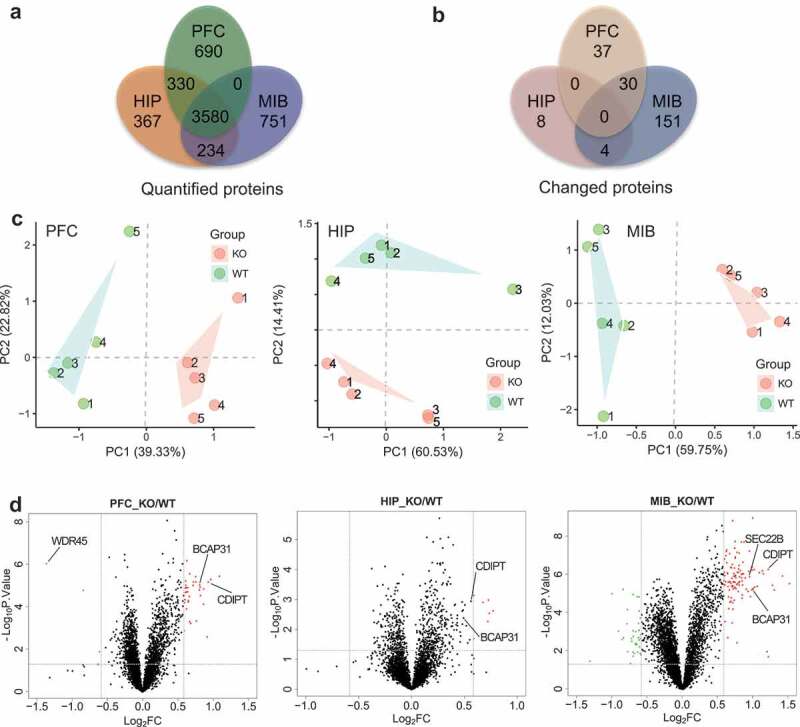
Quantitative proteomic analysis of brain regions from WT and KO mouse. (a) TMT-based mass spectrometry quantification of proteins in PFC, HIP, and MIB (n = 5, male, 6 months). (b) Significantly changed proteins in the three regions (P < 0.05 after Benjamini Hochberg correction, and fold change≥1.5). (c) Principal component analysis of proteins quantified in the three brain regions. (d) Volcano plots of quantified proteins in PFC, HIP and MIB, respectively. PFC: prefrontal cortex, HIP: hippocampus, MIB: mid-brain.
Bioinformatics analysis applying Database for Annotation, Visualization and Integrated Discovery (DAVID) [21,22] on the changed proteins in MIB and PFC reveals that organization of the ER emerged as the most dramatically enriched gene ontology term, followed by lipid metabolic process and endomembrane system organization (Figure 4a). Several other cellular compartments including mitochondria and proteasome complex are also among the top ten enriched terms. Furthermore, weighted gene co-expression network analysis (WGCNA) reveals the accumulation of ER proteins in KO mice (Figure 4b), and a complex network of ER resident proteins as well as ER-associated proteins are shown in the cyan module in Figure 4c.
Figure 4.
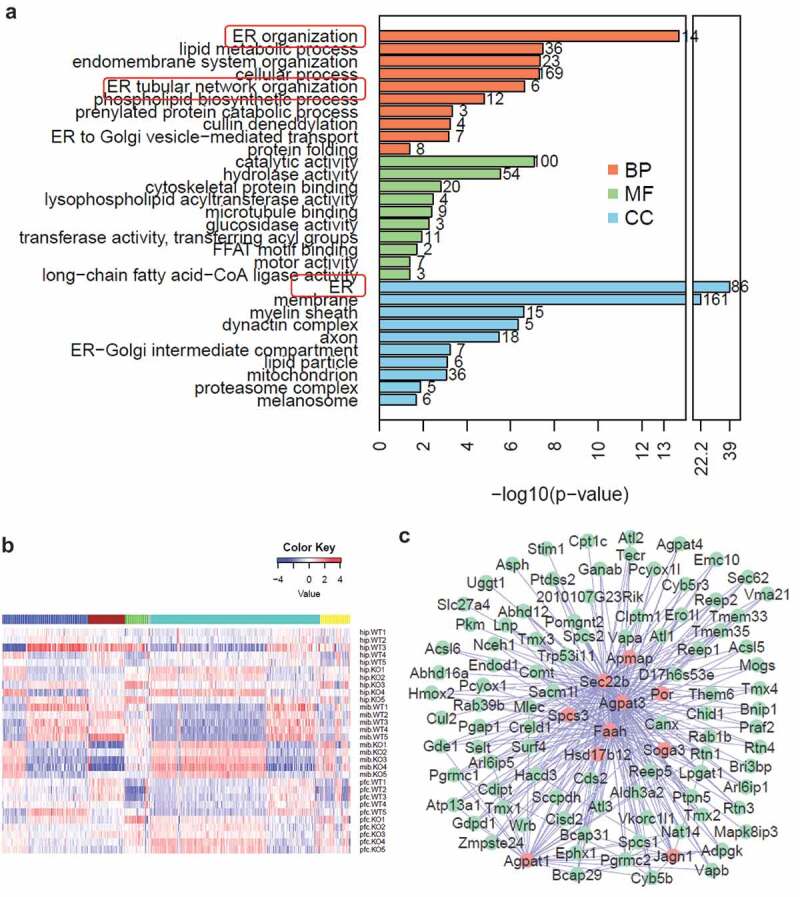
Bioinformatic analysis reveals accumulation of ER proteins in wdr45 KO mice. (a) Top ten significantly enriched gene ontology terms in biological process (BP, brown), molecular function (MF, green) and cellular component (CC, blue). (b) WGCNA of proteins quantified in the three brain regions separates the proteins into five distinct co-expressed modules. Color key represents log2-transformed intensity values of each protein. (c) Protein-protein interaction network of the proteins in green module, with red representing hub proteins that have the most network connectivity.
A recent study applied interaction proteomics to identify 321 unique proteins interacting with six proteins associated with neurodegenerative diseases, including APP (amyloid beta precursor protein), PSEN1 (presenilin 1), HTT (huntingtin), PRKN/PARK2 (parkin RBR E3 ubiquitin protein ligase), and ATXN1 (ataxin 1) [23]. From our quantified 5407 proteins in the three brain regions combined, we applied a less stringent criterion (P < 0.05 and KO:WT ratio > 1.25 or < 0.8) to form a list of 678 significantly changed proteins. We mapped these proteins to the 321 proteins and displayed the overlapped 58 proteins with their normalized expression levels in a heat map (Figure S4a). Among the three brain regions, MIB showed the most striking differences between WT and KO mice for the proteins that interact with these disease-associated proteins. The overlap between our dataset and the interaction proteomics data are shown in Figure S4b. Gene ontology analysis revealed that the ubiquitin-proteosome system was the most significantly enriched process, which is in line with the well-established notion that protein quality control is an underlying molecular mechanism in neurodegeneration. Of note, endoplasmic reticulum is among the top ten significantly enriched terms (Figure S4c).
WDR45 regulated target ER proteins via proteasome and lysosome pathways
We then investigated how WDR45 regulates levels of ER proteins. First, we verified that endogenous SEC22B is significantly upregulated in the midbrain of wdr45 KO mice, consistent with mass spectrometry results (Figure 5a and 5b). We then co-expressed WDR45 and 4 ER proteins in the presence or absence of bafilomycin A1 (BafA1), an autophagy inhibitor that blocks autophagosome-lysosome fusion, or N-benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal (MG132), a proteasome inhibitor. BafA1 but not MG132 effectively blocked WDR45-mediated degradation of SEC22B (Figure 5c and 5d), while both BafA1 and MG132 blocked WDR45 mediated degradation of BCAP31 (Figure S5a and 5b). For SEC61B, MG132 but not BafA inhibited WDR45-mediated degradation (Figure S5c and 5d), while neither of the 2 inhibitors had any effect on CDIPT (Figure S5e and 5f). As a control, only MG132 had a mild effect on GFP stability (Figure S5g and h). These different effects suggest that proteins localize at ER at different compartments are degraded by different mechanisms. Furthermore, cells stably expressing shRNA targeting WDR45 mRNA (sh-WDR45) showed accumulation of all four proteins comparing to cells expressing scrambled (sh-Scrbl) RNA (Figure 5e–g), similar to the effect observed in KO mouse brain. These results indicate that WDR45 negatively influences the stability of a subset of ER proteins. Mechanistically, our co-immunoprecipitation experiments suggest that it is less likely that WDR45 mediated the degradation of ER proteins through interaction with known reticulophagy receptors (Figure S5i-l).
Figure 5.
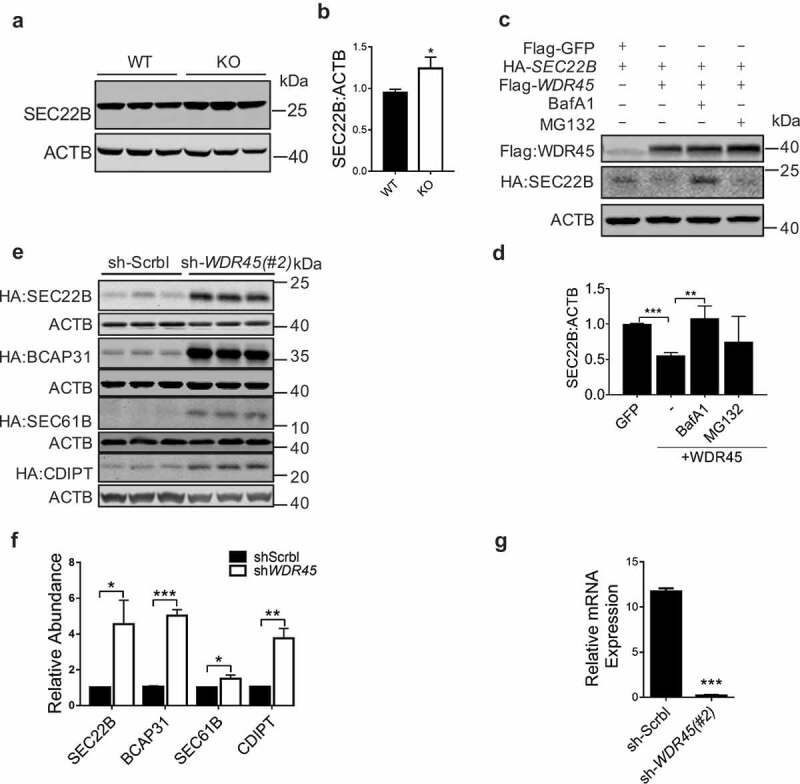
WDR45 mediates degradation of target ER proteins. (a and b) Western blot detection of endogenous SEC22B in mid-brain. Protein abundance was quantified in (b). (c and d) Co-expression of WDR45 and SEC22B in HeLa cells and protein levels were examined by western blot. Some groups of cells were treated with MG132 (20 μM, 12 h) or BafA1 (50 nM, 8 h), respectively. Protein abundances were quantified in (d). (e and f) Stability of exogenously expressed SEC22B, BCAP31, SEC61B and CDIPT in HeLa cells with WDR45 stably knocked down. Bar graph (f) shows statistics of protein expression levels. (g) Real-time PCR quantification of WDR45 mRNA in stable cell lines (#2). Data were expressed as mean ± SEM (n = 3). P values: *P < 0.05, **P < 0.01, ***P < 0.001.
Cells with WDR45 deficiency resulted in ER expansion and increased ER stress
We then investigated whether accumulation of ER proteins correlated to an expanded ER area, a phenomenon that was recently demonstrated to be the result of a reduced selective ER autophagy [24]. Cells expressing shRNA targeting two different WDR45 mRNA sequences showed significant ER expansion, comparing to cells expressing sh-Scrbl, as imaged by labeling the endogenous ER protein CALR (calreticulin) (Figure 6a and b, P < 0.001). Similar results were observed by labeling ER using exogenously expressed BCAP31, CDIPT, and SEC22B (Figure S6a and b). Furthermore, inducing ER stress using tunicamycin (Tm) resulted in exogenous WDR45 being recruited to the ER in WT but not WDR45-deficient cells (Figure S6c). Immunofluorescence analyses revealed that in WT but not KO cortical neurons, CALR (green) accumulated in lysosome (LAMP1, red) after ER stress (Figure 6c and d), suggesting that lysosomal degradation of ER proteins requires WDR45. To test whether this effect of ER autophagy is organelle specific, we treated cells with carbonyl cyanide m-chlorophenyl hydrazone (CCCP) to disrupt mitochondrial membrane potential. In control cells, treatment with CCCP for 24 hours effectively resulted in dramatic loss of PRKN staining and shrinkage of mitochondria, suggesting PRKN-mediated mitophagy. However, in cells expressing sh-WDR45, the mitochondria appeared significantly larger (Figure S6d and 6e). Finally, transmission electron microscope analysis of mouse substantia nigra showed enlarged ER tubules in KO mouse (Figure S7a). Thus, we demonstrated that a defect in macroautophagy resulted in defects in autophagy of ER and mitochondrion, two independent organelles.
Figure 6.
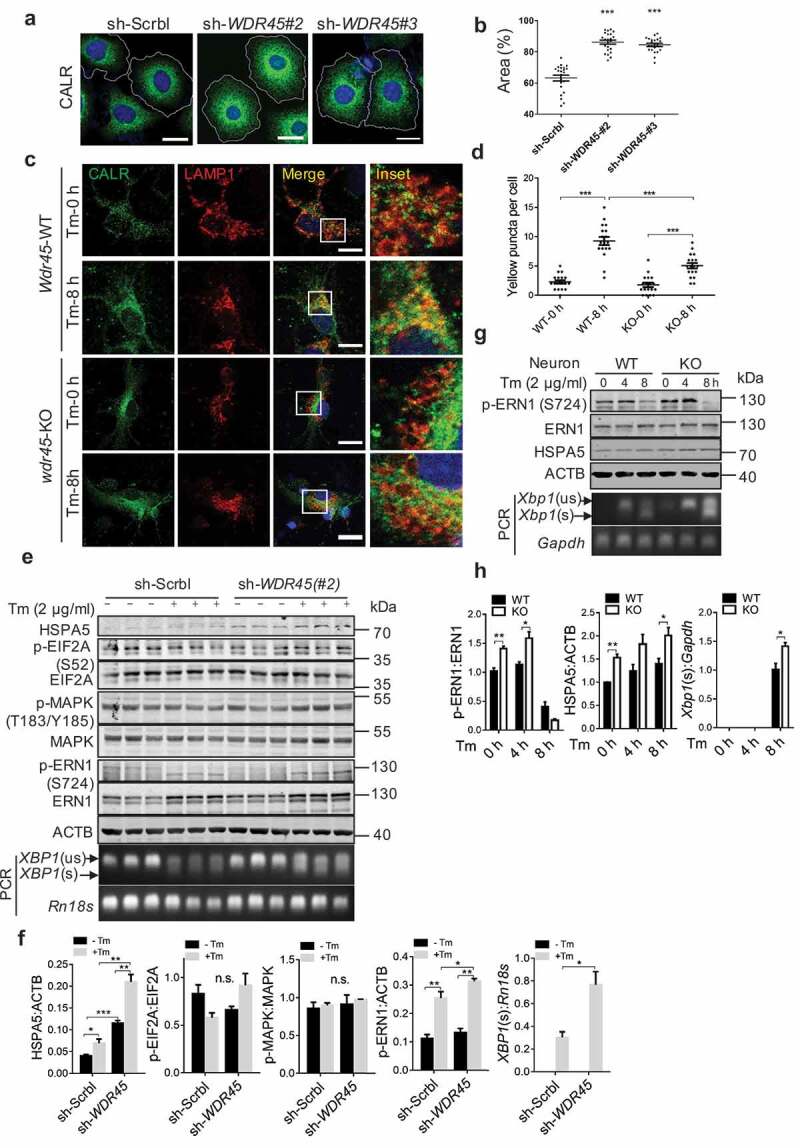
WDR45-deficient cells display increased ER stress and ER area. (a) Representative confocal images of immunofluorescence labeled CALR, an ER resident protein, in HeLa cells. Scale bar: 20 μm. (b) Quantification of ER area, each dot in the graph represents one cell, n = 30. ***P<0.001, unpaired t test. (c) Immunofluorescence double labeling of CALR and LAMP1 in primary cortical neurons and imaged by confocal microscopy. Tm, 2 μg/ml, 8 h. Scale bar: 20 μm. (d) Yellow puncta in lysosome (c) was quantified. Each dot in the graph represents one cell, n = 18. (e and f) HeLa cells, (g and h), primary cortical neurons were treated with Tm, blotted with antibodies in the ER stress pathway. Data were expressed as mean ± SEM (n = 3). **P < 0.05, **P < 0.01, ***P < 0.001. n.s., not significant.
To establish a time line at which defect of autophagy occur, we measured markers of autophagy and ER stress in mouse PFC at 1, 8 and 16 months of age. Western blot of SQSTM1 and MAP1LC3B/LC3B showed that defect of autophagy started to show at 1 month in the PFC of KO mouse, while ER stress did not manifest until 16 months (Figure S8). These in vivo data clearly show that defect of autophagy occurs long before ER stress response, and suggest that WDR45 deficiency-induced impairment of autophagy may results in the inhibition of reticulophagy, as well as mitophagy.
What could be the physiological trigger for compromised organelle integrity? A well-established mechanism to handle ER stress is the unfolded protein response (UPR) [25,26]. We therefore examined this pathway and found that the ER chaperon HSPA5 (heat shock protein 5) was significantly upregulated in human cells expressing sh-WDR45 comparing to control cells, even under basal condition. Inducing ER stress further promoted HSPA5 expression (Figure 6e and f, and Figure S6f-h). Out of the three known UPR pathways [25,26], we found over activation of the ERN1/IRE1 pathway, because phosphorylation of ERN1/IRE1 at serine 724 was significantly upregulated in cells expressing sh-WDR45. In addition, we observed an increased selective splicing product of XBP1 (Figure 6e and f). In contrast, we found no evidence of significant change in EIF2A phosphorylation, an indication of EIF2AK3/PERK activation (Figure 6f). Consistently, in primary neurons from KO mouse, we also found increased HSPA5 expression, ERN1/IRE1 activation and increased selective splicing of Xbp1 (Figure 6g and h).
WDR45 deficiency led to an increased cell death after ER stress
If an abnormally increased ER stress cannot be properly handled by UPR or autophagy, it could eventually leads to apoptosis [26]. Cell viability assay showed that Tm treatment resulted in significantly reduced viability in cells expressing sh-WDR45 than control cells (Figure S9a), the reduced cell viability was further supported by increased CASP3 cleavage (Figure S9b and c). In primary neurons with WDR45 deficiency, there were increased levels of the ER marker CALR, accumulation of the autophagy receptor SQSTM1, and reduced LC3-II:LC3-I ratios (Figure 7a and b). In mouse brain, the defect in autophagy marked by SQSTM1 accumulation and reduced LC3-II:LC3-I levels, increased ER stress marked by HSPA5, DDIT3/CHOP, and activation of the ERN1/IRE1 pathway, and finally increased apoptosis were recapitulated by western blot and immunohistochemistry (Figure 7c–f, Figure S9f-i). ER stress-induced apoptosis is characterized by CASP12 cleavage, consistently, we also found increased CASP12 cleavage in wdr45 KO mouse PFC region at 16 months of age (Figure 7c and d). However, there were differences in the activation of UPR pathway in brain tissues. Increased phosphorylation of EIF2A, an indication of EIF2AK3 activation was significant in KO mouse, while changes in ERN1/IRE1 phosphorylation and ATF6 (N terminal) levels were not significant. Defects in ER stress, UPR, and apoptosis were not observed in 1-month-old KO mouse (Figure S9d and 9e), which is consistent with the idea that aging is one of an important risk factor for neurodegeneration.
Figure 7.
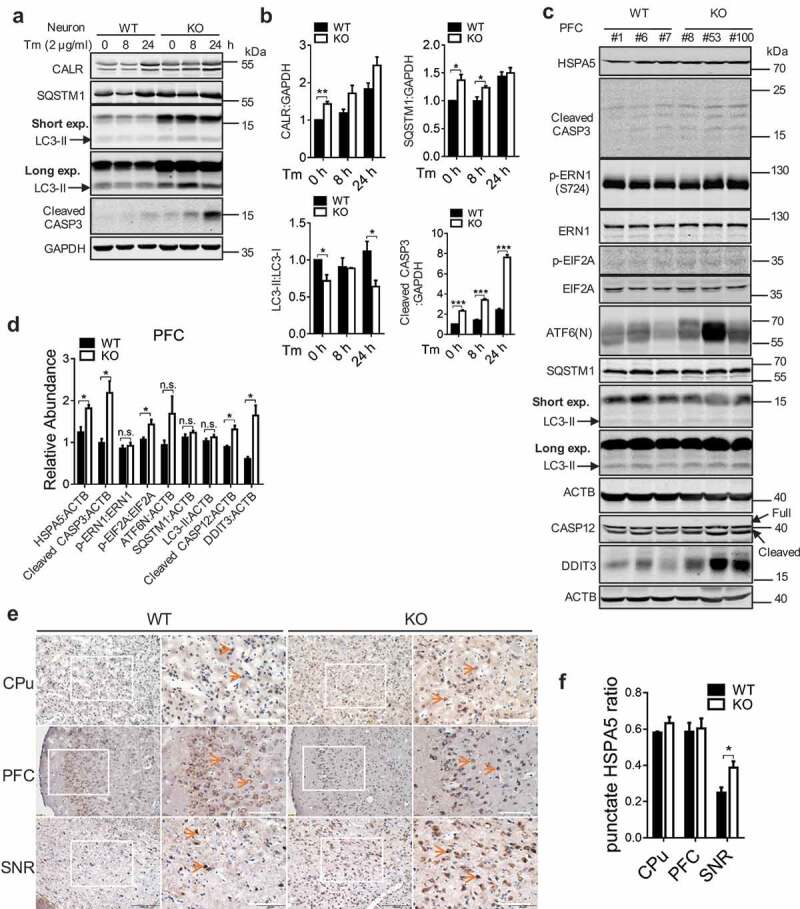
Loss of WDR45 results in increased cell death associated with ER stress. (a) Western blot analysis of CASP3 and autophagy proteins after Tm treatment in WT and KO primary neurons. (b) Bar graph shows statistical analysis of western blot data shown in (a). (c) Western blot analysis of CASP3, ER stress markers and autophagy proteins in the PFC from WT and KO mouse at 18–20 months of age. ‘Short’ and ‘long’ refer to the exposure time. (d) Statistical analysis of (c). (e) Immunohistochemistry of HSPA5 in mouse substantia nigra (SNR), prefrontal cortex (PFC), and caudate putamen (CPu) at 8 months of age. (f) Statistical analysis of (e), n = 3 per group and 3 sections per brain were used for quantification. Data were expressed as mean ± SEM (n = 3) and analyzed by two-tailed unpaired t-test. *P < 0.05, **P < 0.01, *** P < 0.001. n.s., not significant.
Activation of autophagy or inhibition of ER stress rescued apoptosis
We further asked whether pharmacological rescue of apoptosis could be achieved by modulating the levels of autophagy and ER stress. We used rapamycin, an inhibitor of MTOR signaling that could potentially relieve the inhibition of autophagy at basal level by MTOR [27], induction of autophagy with rapamycin may be beneficial by preventing or attenuating pathogenic protein aggregation in neurodegenerative diseases [28]. In addition, we used tauroursodeoxycholic acid (TUDCA), an ER stress inhibitor [29]. Either rapamycin or TUDCA reduced accumulation of exogenously expressed ER proteins SEC22B and SEC61B in WDR45-deficient cells (Figure S10a and 10b). Aggravated ER stress in WDR45-deficient cells was reduced by rapamycin or TUDCA. Increased CASP3 cleavage in WDR45-deficient cells was also reduced by rapamycin or TUDCA (Figure 8a and b). Flow cytometry labeling of cell surface markers ANXA5/annexin V and DNA marker 7-aminoactinomycin D (7-ADD) for apoptosis demonstrated that Tm-induced apoptosis was alleviated by treatment with either rapamycin or TUDCA (Figure 8c and d). Most excitingly, in cultured primary neurons, Tm-induced autophagic activity was defective in KO neurons, as indicated by LC3-II:I ratios. Treatment with rapamycin mildly but significantly increased LC3-II:I ratios, suggesting a partial restore of autophagic activity (Figure 8e and f). Finally, rapamycin and TUDCA significantly reduced CASP3 cleavage in KO neurons (Figure 8e and f). Together, our data provide an integral link between defects in autophagy due to WDR45 deficiency and ER quality control; and suggest an underlying mechanism of β-propeller protein-associated neurodegeneration.
Figure 8.
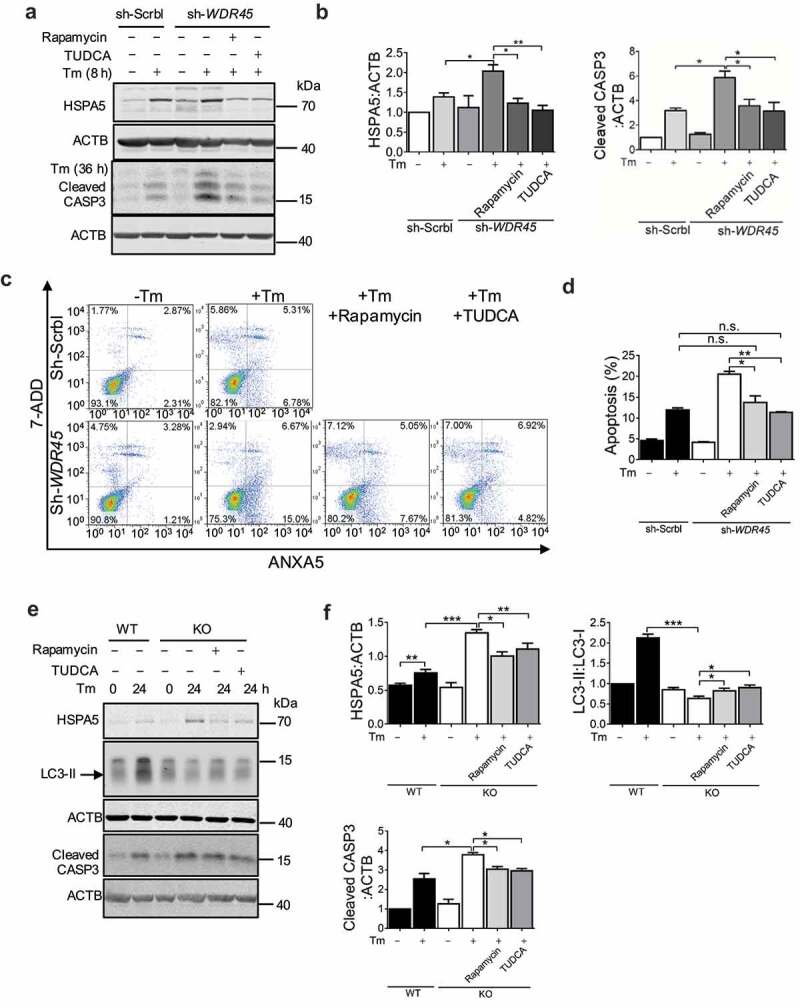
Activation of autophagy or inhibition of UPR rescues abnormal protein processing and increased cell death in WDR45-deficient cells. (a and b) Western blot analysis of endogenous HSPA5 and CASP3 levels after rapamycin (100 nM, 24 h) or TUDCA (100 μM, 24 h) treatment in HeLa cells with or without ER stress. (c and d) Flow cytometry analysis of apoptosis (ANXA5+ 7ADD− and ANXA5+ 7ADD+) in WDR45-deficient HeLa cells after inducing ER stress (2 μg/ml Tm, 36 h). (e and f) Effects of rapamycin or TUDCA on HSPA5 accumulation (2 μg/ml Tm, for 8 h), CASP3 cleavage (2 μg/ml Tm, 24 h), and LC3-II production, in mouse neurons after inducing ER stress. Data were expressed as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, *** P < 0.001. n.s., not significant.w.
Discussion
Although genetic basis of β-propeller protein-associated neurodegeneration has been well established, the molecular and cellular underpinnings of the disease process are currently not yet known. Our study filled this gap by providing compelling evidence showing that a deficit in autophagy due to Wdr45 mutation in mouse leads to improperly handled organelle stress, particularly ER stress, which is accumulated over time during the life span. Accumulation of ER stress triggers UPR through ERN1/IRE1 and EIF2AK3/PERK pathways, and eventually leads to neuronal apoptosis, a hallmark of neurodegeneration. Furthermore, our data suggests that suppression of ER stress, or activation of autophagy through inhibition of MTOR, rescues neuronal death.
A previous study by Zhao et al. generated neuronal-specific knockout of Wdr45 in mouse. The KO mouse showed poor motor coordination and deficits in learning and memory at 9–12 months of age, and axonal swelling at 11–13 months [19]. We generated knockout mouse through germ line mutation. Behaviorally, there was no significant alteration in motor coordination at 6 months as well as 11–13 months of age, which is a discrepancy comparing to Zhao’s study. All the mice used in rotarod test were males in Zhao’s study, while we used mixed number of male and female mice. As Wdr45 locates on the X chromosome, the phenotype of KO male mice was expected to be more serious. Indeed, in our seizure susceptibility test, KO male mice displayed serious seizures while seizures observed in KO female mice were much milder. The gender difference in the rotarod behavior might have increased the P value. On the other hand, we found significant impairment in both spatial and contextual memory as early as 6 months of age (Figure 1e–h). Similarly, the KO mouse showed abnormalities in basal synaptic transmission (Figure S2). Thus, our germ line mutation mouse shares some features with that of conditional knockout mouse. Furthermore, these mice also displayed distinct pathological features such as increased neuronal death at older age (Figure 2 and Figure S2).
Our study provides evidence that a defect in macroautophagy causes defects in autophagy in organelles. To understand the selective vulnerability of ER to WDR45 mutation, we affinity purified WDR45 complexes from cells after induction of ER stress. Mass spectrometry results showed that only ATG2B was the most prominent protein partners of WDR45 (Figure S5i), confirming previous reports [30]. We also attempted to test whether known ER selective autophagy receptors, including RETREG1/FAM134B (reticulophagy regulator 1), SEC62, and RTN3, interacted with WDR45 [24,31,32]. However, none of these receptors showed clear interaction (Figure S5j-l). Furthermore, we isolated ER membrane from cultured mouse neurons and found by mass spectrometry that RTN3 (RETREG1 and SEC62 were not detected) was increased in KO neurons (Figure S7b). This can be explained by the premise that mutation of WDR45 results in low autophagic activity. Accumulation of misfolded proteins in neurons induces ER stress, which can be processed in wild type neurons. ER autophagy maybe achieved through elevated expression of ER-specific receptors to provide a destruction signal [24,31]. Neurons with WDR45 mutation exaggerates ER stress over time, due to a compromised ability to send damaged ER and protein aggregates for autophagic degradation, even though selective ER autophagy signals are intact. Our parallel study of mitochondrial also shows a compromised mitophagy in WDR45-deficient cells, further support this notion that selectivity of organelle autophagy maybe achieved through selective stress of the organelle.
ER stress has emerged as a common cellular pathway underlying neurodegeneration. As the results of mishandled ER stress, protein aggregates in various brain regions have been found in a number of neurodegenerative disorders, including Alzheimer disease, Parkinson disease, frontotemporal dementia, and amyotrophic lateral sclerosis [26]. Through an unbiased quantitative proteomic analysis of mouse brain, we uncovered accumulation of large number of ER proteins in KO mouse. Our functional analysis revealed abnormal UPR pathway that is presumably upregulated to handle increased ER stress. Thus, our study adds another neurodegenerative disorder whose mechanistic onset is at least partly due to increased ER stress and sustained activation of UPR pathway.
Intriguingly, either inhibition of MTOR signaling or inhibition of ER stress is able to alleviate apoptosis in WDR45-deficient cells and neurons. Although inhibition of MTOR could have a wide range of effects on catabolic processes, our data did show an accompanying increase of LC3-II:LC3-I ratio, a maker of increased autophagy. TUDCA is known to exert several effects that lead to neuroprotection, including inhibition of mitochondrial pathway of cell death, reducing oxidative stress, and reducing ER stress [33]; it has been applied in animal models of Alzheimer disease, Parkinson disease, and Huntington disease and show at least some protective effects against neuronal death [34–36]. It is thus not entirely surprising that TUDCA inhibit apoptosis in WDR45-deficient cells to the extent similar to MTOR inhibition. In neurons, however, only MTOR inhibition significantly reduced apoptosis, further highlight the importance of autophagy in maintaining protein and organelle homeostasis in neurons. It is worth noticing that neither inhibition of MTOR nor inhibition of ER stress rescued the ER expansion phenotype in WDR45-deficient cells, at least in the experimental setting similar to rescue of apoptosis. We postulate that the level of ER autophagy induced by rapamycin or TUDCA may not be sufficient to manage the complex ER network. Alternatively, WDR45 may be involved in ER quality control through other pathways. Nevertheless, our study suggests a unified principle underlying neurodegeneration regardless of a variety of etiological origins, and highlight potential cellular pathways that could be modulated to reduce neurodegeneration in BPAN patients.
Materials and methods
Generation of wdr45 knockout mouse
To generate a wdr45-null allele in mice, we targeted exon 6 of the Wdr45 gene which covers the mutated sites found in SENDA patients (Figure 1a) [13]. We used the CRISPAR-Cas9 method as described previously [37]. Briefly, super ovulated female C57BL/6J mice were mated to male C57BL/6J mice, and the embryos were collected from oviducts. Cas9 mRNA (100 ng/μl) and sgRNA (50 ng/μl) (target sequence: 5ʹ-TGACACTCGGGACAACCCCA-3ʹ followed by the PAM sequence AGG) were co-injected into the cytoplasm of one-cell stage embryos. After injection and overnight culture in KSOM medium (Millipore, MR-106-D), the microinjected zygotes were transplanted into pseudopregnant mice. One week after birth, the genomic DNA from the toes or tails of the newborn F0 mice was extracted for sequencing using primers flanking the target site. When male F0 mice grew into 7 weeks and female mice grew into 4 weeks of age, they were mated with wild-type mice. Mice were housed in standard cages in a specific pathogen-free facility on a 12-h light-dark cycle. All mice used for experimental analysis were homozygous.
Behavior
All behavioral experiments were performed on mice at 6 months of age. The gender of the mice used for behavioral tests were indicated in the figure legends.
Motor coordination
The motor coordination was measured using the standard rotarod test. After trained on the rolling rod at 5 rpm, 5–15 rpm and 8–25 rpm for 5 min for three consecutive days, mice were tested for their ability to remain on the rotarod at a speed of 8–40 rpm. The time when the mice fell from the rod was recorded with a maximum of 5 min.
Morris water maze
A round tub was filled with opaque water (20°C) to a depth of 25 cm. A platform was located in the center of one quadrant. In the learning test, each mouse was placed in the maze starting from one of four predetermined spots and was given 60 s to find the fixed platform. The time taken to reach the platform (escaping latency) was recorded by a video tracking system. Each mouse received 4 trials per day for 4 consecutive days. On the 5th day, the probe test was performed without the platform. The mouse was allowed to search for 60 s and the percentage of time spent in the target quadrant was recorded to monitor spatial memory ability.
Eight-arm maze
The mouse was placed into the center of a symmetrical 8-arm maze individually and allowed to freely enter the arms during a 10-min session. The series of arm entries was recorded visually. The percentage of alternation was calculated as the ratio of correct alternations (successive entry into the 8 arms on overlapping triplet sets) to total alternations.
Fear conditioning
The fear conditioning test was performed using the Startle and Fear combined system (Panlab, Harvard Apparatus). For training, after 3 min acclimation, mice were placed in a chamber and an 80 dB tone was delivered for 30 s as a conditioned stimulus. During the last 2 s, a foot shock of 0.4 mA was delivered through a shock generator. Animal movement was recorded through a high-sensitivity weight transducer system by PACKWIN software. On the second day, for the contextual test, mice were placed in the chamber and the freezing response was measured for 3 min without the conditioned stimulus. For the cued test, the freezing response was measured with the conditioned stimulus.
Pilocarpine induced seizure
Pilocarpine (Selleck, S4231) dissolved in PBS (Beyotime, ST476) was administered intraperitoneally (i.p.) at 30 mg/kg 30 min after scopolamine hydrobromide (Selleck, S2508; i.p., 1 mg/kg) administration. Behavioral responses were recorded using a video camera and scored using the following scale: 0, no abnormality; 1, exploring, sniffing, and grooming ceased, becoming motionless; 2, head-nodding, facial and forelimb clonus; 3, myoclonic jerks of the head and neck, with brief twitching movements, or repetitive movements with head-bobbing or tail rigidity; 4, forelimb or forelimb and hindlimb clonus, reciprocal forepaw padding, hindlimb abduction, continuous rearing, and falling, Straub tail response; 5, tonic convulsions; 6, death.
Three-chamber social approach test
The method was described previously [38]. The mice were pseudo-randomly selected from the home cage and placed in the center of a three-chambered apparatus to habituate (each compartment was 20 × 30 × 30 cm). After 5 min, the subject mouse was confined to the central chamber using dividers and a conspecific mouse of the same-sex was placed in either the left or right chamber within a wire framed steel cage. The dividers were removed and the subject mouse was allowed to move freely between the three chambers for 5 min. Next the subject mouse was again confined to the central chamber and a second novel mouse (matched for age and sex) was placed in the remaining wired framed steel cage. The dividers were removed and the subject mouse was able to move freely between the three chambers for a further 5 min. The cumulative duration to enter each zone was measured as well as the amount of time spent in close proximity to the conspecific mouse.
Slice electrophysiology
Recordings were made on hippocampal slices from WT and KO mice as described previously [39]. Briefly, after decapitation, the mouse brain was quickly removed and placed in oxygenated (95% O2:5% CO2) ice-cold artificial cerebrospinal fluid (aCSF) containing (in mM): 125 NaCl (Sigma-Aldrich, V900058), 2.5 KCl (Sigma-Aldrich, V900068), 12.5 D-glucose (Sigma-Aldrich, 47829), 1 MgCl2 (Sigma-Aldrich, M1028), 2 CaCl2 (Sigma-Aldrich, 499609), 1.25 NaH2PO4 (Sigma-Aldrich, V900060) and 25 NaHCO3 (Sigma-Aldrich, V900182) (pH 7.35–7.45). Coronal hippocampal slices (300 mm thick) were obtained with a vibratome (Leica VT 1000S) and incubated at 30 ± 1°C in oxygenated aCSF for at least 1 h before transferred to a recording chamber placed on the stage of an Olympus microscope (BX51WI). The slices were continuously perfused with oxygenated aCSF at room temperature during all electrophysiological studies. Hippocampal CA1 neurons were visualized using an infrared differential interference contrast microscope. Synaptic responses were recorded with an Axon 200B amplifier (Molecular Devices), and the stimulations were delivered with a bipolar tungsten-stimulating electrode placed over Schaffer collateral fibers between 250 and 300 μm from the recorded cells in the CA1 region. Pyramidal neurons were identified based on their ability to exhibit spike frequency adaptation in response to prolonged depolarizing current injection. AMPA receptor-mediated EPSCs were induced by repetitive stimulations at 0.03 Hz, with the neuron being voltage-clamped at – 70mV except where indicated otherwise. The recording pipettes (4–7 MΩ) were filled with a solution containing (in mM): 145 potassium gluconate, 5 NaCl, 10 HEPES, 2 MgATP (Sigma-Aldrich, A9187), 0.1 Na3GTP (Sigma-Aldrich, G8877), 0.2 EGTA and 1 MgCl2 (280–300 mOsm, pH 7.2 with KOH). Paired-pulse facilitation was induced by delivering two consecutive pulses with a 25-, 50-, 75-, 100-, or 200 ms inter pulse interval and calculated as the ratios of the second response peak values over the first response peak values. The miniature EPSCs of CA1 pyramidal neurons in the hippocampus were obtained at −70 mV in the presence of tetrodotoxin (0.5 mM; Sigma-Aldrich, A3109) and picrotoxin (100 mM; Sigma-Aldrich, R284556) without stimulation and analyzed using MiniAnalysis (Synaptosoft). In all of the whole-cell recordings, cell series resistance was monitored throughout experiments and the experiment was excluded from analysis if the resistance changed by more than 20%.
Chemical reagents, antibodies and plasmids
Rapamyicin (Sigma-Aldrich, V900930), tauroursodeoxycholic acid (TUDCA; Shang Hai Yuan Ye, B20921), tunicamycin (Cell Signaling Technology, 12,819), BafA (Sigma-Aldrich, B1793), and MG132 (Selleck, S2619) were used at final concentrations of 100 nM, 100 μM, 2-4 μg/ml, 50 nM and 20 μM, respectively.
The following antibodies were used in the western blot experiments: rabbit anti-HA (1:1000; Cell Signaling Technology, 12698), mouse anti-ACTB/β-actin (1:5000; YEASEN, 30101-ES10), mouse anti-Flag (1:5000, Sigma-Aldrich, 1804), rabbit anti-CALR (calreticulin) (1:1000; Abcam, ab92516), rabbit anti-SQSTM1 (1:1000; Sigma-Aldrich, B0062), rabbit anti-GAPDH (1:5000; Abways, AB0037), rabbit anti-cleaved CASP3/caspase-3 (1:1000; Cell Signaling Technology, 9664), rabbit anti-TOMM20 (1:500 for IF; Santa Cruz Biotechnology, sc11415), rabbit anti-LC3 (1:1000; Sigma-Aldrich, 2775S), and rabbit anti-HSPA5 (1:1000; Abways, CY5166). Rabbit anti-EIF2A, rabbit anti-ERN1, rabbit anti-phospho-ERN1 (S724), rabbit anti-MAPK/JNK, rabbit anti-phospho-MAPK/JNK (T183/Y185), rabbit anti-phospho-EIF2A (S52) are all kind gifts from Dr. Liu Yong (Wuhan University, CHN).
All plasmids were verified by DNA sequencing. The plasmids flag-WDR45, flag-GFP, HA-SEC22B, HA-SEC61B, HA-BCAP31, HA-CDIPT were cloned from pcDNA3.1(+)-flag or HA-CMV vector (a gift from Dr. Weng Jiemin [East China Normal University, CHN]). All primer sequences are listed in Table 1.
Table 1.
Primer sequences for all the clones and real-time PCR experiments used in the study.
| Gene | Primer sequence forward 5ʹ-3’ | Primer sequence reverse 5ʹ-3’ |
|---|---|---|
| For Gene Clone | ||
| Flag-WDR45-human | CGCGGATCCACCATGACTCAACAGCCACTT | CCGGAATTCTTAAAAGTCATCATCATCACAGATG |
| HA-CDIPT-human | GTCCCCGACTACGCCGGATCCATGCCAGACGAAAATATCTT | TGATGGATATCTGCAGAATTCTCACTTCTTCTTGGCGC |
| HA- BCAP31-human | GTCCCCGACTACGCCGGATCCATGGGTGCCGAGGCGTC | TGATGGATATCTGCAGAATTCTTACTCTTCCTTCTTGTCCATGG |
| HA-SEC22B-human | GTCCCCGACTACGCCGGATCCATGGTGTTGCTAACAATGATCG | TGATGGATATCTGCAGAATTCTCACAGCCACCAGAATCGG |
| HA-SEC61B-human | GTCCCCGACTACGCCGGATCCATGCCTGGTCCGACC | TGATA1:C30GGATATCTGCAGAATTCCTACGAACGAGTGTACTTGC |
| For qPCR | ||
| Rtn4-mouse | TCGGGCTCAGTGGATGAGA | ACAGTGTTACCTGGCTGCTC |
| Rtn1-mouse | CGCATCTACAAGTCCGTTCTAC | GCTCCTTCAGAGTGCTGTTCA |
| Reep5-mouse | GGTTCCTGCACGAGAAGAACT | GAGAGAGGCTCCATAACCGAA |
| Cdipt-mouse | TCCTGTTCGTGCCTAACCTTA | AGGAGTCCGCTGAGTAGATAGA |
| Praf2-mouse | GGCGATCCACAACGATGGT | GTGTGCAGCGGACGAATGTA |
| Bcap31-mouse | GCCACCTTCCTCTACGCAG | TGCCATAGGTCACTACCAACTC |
| Sec22b-mouse | AGGAGCCATGACTTTTCACTACA | AGGCCAACTTCTTAGGGAAGG |
| Synpo2-mouse | TGGAAGGCATAACGGACTCTC | GCCCCTCATGTGTAAGATGTTG |
| Syn1-mouse | AGCTCAACAAATCCCAGTCTCT | CGGATGGTCTCAGCTTTCAC |
| Fus-mouse | GCTTCAAACGACTATACCCAACA | GGCCATAACCACTGTAACTCTGT |
| Canx-mouse | CAACAGGGGAGGTTTATTTTGCT | TCCCACTTTCCATCATATTTGGC |
| Fabp7-mouse | GGACACAATGCACATTCAAGAAC | CCGAACCACAGACTTACAGTTT |
| Ewsr1-mouse | GTGGCTTTGGTGGAGGAAGA | CGGTGCTCGCCTTTATCCAT |
| Snrpd3-mouse | AGGGAAGCTCATTGAAGCAGA | TGTGCCACTCGACCATCTCT |
| Wdr45-mouse | CATCTGGAGCAAGATCCAAGGGAAG | GAAATGTAGGCTGGTCACACCT |
| Atg2b-mouse | AACTGCTGACGAATCCTCAGG | GGGGTTCCAGCTAGGTGAGA |
| Gapdh-mouse | CATGGCCTTCCGTGTTCCTA | CCTGCTTCACCACCTTCTTGAT |
| For Xbp1 detection | ||
| XBP1-human | TTACGAGAGAAAACTCATGGC | GGGTCCAAGTTGTCCAGAATG |
| RNA18S-human | CAGCCACCCGAGATTGAGCA | TAGTAGCGACGGGCGGTGTG |
| Xbp1-mouse | GAACCAGGAGTTAAGAACACG | AGGCAACAGTGTCAGAGTCC |
| Gapdh-mouse | GAGTTCTTCAATGGCAAGGA | CCAGTCAGATCAAATGTACCC |
ShRNA knockdown
Annealed oligonucleotides were cloned into pLKO.1puro (a gift from Dr. Xiong Zhiqi (Institute of Neuroscience, SIBS, CHN)) using AgeI and EcoRI cloning sites. The following pairs of oligonucleotides were used to target WDR45: #2: CCGGCAAGATCGTGATCGTGCTGAACTCGAGTTCAGCACGATCACGATCTTGTTTTTTG and AATTCAAAAAACAAGATCGTGATCGTGCTGAACTCGAGTTCAGCACGATCACGATCTTG, #3: CCGGCAAGAACGTCAACTCTGTCATCTCGAGATGACAGAGTTGACGTTCTTGTTTTTTG and AATTCAAAAAACAAGAACGTCAACTCTGTCATCTCGAGATGACAGAGTTGACGTTCTTG. HeLa cells stably expressing shRNA oligonucleotides were generated by lentiviral infection as described previously [40].
RNA extraction, reatl-time PCR
Total RNA was extracted using TRIzol (Gene solution, B140101) and cDNA was synthesized by Prime ScriptIIFirst-Strand cDNA synthesis Kit (Invitrogen, 11752050) according to the manufacturer’s instructions. Quantitative PCR was performed on a real-time PCR system using SYBR Premix (Promega, A6001). Primer sequences are listed in Table 1.
Cell culture and transient transfection
HEK293T and HeLa cells were maintained in DMEM (Invitrogen, 11965) supplemented with 10% FBS (GEMINI, 900–108) and 50 mg/ml penicillin/streptomycin (Gibco, 15140163) at 37ºC in 5% CO2. Cells grown at low confluence in 6 cm, 12- or 6-well tissue culture plates were transfected with 4 μg, 1 μg or 2 μg of plasmid DNA, respectively, using the polyetherimide reagent (Polysciences, 690049). Experiments were performed 24–36 h after transfection.
Primary cortical neurons were harvested from embryonic day 18 mouse pups. Cerebral cortices were collected and dissociated by incubation in Papain (Sigma-Aldrich, 1495005) for 30 min at 37ºC. After termination with FBS, cells were centrifuged at 210 x g for 2 min, and plated with MEM media (Gibco, 41090093) on dishes pre-coated with poly-D-lysine (100 mg/ml; Sigma-Aldrich, P1524). After incubation at 37ºC for at least 4 h, the plating medium was changed to Neurobasal containing B27 (Invitrogen, 17504044), penicillin/streptomycin (Gibco, 15140163) and glutamine (Invitrogen, 35050061). Half the volume of culture medium was changed every 3 days.
Western blot
Cells were lysed with RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% NP40 [Sangon Biotech, A510110]) supplemented with protease inhibitor (Thermo Fisher Scientific, 88,666), sonicated and incubated on ice for 30 min. Homogenates were centrifuged at 15,000 x g for 20 min at 4ºC. Supernatant were collected and protein concentrations were determined by BCA (Thermo Fisher Scientific, 23225). Equal amounts (20 to 50 μg) of proteins were subjected to SDS-PAGE, transferred to the nitrocellulose membrane and then blocked with 5% nonfat milk for 1 h at room temperature. The membrane was incubated with primary antibodies at 4ºC overnight. After washing with TBST (0.1% Tween [Sangon Biotech, A100777] in TBS [Sangon Biotech, B548105]) 3 times, the membrane was incubated with secondary antibodies for 1 h at room temperature.
Immunofluorescence (IF) microscopy
HeLa cells plated on glass coverslips were treated with indicated drugs. Cells were then washed twice in PBS and fixed at room temperature for 30 min with 4% paraformaldehyde (PFA; Sangon Biotech, E672002) in PBS, permeabilized with 1% Triton X-100 (Sangon Biotech, A110694) on ice for 10 min, blocked with 5% BSA (MP Biomedicals, SKU-0215240110) in a 37°C incubator for 60 min and incubated with primary antibodies overnight. The coverslips were washed 3 times with PBST, followed by incubation with a secondary antibody (Life Technology, Alexa Fluor 405-blue, A-31553; 546-red, A-11003; 488-green, A-11001). Confocal pictures were acquired using a Leica TCS SP5 microscope with a 63×/1.4 N.A. objective.
Immunohistochemistry
Mice were trans-cardially perfused, and cerebral tissues were dissected and fixed in 4% PFA for 1 day, dehydrated by 20% and 30% sucrose (Sigma-Aldrich, V900116) in PBS buffer, sectioned at 15 μm using a Leica CM1860.
For immunohistochemistry, brain slices were incubated with 0.01 M sodium citrate, pH 6.02, at 95°C for 10 min, and peroxidase was inactivated with 3% H2O2. Slices were blocked with goat serum and incubated with indicated primary antibodies at room temperature for 2 h. After washing, sections were incubated with biotin-IgG followed by Streptomycin-HRP for 15 min at room temperature. Finally, sections were counterstained with DAB (NEOBIOSCIENCE, ENS004), and DNA was counter stained with hematoxylin. Images were acquired using an Olympus-KCC-REM microscope.
Transmission electron microscopy
Mice were anesthetized by intraperitoneal injection of a mixture of 4% chloral hydrate (0.01 ml/g; Sigma-Aldrich, 135046), and then perfused with 0.1 M phosphate buffer at a flow rate of 6.5 r/min for 5 min, followed by perfusion with 4% PFA solution in 0.1 M phosphate buffer for 15 min. The whole brain was dissected with the BRAIN BLOCKER in Stereotaxic Coordinates and the hippocampus was cut into 1 mm3 blocks(2 blocks,respectively). These slabs were stored in 25% glutaraldehyde (Sigma-Aldrich, G5582) at 4°C for up to 3 months. The slabs were washed in 0.1 M PBS 3 times for 15 min, then treated with 1% OsO4 (Sigma-Aldrich, O5500) for 2 h at 4°C. The slabs were dehydrated with increasing concentrations of acetone and flat embedded in araldite. Ultrathin sections were cut on a microtome, placed onto copper grids, stained with uranyl acetate and lead citrate and examined on a H-7700 electron microscope.
Analysis of apoptosis by FACS
Apoptosis assays on wild type and sh-WDR45 cell lines were performed using ANXA5-FITC apoptosis detection kit (Beyotime, C1062) according to the manufacturer’s protocol. Numbers of ANXA5+ PI− and ANXA5+ PI+ cells were detected using BD FACSCalibur and the data were processed with FlowJo program.
TUNEL assay
Frozen brain sections from WT and KO mice (16 months) were stained with the TUNEL kit (Yeasen, 403006) according to manufacturer’s instructions. Images were acquired using an Olympus-KCC-REM microscope.
Quantitative proteomics
Mouse brains at 6–8 months of age were dissected into PFC (prefrontal cortex), HIP (hippocampus) and MIB (middle brain). Each sample was lysed in urea buffer (7 M urea, 2 M thiourea, 50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 7.5, with protease [Sigma-Aldrich, P8340] and phosphatase inhibitors [Sigma-Aldrich, 4906845001]). For TMT labeling, 50 μg protein lysate per sample were reduced with 10 mM dithiothreitol (DTT; Sigma-Aldrich, 43217) at 55°C for 30 min, alkylated with 50 mM iodoacetamide (Sigma-Aldrich, I1149) in the dark for 30 min, precipitated with six volumes of pre-chilled acetone overnight, and digested with trypsin (1:100; Promega, V5113) overnight at 37°C in 100 mM tetraethylammonium bromide (TEAB; Sigma-Aldrich, 140023). Peptides from the ten conditions were labeled with 0.8 mg of 10-Plex TMT (Thermo Scientific, 90110) respectively, and combined afterward following the manufacturer’s instructions.
A total of 500 μg tryptic peptides in each brain region were separated into 60 fractions using high-pH RPLC (Waters XBridge C18 column 5 μm, 250 mm × 4.6 mm; mobile phase A [5 mM NH4COOH, 2% acetonitrile, pH 10.0] and B [5 mM NH4COOH, 98% acetonitrile, pH 10.0]) at a flow rate of 1 ml/min using the following linear gradient: 0–6% phase B for 6 min, 6%-28.5% phase B for 30 min, 28.5%-34% phase B for 5 min, 34%-60% phase B for 10 min, 60% phase B for 9 min. The eluate was collected 1 minute into vials (1 ml/tube). Finally, 60 fractions were combined into 12 components in zigzag fashion. Peptides were concentrated in a speed Vac, desalted in C18 STAGE-tips and primed for LC−MS analysis.
Peptides were analyzed on an EASY-nLC1000 LC (Thermo Scientific, USA) coupled on line with the Q-Exactive Orbitrap mass spectrometer (Thermo Scientific, USA). Peptides were separated on an in-house packed C18-column (15 cm, 75 μm I.D., 3 μm p.s.) with a 120-minutes gradient from 12% to 32% buffer B (98% ACN, 0.1% formic acid) at a flow rate of 250 nl/min. One full scan MS from 400 to 1600 m/z followed by 12 MS/MS scan were continuously acquired. The resolution for MS was set to 70000 and for data-dependent MS/MS was set to 35000. The nano-electrospray source conditions were: spray voltage, 1.8 KV; no sheath and auxiliary gas flow; heated capillary temperature, 275°C. For higher-energy C-trap dissociation (HCD), the isolation window was set to 2 m/z and the normalized collision energy of 27 was applied.
Analysis of mass spectrometry data
The MaxQuant (version 1.4.1.2) software was used to analyze the MS/MS raw data. For the database searching parameters, the precursor mass tolerance was set to 15 ppm. Trypsin/P was set as the protease, accounting for in-source fragmentation of lysine or arginine residues followed by proline. Two missed cleavages were allowed. All data were searching against with the UniPort Mouse database (sequences) including oxidation (+15.9949 Da) of methionine, and TMT 10-plex (+229.1629 Da) on lysine and carbamidomethylation (+57.0215 Da) of cysteine as static modifications.
Parallel reaction monitoring
Protein candidates with more than 2-fold change and adjust P-value of less than 0.05 were selected for further validation by targeted liquid chromatography-parallel reaction monitoring (LC-PRM) MS. Peptides were separated on C18-column with a 75-minute gradient from 12% to 32% buffer B (98% ACN, 0.1% formic acid) at a flow rate of 250 nl/min. PRM MS2 spectra were collected at a resolution of 17500 with AGC target value of 2e4. Raw data were analyzed by Skyline using a data-independent acquisition method (version 3.6.1), with a q value of 0.1 [41].
Bioinformatics analysis
Principal component analysis (PCA) was conducted to demonstrate overall differences of samples with the function princomp in R. Functional annotation was performed with the David bioinformatics resource v6.7 (https://david.ncifcrf.gov/). The functional annotation term categories of biological processes, cellular components and molecular functions are analyzed.
Statistics
Data are presented as standard error of the mean (SEM) of multiple replicates. All data presented are representative of at least three independent experiments. P-values were generated by two-tailed, unpaired t-test assuming equal variance using Prism Graphpad Software (San Diego, CA). The value 0.05 (*), 0.01 (**) and 0.001 (***) was assumed as the level of significance for the statistic tests.
Study approval
All experiments involving animal treatment and care were performed following the Institutional Animal Care and Use Committee protocols from the East China Normal University (ECNU).
Funding Statement
This work was supported by the East China Normal University National 985 Project Fund (CN) [11300-120215-10321].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary material for this article can be accessed here.
References
- [1].Choi AMK, Ryter SW, Levine B.. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. [DOI] [PubMed] [Google Scholar]
- [2].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006. 04/19/online;441:885. [DOI] [PubMed] [Google Scholar]
- [3].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006. 04/19/online;441:880. [DOI] [PubMed] [Google Scholar]
- [4].Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93(5):1015–1034. [DOI] [PubMed] [Google Scholar]
- [5].Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases [Review Article]. Nat Rev Neurosci. 2015. 05/20/online;16:345. [DOI] [PubMed] [Google Scholar]
- [6].Yamamoto A, Yue Z. Autophagy and its normal and pathogenic states in the brain. Annu Rev Neurosci. 2014. July 08;37(1):55–78. [DOI] [PubMed] [Google Scholar]
- [7].Haack Tobias B, Hogarth P, Kruer Michael C, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91(6):1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Stige KE, Gjerde IO, Houge G, et al. Beta-propeller protein-associated neurodegeneration: a case report and review of the literature. Clin Case Rep. 2018;6(2):353–362. PubMed PMID: 29445477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hayflick SJ, Kruer MC, Gregory A, et al. Beta-propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain. 2013. 05/17 12/03/received 02/22/revised 02/24/accepted;136(6):1708–1717. PubMed PMID: PMC3673459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Araújo R, Garabal A, Baptista M, et al. Novel WDR45 mutation causing beta-propeller protein associated neurodegeneration (BPAN) in two monozygotic twins. J Neurol. 2017. May 01;264(5):1020–1022. [DOI] [PubMed] [Google Scholar]
- [11].Gregory A, Hayflick SJ. Genetics of neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Rep. 2011. June 01;11(3):254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kasai-Yoshida E, Kumada S, Yagishita A, et al. First video report of static encephalopathy of childhood with neurodegeneration in adulthood. Mov Disord. 2013;28(3):397–399. [DOI] [PubMed] [Google Scholar]
- [13].Saitsu H, Nishimura T, Muramatsu K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013. 02/24/online;45:445. [DOI] [PubMed] [Google Scholar]
- [14].Wynn DP, Pulst SM. A novel WDR45 mutation in a patient with β-propeller protein-associated neurodegeneration. Neurol Genet. 2016. PubMed PMID: PMC5141522. DOI: 10.1212/NXG.0000000000000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bakula D, Müller AJ, Zuleger T, et al. WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy [Article]. Nat Commun. 2017. 05/31/online;8:15637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dall’Armi C, Devereaux KA, Di Paolo G. The role of lipids in the control of autophagy. Curr Biol. 2013;23(1):R33–R45. PubMed PMID: PMC3587843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tooze SA, Yoshimori T. The origin of the autophagosomal membrane [Perspective]. Nat Cell Biol. 2010. 09/01/online;12:831. [DOI] [PubMed] [Google Scholar]
- [18].Lu Q, Yang P, Huang X, et al. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell. 2011. August 16;21(2):343–357. [DOI] [PubMed] [Google Scholar]
- [19].Zhao YG, Sun L, Miao G, et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy. 2015. 05/22 10/31/received 04/16/revised 04/27/accepted;11(6):881–890. PubMed PMID: PMC4502681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Webster HD. TRANSIENT, FOCAL ACCUMULATION OF AXONAL MITOCHONDRIA DURING THE EARLY STAGES OF WALLERIAN DEGENERATION. J Cell Biol. 1962. 08/14/received;12(2):361–383. PubMed PMID: PMC2106025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008. 12/18/online;4:44. [DOI] [PubMed] [Google Scholar]
- [22].Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009. 11/25 09/10/received 10/24/revised 11/03/accepted;37(1):1–13. PubMed PMID: PMC2615629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hosp F, Vossfeldt H, Heinig M, et al. Quantitative interaction proteomics of neurodegenerative disease proteins. Cell Rep. 2015. May 19;11(7):1134–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Khaminets A, Heinrich T, Mari M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015. 06/03/online;522:354. [DOI] [PubMed] [Google Scholar]
- [25].Frakes AE, Dillin A. The UPRER: sensor and coordinator of organismal homeostasis. Mol Cell. 2017. June 15;66(6):761–771. [DOI] [PubMed] [Google Scholar]
- [26].Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration [Review Article]. Nat Rev Neurol. 2017. 07/21/online;13:477. [DOI] [PubMed] [Google Scholar]
- [27].Kim YC, Guan K-L. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015. January 02;125(1):25–32. PubMed PMID: PMC4382265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bové J, Martínez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights [Review Article]. Nat Rev Neurosci. 2011. 07/20/online;12:437. [DOI] [PubMed] [Google Scholar]
- [29].Keestra-Gounder AM, Byndloss MX, Seyffert N, et al. NOD1/NOD2 signaling links ER stress with inflammation. Nature. 2016. March 23;532(7599):394–397. PubMed PMID: PMC4869892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system. Nature. 2010. June 20;466(7302):68–76. PubMed PMID: PMC2901998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fumagalli F, Noack J, Bergmann TJ, et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery [cited 1173-+-1173-+ p.]. Nat Cell Biol. 2016;18:1173–1184. [DOI] [PubMed] [Google Scholar]
- [32].Grumati P, Morozzi G, Hölper S, et al. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife. 2017;6:e25555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Özcan U, Yilmaz E, Özcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes [10.1126/science.1128294]. Science. 2006;313(5790):1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Castro-Caldas M, Carvalho AN, Rodrigues E, et al. Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson’s Disease. Mol Neurobiol. 2012. October 01;46(2):475–486. [DOI] [PubMed] [Google Scholar]
- [35].Keene CD, Rodrigues CMP, Eich T, et al. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease [10.1073/pnas.162362299]. Proc Nat Acad Sci. 2002;99(16):10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nunes AF, Amaral JD, Lo AC, et al. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol Neurobiol. 2012. June 01;45(3):440–454. [DOI] [PubMed] [Google Scholar]
- [37].Shao Y, Guan Y, Wang L, et al. CRISPR/Cas-mediated genome editing in the rat via direct injection of one-cell embryos. Nat Protoc. 2014. 09/25/online;9:2493. [DOI] [PubMed] [Google Scholar]
- [38].Langguth M, Fassin M, Alexander S, et al. No effect of prenatal vitamin D deficiency on autism-relevant behaviours in multiple inbred strains of mice. Behav Brain Res. 2018. August 01;348:42–52. [DOI] [PubMed] [Google Scholar]
- [39].Zhou Z, Liu A, Xia S, et al. The C-terminal tails of endogenous GluA1 and GluA2 differentially contribute to hippocampal synaptic plasticity and learning. Nat Neurosci. 2018. January 01;21(1):50–62. [DOI] [PubMed] [Google Scholar]
- [40].Popovic D, Akutsu M, Novak I, et al. Rab GTPase-activating proteins in autophagy: regulation of endocytic and autophagy pathways by direct binding to human ATG8 modifiers. Mol Cell Biol. 2012. 12/14/received 01/14/rev-request 02/11/accepted;32(9):1733–1744. PubMed PMID: PMC3347240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].MacLean B, Tomazela DM, Shulman N, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010. 02/09 12/18/received 01/11/revised 02/03/accepted;26(7):966–968. PubMed PMID: PMC2844992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.