

ABSTRACT
The discovery of a large number of long noncoding RNAs (lncRNAs), and the finding that they may play key roles in different biological processes, have started to provide a new perspective in the understanding of gene regulation. It has been shown that the testes express the highest amount of lncRNAs among different vertebrate tissues. However, although some studies have addressed the characterization of lncRNAs along spermatogenesis, an exhaustive analysis of the differential expression of lncRNAs at its different stages is still lacking. Here, we present the results for lncRNA transcriptome profiling along mouse spermatogenesis, employing highly pure flow sorted spermatogenic stage-specific cell populations, strand-specific RNAseq, and a combination of up-to-date bioinformatic pipelines for analysis. We found that the vast majority of testicular lncRNA genes are expressed at post-meiotic stages (i.e. spermiogenesis), which are characterized by extensive post-transcriptional regulation. LncRNAs at different spermatogenic stages shared common traits in terms of transcript length, exon number, and biotypes. Most lncRNAs were lincRNAs, followed by a high representation of antisense (AS) lncRNAs. Co-expression analyses showed a high correlation along the different spermatogenic stage transitions between the expression patterns of AS lncRNAs and their overlapping protein-coding genes, raising possible clues about lncRNA-related regulatory mechanisms. Interestingly, we observed the co-localization of an AS lncRNA and its host sense mRNA in the chromatoid body, a round spermatids-specific organelle that has been proposed as a reservoir of RNA-related regulatory machinery. An additional, intriguing observation is the almost complete lack of detectable expression for Y-linked testicular lncRNAs, despite that a high number of lncRNA genes are annotated for this chromosome.
KEYWORDS: LncRNAs, spermatogenesis, testis, RNAseq, transcriptome
Introduction
Mammalian testes are very complex organs, composed of over 30 different cell types. As a consequence of this complexity, gene expression studies have required the development of methods for spermatogenic-specific cell types isolation such as unit gravity sedimentation (Staput) and centrifugal elutriation, that allow an enrichment in certain cell types [1,2]. Later on, FACS (Fluorescence-Activated Cell Sorting) appeared as the dominant technology as it enables the obtainment of highly pure male germline-specific cell populations [3,4].
Spermatogenesis is an intricate differentiation process that can be divided into three main consecutive yet overlapping phases (Supplementary Fig. S1A): the mitotic proliferation of spermatogonia (the precursors of meiotic cells), meiotic divisions, and spermiogenesis. Meiosis is characterized by significant unique events such as the alignment and pairing of homologous chromosomes that take place during early meiotic prophase I (i.e. leptotene and zygotene stages), and recombination (crossing-over) at the following prophase stage (i.e pachytene) [5]. On the other hand, during spermiogenesis round spermatids (the cells that result from the meiotic divisions) go through several differentiation stages until becoming sperm, and this process is accompanied by dramatic changes: acquisition of a flagellum, loss of most cytoplasm, nuclear elongation, acrosome formation, reorganization of mitochondria, and chromatin compaction. In particular, chromatin compaction is achieved through the replacement of most histones first by transition proteins and then by protamines, and this replacement results in transcriptional silencing at the later spermiogenesis stages [6].
Due to transcriptional silencing, spermatogenic cells at earlier stages have developed a panoply of mechanisms for post-transcriptional regulation and translational delay, as a strategy to regulate the time of synthesis for proteins that are required later on by elongated spermatids and sperm [6–8]. The involved regulatory mechanisms may be as diverse as mRNA sequestration as free ribonucleoprotein particles, binding of repressors to UTRs of testis-specific transcripts, regulation of poly(A) tails length, and others [6]. Another such strategies may be the retention of RNAs in the chromatoid body, a membrane-less spermatid-specific perinuclear granule that contains lncRNAs, piRNAs, mRNAs, and proteins for mRNA processing, and whose role is probably linked to the control of RNA-related processes [9]. In addition, translational control mechanisms may involve noncoding RNAs. In fact, a distinctive feature of testis is the extensive transcription of noncoding RNAs, among which long noncoding RNAs (lncRNAs) stand out [10].
LncRNAs have been defined as RNAs longer than 200 nucleotides (as opposed to small noncoding RNAs) that are not translated into proteins [11,12]. However, as the definition is arbitrary, some lncRNAs may be smaller than 200 nt [13], and, moreover, a minority of them might actually encode short functional peptides [14]. In general, lncRNAs share some traits among vertebrates, such as the fact that they are mostly transcribed by RNA polymerase II, are often alternatively spliced, and many of them are polyadenylated and capped [15–18]. Besides, some of their characteristics comprise low abundance, low sequence conservation, and highly tissue- and developmental-specific expression patterns [17–21]. According to their location, they have been classified as overlapping protein-coding genes in sense or antisense direction, corresponding to bidirectional promoter transcripts, intronic, intergenic (lincRNAs), associated with enhancers or repetitive regions, or 3ʹ- overlapping [11,17,22]. Some lncRNA genes may host miRNAs [22]. Besides, some long, unspliced macro lncRNAs have been described [23].
The high representation of lncRNAs in mammalian genomes, together with their restricted expression patterns, suggests that at least part of them should be functional [24,25]. In fact, roles have been elucidated for different lncRNAs in relation to transcriptional activation or repression; chromatin remodelling; regulation of mRNAs splicing, editing and degradation; modulation of the activity or abundance of proteins or RNAs; regulation of miRNA activity; and other functions [11,22,26,27]. So far, lncRNAs have been implicated in processes as diverse as the maintenance of pluripotency, embryonic development and organ differentiation, imprinting, organization of cell architecture, and apoptosis, among others. Moreover, several lncRNAs have been related to the development of different diseases, including cancer [20,26–28].
Different reports coincide with the fact that the testes express the largest numbers of lncRNAs among different tissues [10,17,20,21,29]. A number of studies have addressed the characterization of testicular lncRNAs in different mammalian species, disclosing that they tend to share the general traits described for lncRNAs [30–32], and revealing that at least some testicular lncRNAs are functional during spermatogenesis [33,34]. A handful of papers addressed differential lncRNAs expression along mouse spermatogenesis, using microarrays [35,36] or RNAseq [10,37–39]. These studies employed whole testes of animals of increasing ages [35,37], or spermatogenic stage-specific cell populations isolated by Staput or elutriation [10,36,38,39] that only allow the obtainment of enriched (but not pure) cell populations [4]. In some of these studies, lncRNAs were not the main research objective but they arose as a by-product, and therefore their characterization was not exhaustive [37]. Additionally, concerning RNAseq studies, data analysis has recently incorporated more accurate tools for the study of lncRNAs [40] than the ones used in most previous papers. An interesting recent paper reported single-cell RNA-sequencing of 20 different spermatogenic cell subtypes [41]. While this report represents an important advance as it included some previously unpurified cell types, this was achieved by manipulating the spermatogenic process through combining transgenic labelling with synchronization of the cycle of the seminiferous epithelium by means of WIN 18,446/retinoic acid. Besides, lncRNAs were not extensively characterized.
In the past, we have reported the collection of highly pure stage-specific spermatogenic cell populations from mouse by flow cytometry sorting [42,43]. We used these cell populations for transcriptome analyses, establishing stage-specific gene expression signatures along spermatogenesis with an unprecedented reliability in the profiling [4]. However, the employed RNAseq libraries were not strand-specific, thus limiting their usefulness to characterize the whole repertoire of lncRNAs as a high proportion of them are antisense to coding transcripts and therefore require strand-specific libraries for their reliable detection [44].
In the present work, we aimed to characterize the lncRNAs expressed in highly pure stage-specific testicular cell populations obtained with our tailored flow cytometry purification protocols, using deep sequencing of strand-specific RNAseq libraries and current bioinformatics tools. We hereby present a comprehensive analysis of the differential expression of lncRNAs at distinct stages of mouse spermatogenesis. The obtained data provide a highly reliable information set available to the community for future studies in the field.
Results
To address the characterization of differential gene expression of lncRNAs throughout spermatogenesis, we performed transcriptome analysis of strand-specific RNAseq libraries generated from highly purified testicular cell populations by flow sorting (Supplementary Fig. S1B). The cell populations, representative of landmark stages along mouse spermatogenesis, were the same as in a previous report from our group, in which the coding transcriptome was disclosed [4]: 2C (a heterogeneous population with 2C DNA content, consisting of spermatogonia and testicular somatic cells); LZ (lepto-zygotene spermatocytes); PS (pachytene spermatocytes); and RS (round spermatids).
Sequence analysis of the different libraries yielded on average 150 million reads per sample. Between 84.21% and 94.15% of the reads conserved after trimming aligned with the reference genome (Supplementary Table S1). Up to 0.22% reads mapped on ribosomal RNA and 3.28% on mitochondrial RNA, indicating that the depletion step was successful. Principal component analysis and correlation matrix showed high reproducibility between biological replicas (Supplementary Fig. S2), as well as with our previous RNAseq study (data not shown). Overall, the obtained data is a very deep set of reads with robust reproducibility, useful to characterize even lowly expressed lncRNAs.
Round spermatids contain the highest number of lncRNAs among different testicular cell populations
The obtained data was analysed in parallel with two pipelines, CLC Genomics Workbench and Hisat/StringTie/Ballgown. We only considered mappings onto previously annotated lncRNAs that arose from the intersection of both pipelines, had a variance across samples of more than 1, and presented more than 0.1 FPKM. Although this may turn out to be a strict criterion excluding a high number of reads, it yields very reliable results. Notably, 83.5% of the lncRNA genes detected as expressed with CLC, coincided with those detected with Hisat/StringTie/Ballgown (Fig. 1A). Our stringent approach detected the presence of transcripts corresponding to 878 annotated lncRNA genes in the full dataset that includes the four studied cell populations (See Fig. 1A, and Supplementary Table S2). This represents 10% of all annotated lncRNA genes in Ensembl database (Gm38.p6 release 93). Among them, 28.4% (249 lncRNAs) were common to all the analysed cell populations. Two hundred and eighty-four lncRNAs were present in the 2C cell population, 595 in LZ, 528 in PS, and 863 in RS (Fig. 1B).
Figure 1.
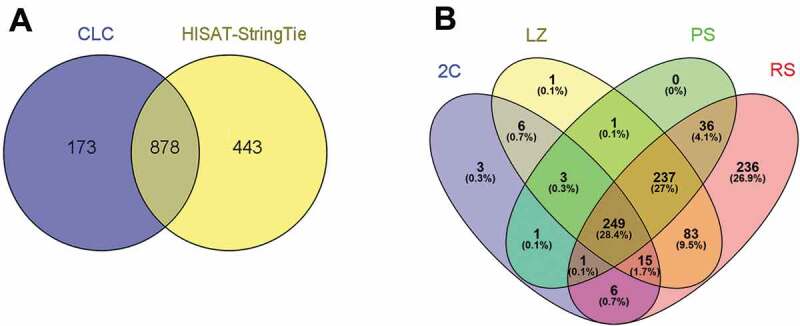
LncRNA genes expressed in the four testicular cell populations. (A) Venn diagram showing the annotated lncRNA genes detected as expressed in the four cell populations with two different pipelines, and that passed all the filters. Only lncRNAs whose variance across samples was more than 1 and FPKM ≥ 0.1 in the three biological replicas, were considered. (B). Venn diagram showing the lncRNA genes expressed in each of the cell populations (2C, LZ, PS, and RS). Separate and overlapping expression between samples is shown. The considered parameters were the same as in (A).
Concerning differentially expressed (DE) lncRNA genes, pairwise comparisons between cell populations in chronological order along the progression of the spermatogenic wave rendered 96 annotated DE lncRNA genes in LZ vs 2C population, 58 in PS vs LZ, and 411 in RS vs PS (Fig. 2, and Supplementary Table S2), considering a log2 fold-change ≥ │2│cut-off and an FDR p-value ≤ 0.05 as significant. Importantly, these numbers did not change when the expression cut-off was raised from 0.1 FPKM to 1 FPKM. Thus, the highest number of testicular lncRNA genes is expressed in spermiogenesis, and much more so with regards to the DE ones.
Figure 2.
Representation of the DE lncRNA genes between pairwise sample comparisons of the four testicular cell populations in chronological order along spermatogenesis. The following comparisons were performed: LZ vs 2C; PS vs LZ; RS vs PS (log2 fold-change ≥ │2│, FDR p-value correction ≤ 0.05). Only those genes whose variance across samples was more than 1 and FPKM ≥ 0.1, were considered. (A) Venn diagram of upregulated lncRNA genes between stage transitions. (B) Venn diagram of downregulated lncRNA genes between stage transitions. (C) Heat map of expression levels of lncRNA genes in the four cell populations, with three biological replicas each. All lncRNA genes detected as differential in at least one stage transition were included. Z-score values are coded on the green-to-red scale (high expression: red; low expression: green).
Among the DE lncRNAs, the number of upregulated genes (Fig. 2A) was notoriously higher than that of downregulated genes (Fig. 2B), for all the pairwise comparisons. Despite this, RS exhibited the highest number of upregulated and downregulated DE lncRNA genes as clearly depicted in the heatmaps, where the highest number of DE lncRNAs in RS is evident (Fig. 2C).
Then, we set out to contrast our data with those from other studies. In order to use the most comparable available datasets, we chose the results from Lin et al. [38] and Wichman et al. [39], as both studies employed strand-specific RNAseq of cell populations enriched in different stages of mouse spermatogenesis. However, on the other hand, notorious differences exist between these works and ours in the method for cell purification (both these studies used Staput, which involves longer cell collection times and renders lower purity), the age of the animals selected for the collection of the different fractions, and the pipelines used for data analysis. In spite of this, after re-analysing their raw data we found a significant correlation between expression data from their experiments and ours for the different cell populations (r = 0.49–0.67, p < 10−5 for the dataset from ref [39]; r = 0.38–0.47, p < 10−5 for ref [38]; Supplementary Fig. S3).
Characterization of lncRNAs in the different testicular cell populations
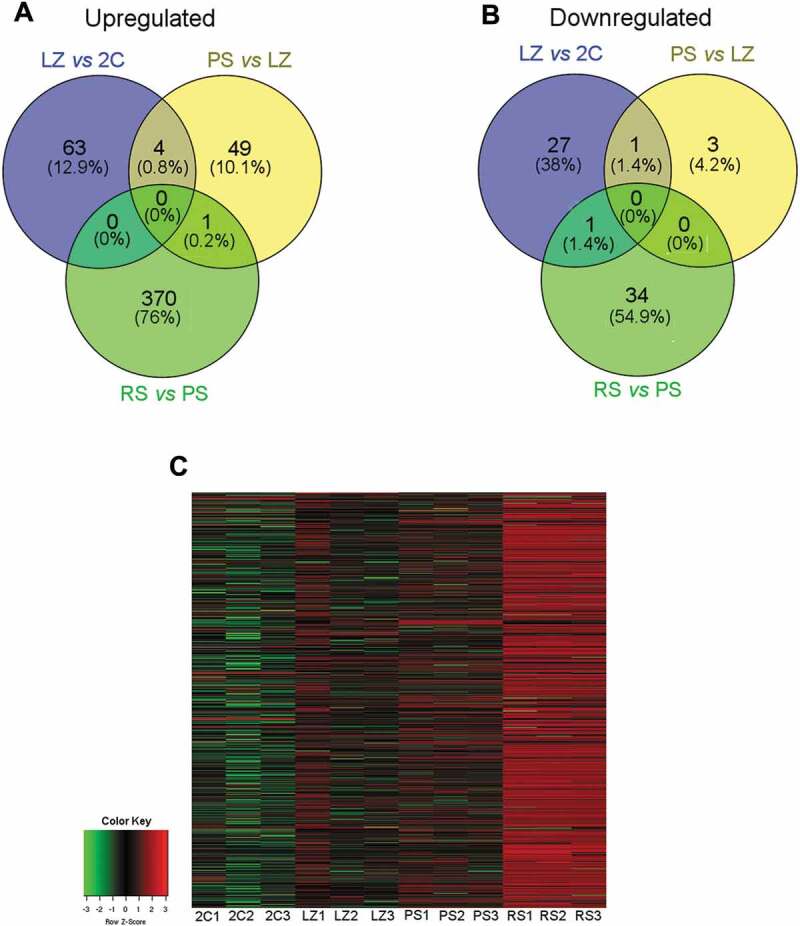
In order to characterize the spermatogenic lncRNAs, we first analysed the chromosomal distribution of the lncRNA genes appearing as expressed in our lists (see Supplementary Table S2). In particular, it is noticeable that in spite of the small size of the Y chromosome, a very high number of lncRNAs are annotated on this chromosome in Ensembl database. Strikingly, we observed a very strong depletion in the number of Y-linked lncRNA genes expressed in testis (hypergeometric test p < 10−9; see Methods), and this was true for the lncRNAs from the four cell populations (Fig. 3A). On the other hand, a relatively low number of lncRNA genes are annotated on the X chromosome. In relation to testicular X-linked lncRNAs, we observed a switch off of lncRNAs in PS, and their switch on in RS (Supplementary Fig. S4). Nevertheless, the numbers are too small to attribute this finding to meiotic sex chromosome inactivation (MSCI). Moreover, the time course of testicular X-linked lncRNAs expression could simply reflect the general behaviour of most testis-expressed lncRNAs, i.e. upregulation in RS.
Figure 3.
Characterization of lncRNAs in the different testicular cell populations. (A) Chromosomal distribution of lncRNAs. The total number of lncRNA genes annotated for each chromosome, and the number of lncRNA genes identified as expressed per chromosome in our lists for the 2C, LZ, PS and RS cell populations are shown. A mouse karyotype is presented below the graphic, as a reference of chromosome size. *p < 10−9. (B) Length distribution of the expressed lncRNA genes in 2C, LZ, PS and RS cell populations. (C) Exon number distribution of the expressed lncRNA genes in the four testicular cell populations. (D) Biotype distribution of the expressed lncRNA genes in the four testicular cell populations.
We then analysed some features of the lncRNAs in the four cell populations, such as the average transcript length, number of exons, and biotypes (see Supplementary Table S2). Less than 20% of the expressed lncRNAs were smaller than 500 nt, around 75% had between 501 and 3,000 nt, and only a small proportion (between 6% and 13%) were larger than 3,000 nt, for the four populations (Fig. 3B). Concerning exon number, most lncRNAs had less than five exons, with a high proportion of them (around 40%) containing only two exons (Fig. 3C). When only the DE lncRNA genes were considered, these characteristics were observed for the different stage transitions as well (Supplementary Fig. S5 A, B).
Regarding biotypes, we used the categorization from Ensembl that classifies lncRNAs into lincRNAs (long intergenic non-coding RNAs), antisense (AS) lncRNAs (overlapping a protein-coding gene on the opposite strand), sense overlapping, sense intronic, macro lncRNAs (unspliced ncRNAs of several Kb in size), 3ʹ- overlapping ncRNAs (on the 3ʹ-UTR of a protein-coding locus on the same strand), and bidirectional promoter lncRNAs (originating from promoter regions of protein-coding genes, in the opposite direction on the other strand).
LincRNA genes were the most abundantly expressed in our lists, followed by AS lncRNAs (Fig. 3D). This is not unexpected, as these have been reported to be the two most highly represented categories among lncRNAs in general [17], and particularly in testis as well [29,35,38]. However, the proportion of AS lncRNAs overlapping protein-coding genes in our lists was surprisingly high in relation to the small proportion of the genome occupied by protein-coding genes (see Fig. 3D). As stated above for other features, this was true for the four cell populations, and both for the total expressed and DE lncRNA genes (Supplementary Fig. S5C).
In particular, concerning lincRNAs, Lagarde et al. (2017) developed an experimental reannotation of the GENCODE intergenic polyA+ lncRNAs by means of RNA Capture Long Seq (CLS) [18]. We then re-analysed our sequencing data for lincRNAs, employing CLS annotation as a reference. Using Hisat/Stringtie/Ballgown, we detected 1,020 lncRNAs expressed in the 2C cell population, 1,497 in LZ, 1,392 in PS, and 2,036 in RS (Supplementary Table S3). However, it is important to point out that due to existant redundancies, the number of unique lincRNA species at each stage must be lower (see Discussion). The analysis of our data with both annotations showed coincidence levels that ranged from 34.1% to 36.9% (Supplementary Fig. S6A) with high correlations (Supplementary Fig. S6B) for the four testicular cell populations. This is a good coincidence level, considering that the overall coincidence between CLS and GENCODE transcript catalogues from mouse is 20% [18]. In relation to DE lincRNAs, using CLS annotation we observed 52 DE lincRNAs at the LZ vs 2C transition, 54 in PS vs LZ, and 572 in RS vs PS. Thus, in general terms, the re-analysis of our data with CLS annotation confirms for the case of lincRNAs, the highest number in RS, both for the expressed and for the upregulated genes. On the other hand, as expected, transcripts identified with CLS reference were, in general, longer than those identified using Ensembl (median = 1,032 and 808 for CLS and Ensembl, respectively).
Co-expression of lncRNAs with overlapping and neighbouring coding genes
Several studies have shown that AS transcripts can interfere with sense transcription of protein-coding genes by regulating gene expression and/or genome integrity, and exerting their effect in cis or trans, either locally or distally. Nevertheless, due to their genomic arrangement, it is believed that more frequently they act locally, in cis (e.g. by a sense-AS self-regulatory mechanism) [28,45].
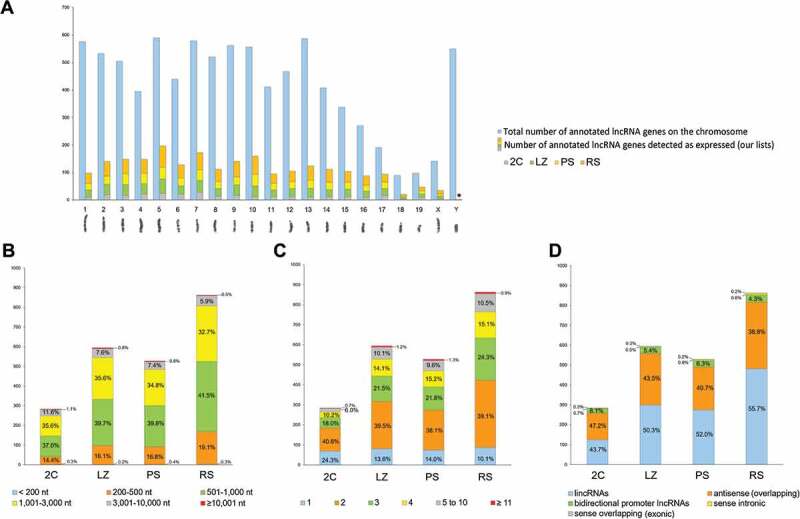
The high representation of AS lncRNAs in our lists, prompted us to analyse their co-expression with overlapping protein-coding genes. We found that for 85.5% of the expressed AS lncRNAs, the host protein-coding gene also appeared as expressed in our lists (Supplementary Table S4). Moreover, 81.5% of the DE AS-overlapping lncRNAs followed the same expression pattern as their cognate protein-coding genes (i.e. both either upregulated or downregulated at the same spermatogenic stage transition/s). Interestingly, for over 72% of the co-expressed gene pairs (i.e. 88.75% among those following the same expression pattern), their expression pattern was ‘up-up’ (both the coding gene and the overlapping AS were upregulated; r = 0.81, p < 10−5) (Fig. 4A, and Supplementary Table S4). In 13% of the gene pairs, an inverse correlation between the AS and its host mRNA gene was observed (i.e. one was upregulated and the other was downregulated at the same stage transition/s; see Fig. 4A; r = −0.70, p < 10−4), while in only 5% of the cases the expression pattern between the AS lncRNA and its host coding gene could not be correlated (r = 0.20, p = 0.52).
Figure 4.
Co-expression of testicular lncRNA genes with protein-coding genes along the different spermatogenic stage transitions. (A) Representation of the percentages of the different co-expression patterns of antisense (AS) lncRNAs and their host protein-coding genes. The schematic diagram shows different positions of overlapping AS lncRNAs (white arrows) in relation to their cognate co-expressed mRNAs (black arrow). Expression patterns were classified as: up-up (both the AS and the coding gene are upregulated); down-down (both the AS and the coding gene are downregulated); same combined pattern (both the AS and the coding gene follow the same combined behaviour along spermatogenesis, e.g. both upregulated at a certain stage transition and then both downregulated at another stage transition, or vice versa); inverse pattern (the AS is upregulated when the coding gene is downregulated, or vice versa); and no correlation (the expression patterns of the AS and the coding gene were not correlatable). (B) Representation of the percentages of the different co-expression patterns of lincRNAs and neighbouring protein-coding genes. The results for gene pairs where the lincRNA is located at <300 Kb, <100 Kb, and <30 Kb distance from the co-expressed coding gene are shown. The top diagram illustrates the different distances at which co-expression patterns between lincRNAs (white rectangles) and protein-coding genes (black rectangle) were analysed. The categories of the expression patterns are the same as in (A). (C) Distribution of the distance of lincRNAs from their co-expressed neighbour coding genes. The number of gene pairs located at <300 Kb from the coding gene is taken as 100%. The percentage of lincRNAs located at varying distances from the coding gene is shown. The expected percentages are those that would be observed at <100 Kb, <30 Kb and <10 Kb, if the distribution was even. For all the co-expression analyses, only those pairs in which both genes were expressed in our transcriptomes and at least one of them was DE, were considered.
Additionally, in at least 61% of the differentially co-expressed gene pairs, a testis-specific role or testis-restricted expression pattern for the protein-coding gene has been described (see Supplementary Table S4). Gene ontology (GO) analysis of the coding genes in the DE gene pairs showed a moderate enrichment (p < 0.05) in the terms ‘spermatogenesis’, ‘sperm motility’, and ‘microtubule-based movement’ for the biological process category, and ‘microtubule motor activity’ for the molecular function category (data not shown).
We then evaluated the co-expression of lincRNAs with their neighbour protein-coding genes, as lincRNAs were the most highly represented lncRNA biotype in our lists. An increasing number of studies have shown that lincRNAs can affect the expression of their neighbouring protein-coding genes or other target genes by acting through different mechanisms, either in cis or in trans [27,46,47]. Thus, in order to identify possible functional relations, different studies have correlated lincRNAs with neighbour mRNA loci located at varying distances such as <300 Kb [47], <100 Kb [48,49], <30 Kb [35], or <10 Kb [29], depending on the study.
In this work we have analysed the co-expression of lincRNAs with coding genes (Supplementary Table S5), starting at a distance of <300 Kb. For 43% of the testis-expressed lincRNAs, there was at least one neighbour protein-coding gene whose transcript was also present in our lists of expressed genes. 41.7% of the DE gene pairs showed the same expression pattern, among which 77.7% were ‘up-up’ (32.41% of all the co-expressed pairs; r = 0.78¸ p < 10−5). On the other hand, 15.33% of the gene pairs showed an inverse correlation (r = −0.71¸ p < 10−5). Conversely, 43% of the gene pairs showed a very low correlation coefficient all along the stage transitions (r = −0.25, p < 10−5; Fig. 4B). When only pairs located at <100 Kb were considered, the proportion of pairs with the same expression pattern along stage transitions raised to 53% (80% of which were ‘up-up’, i.e. 42.38% of the total; r = 0.75, p < 10−5), while the percentage of gene pairs whose expression pattern could not be correlated (r = −0.07, p = 0.51) turned out to be 35% (see Fig. 4B). When we further narrowed the distances to <30 Kb, the percentage of pairs showing the same behaviour increased to 61.49% (of which almost 88% were ‘up-up’, i.e. 53.73% of the total; r = 0.85, p < 10−5), although the percentage of those with no correlation (r = −0.06, p = 0.64) remained unchanged (see Fig. 4B). No significant differences were found when the distances were further reduced to <10 Kb (not shown). GO analysis of the coding genes co-expressed with neighbouring lincRNAs showed an enrichment (p < 0.01) in ‘nucleosome assembly’, ‘positive regulation of gene expression’, and ‘gene silencing’ (biological process), and ‘nucleosomal DNA binding’ (molecular function), among other categories (not shown).
In summary, the smaller the distance between lincRNAs and their neighbouring coding genes, the greater the trend to follow the same expression patterns along the different spermatogenic stage transitions. Anyway, the percentage of gene pairs that exhibited a similar behaviour was much higher for sense/AS pairs than for those involving lincRNAs. Likewise, while the proportion of the AS lncRNAs whose expression pattern could not be correlated with that of their cognate coding genes was very small, this percentage was significantly higher for lincRNAs. Despite this, most co-expressed lncRNA/coding gene pairs – both for AS and lincRNAs – were upregulated in RS. When we performed GO analysis for the coding genes in the RS-upregulated co-expressed pairs (including both AS and lincRNAs), we observed an enrichment in basically the same categories as above, while some other spermiogenic-specific categories such as ‘acrosomal vesicle’, also appeared significantly enriched (p < 0.01). Above all, in all GO analyses there was a highly significant enrichment in the term ‘testis’ for the tissue category (p < 10−27), indicating that most DE coding genes that are co-expressed with lncRNAs along spermatogenesis are testis-specific.
We also observed that the distribution of lincRNAs in relation to their co-expressed neighbour coding genes was not uniform: 44.37% of the lincRNAs positioned at <100 Kb from a co-expressed coding gene were within <30 Kb distance, and 64.17% of those at <30 Kb were actually located at <10 Kb. Thus, testis-expressed lincRNAs are more concentrated within 10 Kb of co-expressed protein-coding genes, in accordance with what was observed in a couple of previous reports for lincRNAs in general [20,50] (Fig. 4C).
Next, we asked if the lncRNAs that are co-expressed with coding genes along spermatogenesis are conserved in humans, which would reinforce the idea of a possible functional relationship. For this purpose, we first used our co-expression lists of testicular AS lncRNAs and coding genes, and searched for annotated AS lncRNAs for the homologous coding genes in the human genome. By using two different human databases in parallel (Ensembl and Chess), we found annotated AS lncRNAs in human for 46% and 49% of them, respectively (see Supplementary Table S4). These percentages would be higher than the overall proportion of orthologous sense-AS pairs between mouse and human, which has been suggested to be less than 20% [51].
We then selected our co-expression lists of coding genes within <30 Kb from lincRNAs in mouse, and searched for the existence of homologous lincRNAs at syntenic positions in the human genome, as it has been suggested that the maintenance of the genomic position of lncRNAs relative to protein-coding genes might be important in determining their function [52]. Despite the fact that in most cases we located highly homologous DNA sequences in the human genome and near the same coding genes as in mouse, in general, we found no evidence of transcription for those specific sequences in human. On the other hand, in 59% of the cases, we found an annotated lincRNA within <30 Kb of the homologous coding gene in the human genome (see Supplementary Table S5), although blast searches showed no significant sequence homology with the mouse lincRNAs located at syntenic positions. In this regard, it has been stated that some lincRNAs can be orthologs located at conserved genomic locations, yet perhaps their sequences may be too divergent to be detected with the existing tools, or their function may not depend on the strict nucleotide sequence [50,53]. The observed percentage of synteny is also higher than the ones suggested for lncRNAs between mouse and human in other contexts. As an example, it has been reported that only 10% of the DE lncRNAs upon activation of the innate immune response in human showed syntenic versions in mouse [52]. Although our findings could seem to go against previous reports suggesting low evolutionary conservation of testicular lncRNAs [21,38], those studies did not specifically refer to the conservation of testicular co-expressed pairs. Anyway, in principle, we cannot draw definitive conclusions about the existence of a significant conservation of co-expression patterns along spermatogenesis, as we do not know whether human syntenic lncRNAs exhibit the same co-expression patterns as in mouse.
Validation of the co-expression patterns between lncRNAs and protein-coding genes
In order to validate the co-expression patterns of lncRNAs with coding genes from our RNAseq data, we selected 13 pairwise lncRNA-coding gene combinations for the analysis of their expression levels via RT-qPCR. On one side we tried to choose genes that would be upregulated at the different stage transitions and, on the other side, we aimed at selecting genes representative of different expression profiles (e.g. ‘up-up’, ‘down-up’, etc.). However, as for the vast majority of co-expressed pairs – both for those including AS and lincRNAs – their expression pattern was ‘up-up’ in RS, most of the selected pairs for confirmation were of this type. To exemplify this correlation, and focusing on pairs whose coding genes would encode proteins with known spermatid- or sperm-specific roles, we chose Tnp1 (transition protein one), Lyzl4 (Lysozyme-like 4), Spata2l (Spermatogenesis associated 2 like), Akap1 (A-kinase anchoring protein 1), Pdzk1 (PDZ domain containing 1), Nkx2-6 (NK2 homeobox 6), and Lrp8 (LDL receptor related protein 8, also known as apoER2) (Fig. 5 A-G). TNP1 participates in the replacement of histones by protamines in elongating spermatids [6]. LYZL4 is a sperm-related protein involved in fertilization [54], while SPATA2L is a paralog of SPATA2 (a necroptosis-involved protein), which is required for full fertility in mouse [55]. AKAP1 anchors protein-kinase A to mitochondria in sperm [56], while PDZK1 (localized at the middle piece of the sperm tail) and LRP8 are epididymal proteins required for normal sperm morphology and motility [57,58]. Although no specific bibliographic information is available for Nkx2-6, its expression pattern is highly restricted to adult testis [59] (https://www.ncbi.nlm.nih.gov/gene/18092#gene-expression), as has been also shown for a closely related member of its same gene family [60]. Tnp1, Lyzl4, Spata2l, Pdzk1, and Nkx2-6 have a co-expressed AS, while Akap1 has a co-expressed neighbour lincRNA, and Lrp8 has both a co-expressed AS and a neighbour lincRNA (see Supplementary Tables S4 and S5).
Figure 5.
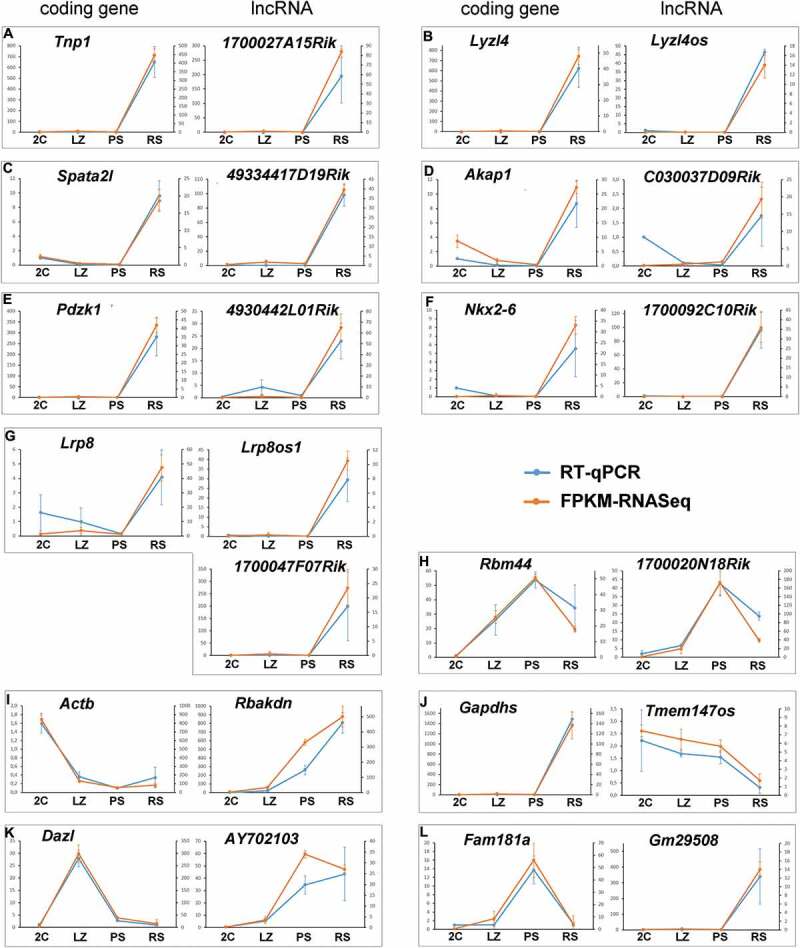
Validation of the dynamic co-expression patterns of 13 selected gene pairs of lncRNAs and protein-coding genes. Gene pairs representative of the different co-expression profiles are shown. The graphics show the consistency between RNAseq data and RT-qPCR. The left axis indicates the relative quantification (RQ) of RT-qPCR, while the right axis indicates the FPKM values for RNAseq. The error bars correspond to ±SD.
To exemplify a positive correlation for gene pairs with an expression peak in PS we chose Rbm44 (RNA-binding protein 44), an intercellular bridge component of pachytene and secondary spermatocytes [61], and its neighbour lincRNA (Fig. 5H).
In order to show some inverse correlations, we selected Actb (beta-actin), Gapdhs (Glyceraldehyde-3-phosphate dehydrogenase, spermatogenic), Dazl (Deleted in azoospermia like), and Fam181a (Family with sequence similarity 181 member A).
Actb was chosen as an example of an inverse correlation where the coding gene is downregulated from the 2C population while its neighbour lincRNA, Rbakdn, shows an opposite expression pattern (Fig. 5I). Regarding Rbakdn, an interesting feature we found is that this lincRNA is conserved between mouse and human, and in both species its expression is testis-restricted [59,62] (see https://www.ncbi.nlm.nih.gov/gene/100042605 and https://www.ncbi.nlm.nih.gov/gene/389458, respectively). Moreover, both in mouse and human Rbakdn is located near the same genes, including Act b.
Gapdhs, a spermiogenesis-specific counterpart of Gapdh that is required for sperm motility and male fertility [63], was selected to exemplify an inverse correlation between a coding gene that is upregulated from PS to RS and its AS, with the opposite expression pattern (Fig. 5J).
We have previously shown that Dazl, which encodes a germ cell-specific RNA-binding protein required for the differentiation of germ cells, shows a marked expression peak in LZ and abruptly decreases at the LZ-to-PS transition [4]. Conversely, its neighbour lincRNA is upregulated at the LZ-to-PS transition, coinciding with the decline of Dazl mRNA (Fig. 5K). Finally, we selected Fam181a because it is overexpressed both in mouse and human testes [59,62] (https://www.ncbi.nlm.nih.gov/gene/100504156 and https://www.ncbi.nlm.nih.gov/gene/90050), and a QTL related to fertility in cattle that overlaps this gene has been detected [64]. Fam181a exemplifies an inverse correlation for a gene with an expression peak in PS and downregulated at the PS-to-RS transition, and whose AS expression starts coincidentally with the mRNA decline (Fig. 5L).
The dynamic co-expression patterns of all genes were highly consistent with RNAseq analyses (see Fig. 5), showing the high reliability of the data in our lists.
An antisense lncRNA and its overlapping mRNA co-localize in the chromatoid body of round spermatids
Next, in order to characterize a possible relation between AS lncRNAs and host mRNAs, we chose one such pairs for in situ hybridization using Stellaris® RNA-FISH probes [65]. The selected RNAs were Kcnmb4 mRNA, which encodes a regulatory subunit of a calcium-activated potassium channel [66] and its overlapping AS lncRNA, Kcnmb4os1 (Fig. 6A). The expression of both the sense and AS transcripts is differential of testis compared to other mouse tissues [59] (https://www.ncbi.nlm.nih.gov/gene/58802 and https://www.ncbi.nlm.nih.gov/gene/?term=kcnmb4os1, respectively). Both probes gave positive signals that co-localized in the chromatoid body of RS (Fig. 6B; colocalization Pearson´s correlation coefficient 0.989), as shown by co-staining for MVH (DDX4), a well-characterized chromatoid body marker [9] (Fig. 6C). On the other hand, a probe against widely studied lncRNA Malat1 [67] that was used as a positive control, gave the expected localization pattern in the nuclei of somatic testicular cells (Supplementary Fig. S7).
Figure 6.
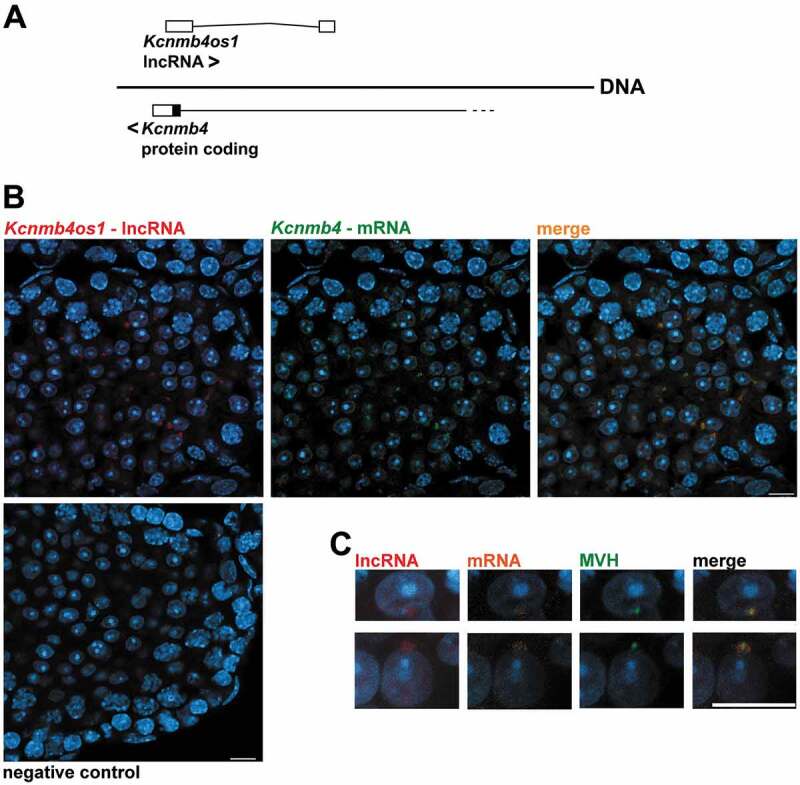
Fluorescent in situ hybridization (FISH) analysis of Kcnmb4 mRNA and its overlapping Kcnmb4os1 AS lncRNA in mouse testis. (A) Diagram showing the structure and orientations of Kcnmb4 and Kcnmb4os1 transcripts. White rectangles represent non-coding exons. For the sense transcript, only the last coding exon is shown (coding region in black, 3ʹ UTR in white). (B) Co-localization of the sense and AS transcripts in the chromatoid body of RS. Red: AS lncRNA; green: mRNA. The right frame shows co-staining with both probes. (C) Co-immunostaining with anti-MVH antibody (green) in RS as a marker for the chromatoid body. The AS is shown in red, while the mRNA is shown in orange in this case. The image to the right shows the triple staining. All the sections were co-stained with DAPI. Bars: 10 µm.
Discussion
Recent studies have identified thousands of testis-expressed lncRNAs in mouse [10,29,35,38]. However, the estimation of the exact number of lncRNAs is far more complicated than for coding transcripts. Besides the fact that coding potential cannot be taken into account for evaluation, lncRNAs are in general less abundant compared to mRNAs [17,20], and therefore it is difficult to set a baseline above which a lncRNA gene is considered as transcribed. Another drawback is that the annotation of lncRNAs is much less refined than that of coding transcripts. In particular, a disadvantage of microarray-based studies is that due to the lack of complete annotations microarray probes are highly redundant [38], which may lead to overestimations of lncRNA numbers. Concerning RNAseq, although most analyses have employed dedicated software tools, more recently new software tools that provide more accurate overall results have been developed [40]. Another fact that complicates the scene is that in most studies it is not clearly specified whether lncRNA species (including transcript variants) or lncRNA genes were accounted for. In a different study (to be published elsewhere), we found around 60,000 unannotated transcripts that corresponded to non-coding RNAs (including splice variants) expressed in all the cell populations.
In this paper, we decided to work with annotated lncRNA genes. Besides, we deliberately decided to privilege reliability at the expense of amount of detected genes. The reliability of our data is based in the first place, in the method for cell classification, which yields highly pure stage-specific testicular cell populations [4]. These were combined with strand-specific RNAseq, which is essential for the accurate identification of AS lncRNAs [44,45]. This differentiates our stage-specific data from those from some other reports that used methods for cell-type enrichment [10,36,38,39] or whole testes [35,37], and some of them in combination with microarrays for transcriptome analysis [35,36]. Moreover, we have used the newest available bioinformatics tools for RNAseq analysis [40], and we have only selected those genes that arose from the intersection of two different pipelines. Besides, we have focused on lncRNA genes and not on lncRNA transcripts (i.e. we have not considered splice variants); this highly reduces the number of identified species. In addition, all DE genes were selected using FDR p-value correction, which corrects type I error thus reducing the number of false positives in the reported lists [68,69]. Hence, although we are aware that we are dealing with only a subset of all testis-expressed lncRNAs, we are convinced of the reliability of the results presented here, that correspond to those lncRNAs for which the evidence of their expression patterns is robust. We want to denote that despite the marked differences in the sampled cell populations between our study and others (see Results), we found significant correlations of detected genes with those studies we chose for comparison [38,39].
Additional support for the robustness and reproducibility of our data also comes from RT-qPCR analyses, which allowed confirming all the lncRNA expression patterns we chose for validation, as well as their co-expression with coding genes. Needless to say, as the raw data is deposited at the SRA, it is available for re-analysis by means of new or less conservative approaches.
A very remarkable result of our study is that the great majority of lncRNA genes in mouse testis are expressed in RS, which is in accordance with a couple of previous reports [10,38], but not with others [36,39]. Interestingly, we found that the difference in favour of RS was even much greater for the case of the DE lncRNA genes, indicating that for most lncRNAs that are present at different spermatogenic stages, their expression levels significantly raise after meiotic prophase.
We have analysed the lncRNA populations in the different spermatogenic cell types. Our analyses show that although the molecules that compose the lncRNA populations significantly vary at each spermatogenic stage, they all share the same general characteristics, with most lncRNAs being between 500 and 3,000 nt in length, and having less than 5 exons. These features are shared with testicular lncRNAs from other species, such as pig [31] and chicken [49]. On the other hand, these results differ from those of Chalmel et al. [30] that reported that in rat lncRNAs with an expression peak in meiosis were exceptionally long.
Most testicular lncRNAs in our lists are lincRNAs followed by AS, which has been also observed in some other studies, both for mouse [35,38] and chicken [49]. Interestingly, however, we have found that in all the cell populations the percentage of AS lncRNAs was surprisingly high, most probably due to the fact that strand-specific RNAseq contributes to their reliable identification. In this regard, it has been estimated that about 32% of lncRNAs in human would be AS to protein-coding genes [17], suggesting that regulation through AS lncRNAs is a commonly used mechanism [22,45].
Our results indicate that for the vast majority of AS lncRNAs that were co-expressed with their host coding genes in testis, there was a high correlation between the expression pattern of the sense and AS, all along the analysed spermatogenic stage transitions. Moreover, in most cases there was a positive correlation, and both the coding gene and the AS were upregulated. This, in the first place, suggests that the existing permisive, transcription-compatible chromatin state, would facilitate transcription from the other strand [28]. On the other hand, some studies in other tissues have revealed that AS lncRNAs transcription/transcripts can interfere with sense coding transcripts at different levels and in different ways, i.e. by acting at the initiation of transcription, co-transcriptionally, or post-transcriptionally, and exerting either activating or repressing effects [28,45]. In case at least some of the overlapping AS lncRNAs in testis modulate the expression of their host coding genes, the fact that most pairs are positively co-expressed could suggest a mechanism for regulation. A possibility is that AS lncRNAs transcription/transcripts mostly carry out a positive regulation on the expression of coding genes. Although we cannot exclude this possibility and, indeed, regulatory mechanisms of this type have been described in other tissues [70–72], the fact that the great majority of the co-expression of testicular AS lncRNAs with their cognate coding genes takes place in RS, raises another attractive hypothesis. As stated above, RS are characterized by extensive post-transcriptional regulation, among which translational delay stands out as a strategy through which a high amount of mRNAs are sequestered by diverse mechanisms [4,6]. This allows to regulate the translation time for sperm-related proteins [73], whose premature production in many cases would be detrimental [74,75]. Furthermore, an associated trait to the high post-transcriptional regulation levels in spermatogenic cells, and mainly of RS, is the existence of widespread transcriptional activity [10], which may be accompanied by inefficient translation as a mechanism to prevent protein overexpression [6]. Thus, it is tempting to speculate that at least in some cases, testicular AS lncRNAs could somehow act on their complementary coding transcripts by sequestering them for translational repression, and/or eventually stabilizing them for delayed translation. An example of an AS lncRNA that enhances the stability of its sense mRNA in the brain is BACE1-AS, which has been associated with Alzheimer’s disease [76].
The idea that at least some AS lncRNAs could modulate gene expression in RS by sequestering and/or stabilizing mRNAs for delayed translation, may be supported by the fact that many of the coding genes that appeared in our lists as upregulated in RS and co-expressed with their AS, encode proteins that are used in elongated spermatids or mature sperm. One such proteins is TNP1; in this regard, it is well known that the mRNAs for protamines and transition proteins are translationally repressed in RS, and their premature translation causes spermatogenic arrest and infertility [74,75].
It is interesting that we identified an AS lncRNA (Kcnmb4os1) that co-localizes with its overlapping sense mRNA (Kcnmb4) in the chromatoid body of RS. While the true functions of this spermatid-specific structure remain intriguing, the available evidence points to a role in RNA-related processes such as the storing of repressed mRNAs and, more recently, also in the degradation of transcripts via nonsense mediated decay (NMD) [9,77]. However, how RNAs are targeted to the chromatoid body is presently unclear [77]. In particular, KCNMB4 is a regulatory subunit of a calcium-activated potassium channel, which modulates calcium sensitivity [66]. It is well established that ion channels play essential roles in sperm motility, sperm activation, acrosome reaction, and fertilization [78]. Moreover, although thus far the specific function of KCNMB4 in testis is not clear, its modulatory role in spermatogenesis has been suggested [79]. A nice hypothesis that deserves to be explored would be that Kcnmb4os1 somehow interacts with the sense transcript, with the consequence of its targeting to the chromatoid body. If this were true, maybe it could be part of a tuning mechanism to modulate ion channel function in germ cells. It will be interesting to analyse the localization patterns of more AS/sense RNA pairs in RS, in order to determine if co-localization in the chromatoid body is an extended phenomenon.
Regarding lincRNAs co-expressed with neighbour coding genes, the proportion of them whose expression pattern was consistent with that of a coding gene in the four testicular cell populations was significantly lower than that for AS lncRNAs. This most probably indicates that many lincRNAs that are expressed in the same tissue and even at the same time as nearby coding genes, are not really co-regulated with them. In many cases, the co-expression pattern could simply represent transcriptional noise. This could be particularly so in the male germline, as a consequence of the widespread promiscuous transcription that operates in these cells [10]. On the other hand, the role of an important number of lincRNAs in modulating gene expression in different tissues has been undoubtedly shown [27,50,70,80]; however, we still have no clues about what proportion of them may have a biological role.
Coincidentally with our observations concerning AS, most of the lincRNAs/neighbour gene pairs whose expression pattern followed a similar behaviour in all the stage transitions, showed upregulation in RS. The proportion of pairwise co-expressed lincRNAs/neighbour coding genes whose pattern was ‘up-up’, increased as the distance between the neighbouring genes decreased. Interestingly, our results both regarding the impact of the distance on co-expression patterns, and the positive correlation of AS with coding genes, are strikingly coincident with those obtained by Derrien et al. [17], through a bioinformatic analysis of the human lncRNA GENCODE annotation.
Specifically, in relation to lincRNAs, we also used our RNAseq data for re-analysis with a new, long-transcript annotation of intergenic lncRNAs (CLS) [18]. Although we found some redundancies that make direct comparisons difficult (e.g. many IDs from CLS correspond to the same GENCODE ID; many testicular transcripts also appear in other tissue/s with a different ID), this analysis produced an interesting wealth of new information. In particular, it is allowed to assign over 2,000 gene IDs corresponding to long-read intergenic transcripts, to specific spermatogenic stages.
Another intriguing fact is the almost complete depletion of testicular lncRNAs from the Y chromosome, despite the high number of annotated lncRNAs from this chromosome. It is interesting that a transcriptomic analysis of the chromatoid body of RS detected non-coding transcripts derived from all the chromosomes but the Y chromosome [9]. We here extend this finding to the whole transcriptome from RS and, moreover, to testicular lncRNAs in general. We currently lack an explanation for this, but we do not relate it to MSCI as we observed this depletion in the four testicular cell populations, and not specifically in relation to the pachytene stage. Definitely, this curious fact will deserve further investigation.
Finally, we have noted that in a number of cases, lncRNAs annotated as AS overlapping, are in fact very close adjacent, but non-overlapping neighbours to coding genes (and therefore, sensu stricto they should be classified as lincRNAs). Furthermore, while attempting to conduct conservation studies, we have observed that although for many mice lncRNAs the homologous DNA sequences exist in other mammalian species, there are no lncRNAs annotated for those specific sequences in the other species. This raises the question of whether none of those homologous sequences is transcribed in the other species, or if at least for some of them, the homologous lncRNAs may have not been annotated yet. No doubt, the years to come will represent a breakthrough in the research of lncRNAs in testis as annotations are optimized and, most importantly, as their roles in relation to the modulation of gene expression in spermatogenesis and fertility start to be elucidated.
Methods
Ethics statement
All animal procedures were performed following the recommendations of the Uruguayan National Commission of Animal Experimentation (CNEA), approved experimental protocol 001/02/2012 (code: 008/11; http://www.cnea.org.uy/index.php/instituciones/registro/10). Animals were euthanized by cervical dislocation, in accordance with the National Law of Animal Experimentation 18,611 (Uruguay).
Animals
Male CD-1 Swiss mice (Mus musculus) at different ages were obtained from the animal facility at Instituto de Higiene of Facultad de Medicina (UdelaR, Montevideo, Uruguay). Immediately after euthanasia, testes were dissected and tunica albuginea was removed before proceeding to the preparation of testicular cell suspensions. Testes for in situ hybridization were processed with the tunica albuginea.
Preparation of cellular suspensions and sorting by flow cytometry
Testicular cell suspensions were prepared by a procedure described earlier in our laboratory [42,43,81]. We introduced a brief modification to the preparation of the 2C cell population, i.e. a treatment with 0.6 U/mL collagenase for 15 min at room temperature before mechanical disaggregation step. Cells were counted in a Neubauer chamber and resuspended at a concentration of 1 × 106 cells/mL in Dubelcco’s Modified Eagle’s medium (DMEM) supplemented with 10% foetal calf serum. Testicular cell suspensions were stained with Vybrant DyeCycle Green (Invitrogen, Life Technologies, Carlsbad, CA), as previously described [43]. Samples were analysed and sorted with a FACSVantage flow cytometer (Beckton Dickinson, CA) furnished with an argon ion laser (Coherent, Innova 304) tuned at 488 nm of excitation wavelength (100 mW), and using a 70 µm nozzle. The protocol for cell analysis and sorting was the same as reported earlier [4], with a slight difference in the ages of the animals used for the obtainment of each cell population. The 2C cell population was classified from a testicular cell suspension of a pool of up to five individuals ageing 12–14 days post-partum (dpp), LZ cell population was obtained from 15 to 18 dpp animals, PS from 16 to 19 dpp, and RS from 22 to 24 dpp animals. Of note, due to the age of the animals employed for the classification of the 2C population, this fraction does not include spermatocytes II. Sorting was set in Counter mode that yields the highest purity achieved by the equipment (>95%), with three sorted drops as envelope. Twelve samples were obtained (four different cell populations, with three biological replicates each), collected in PBS treated with 0.1% DMPC, spun down (500 g, 10 min, 4ºC), deep frozen in liquid nitrogen, and stored at −80ºC until use.
The purity of each sorted fraction was assessed, first using laser confocal and differential interference contrast (DIC) microscopy for the estimation of chromatin characteristics (based on VDG green fluorescence) as well as cell and nuclear size and morphology, as in previous reports from our group [42,43]. Besides, the purity of the LZ and PS sorted fractions was confirmed by immunodetection with an antibody against SYCP3 (a marker of the synaptonemal complex; Acris Antibodies, 1:100) on spread cells as instructed [82], to monitor the advance of meiotic prophase. The purity of the RS fraction was confirmed with an antibody against MVH, a marker of the chromatoid body (Abcam ab13840, 1:2,000) (see Supplementary Fig. S1).
RNA extraction and construction of sequencing libraries
Total RNA from each of the 12 samples was extracted with PureLink RNA Mini Kit (Ambion, Life Technologies), following manufacturer’s recommendations. RNA quantitation was performed by fluorometry using Qubit 2.0 and RNA HS Assay kit (Life Technologies). We used Ovation RNA-Seq System 1–16 for Mouse kit (NuGEN) to generate strand-specific sequencing libraries. In brief, 60 ng of total RNA was used as input, and the libraries were constructed without fragmentation of the RNA samples. The material was amplified for 16 PCR cycles, according to the instructions from the kit. Library concentration was measured by fluorometry with Qubit 2.0 and dsDNA Assay Kit (Life Technologies), and library quality was assessed on a 2100 Bioanalyzer system (Agilent, Santa Clara, CA). Libraries were sequenced at Fasteris (Switzerland) on an Illumina Hiseq4000 platform, and 150 bp paired-end reads were generated.
LncRNA data analysis using Ensembl database
Raw data was processed using two different pipelines in parallel. The first used software package was CLC Genomics Workbench 10.1.1 (CLC bio). Raw reads were trimmed by quality (Q > 20), length (more than 50 bp), and Illumina adaptors were removed. RNAseq analysis was performed with CLC to obtain lists of differentially expressed (DE) genes among spermatogenic stages, using the M. musculus Ensembl database (Gm38.p6 release 93) as reference genome. Mapping to the reference genome was performed using the following parameters: mismatch cost 2; insertion cost 3; deletion cost 3; length fraction 0.8; similarity fraction 0.8; and maximum number of hits per read 10. Our analyses were based on expressed lncRNA genes and not on lncRNA species (i.e. lncRNA splice variants were not considered). Differential gene expression between the four testicular cell populations was obtained by means of ‘DE for RNAseq analysis’ tool included in CLC Genomics Workbench (based on TMM normalization, assuming a negative binomial distribution, and modelling each gene by a Generalized Linear Model [GLM]). The statistical analysis across all group pairs (which relies on Wald test) was done by pairwise comparisons in chronological order of appearance along the first spermatogenic wave (LZ vs 2C; PS vs LZ; RS vs PS), retaining genes with log2 fold-change ≥ │2│, and False Discovery Rate (FDR) p-value correction ≤ 0.05. We also filtered by variance, making a selection of those genes whose variance across samples was more than 1, and RPKM ≥ 0.1.
The second pipeline used in parallel was based on free access software. We trimmed the sequences using Trim Galore, and employing the same parameters as in CLC. Clean reads were aligned to the M. musculus reference genome of Ensembl database (Gm38.p6 release 93; index was created using a masked reference), with Hisat 2.0 [83]. Aligned reads were assembled into transcripts and counted with StringTie 1.3 [84]. Differential gene expression (again, by pairwise comparisons in chronological order of appearance along the first spermatogenic wave) was analysed with Ballgown [85]. DE genes were identified with the following parameters: log2 fold-change ≥ │2│, FDR p-value correction ≤ 0.05, and again considering only those genes whose variance across samples was more than 1 and FPKM ≥ 0.1. All RNAseq raw data were deposited in SRA repository, PRJNA548952.
A list of annotated lncRNAs available in Ensembl database (release 93) was downloaded. This list was used for filtering the DE gene results, in order to obtain a sub-list of annotated DE lncRNAs. LncRNAs that passed all the filters, and were detected as expressed or DE by both pipelines, were kept for further analysis.
Principal Component Analysis (PCA) was generated using 'PCA for RNAseq' CLC tool (that uses normalized log CPM [Counts Per Million] values as input). Matrix correlation was constructed in R bioconductor, calculating Pearson’s correlation coefficient between FPKM expression of every transcript in each of the 12 samples.
Venn diagrams were constructed using Venny 2.1 (http://bioinfogp.cnb.csic.es/tools/venny/). For heatmaps construction, we used R bioconductor version 3.4.4 (http://www.R-project.org).
Chromosomal distribution
To determine whether lncRNAs had preferential chromosome location, hypergeometric tests were performed in R bioconductor for the lncRNA genes expressed in each cell population, and enrichment/depletion p-values were calculated.
Co-expression analysis
We analysed the co-expression of overlapping antisense (AS) lncRNAs with their host coding genes, and lincRNAs with their neighbour coding genes. For this purpose, we searched the coordinates of our lists of expressed coding- and lncRNA genes by means of BioMart tool in Ensembl database. We generated two lists (coding and non-coding) in BED file format. For lincRNAs, we added 300 Kb, 100 Kb, 30 Kb, or 10 Kb distances to both ends. BEDTools Intersect was applied over both lists (https://github.com/arq5x/bedtools/blob/master/docs/content/tools/intersect.rst) to obtain neighbour coding genes. The results were merged with the tables of DE genes. We only kept those pairs in which both genes were expressed in our transcriptomes, and at least one of them was DE. We used the Pearson’s correlation test to calculate the correlation coefficients with their corresponding p-values for the lncRNA/coding gene pairs.
Gene ontology analyses for the coding genes overlapping AS lncRNAs or neighbouring lincRNAs were conducted using the functional annotation present at David Bionformatics Resources 6.8 website (https://david.ncifcrf.gov/).
For syntenic analysis of lncRNAs in human, we used Ensembl Human Genome (GRCh38.p12, release 93). Chess database (http://ccb.jhu.edu/chess/) [86] was used in parallel for the analysis of gene pairs involving AS lncRNAs. As Chess does not discriminate between lncRNA subtypes except for AS, it was not employed for the syntenic analysis of gene pairs involving lincRNAs.
Data comparison with other RNAseq studies
For comparison of our lncRNA lists with those from other reports, we downloaded datasets from refs [38] and [39] We re-processed the raw data from both studies using CLC, as the study from ref [38] had been only performed with two biological replicas, and the use of Ballgown is not recommended for less than three replicas. In order to make data comparable, the same parameters as indicated above for our sequencing data were employed. The generated lists of lncRNAs expressed at each stage were crossed with our CLC-generated lists. We used the Pearson´s correlation test to calculate the correlation coefficients with their corresponding statistical significance for the coincidences.
LncRNA data analysis using CLS database
For the study with CLS annotation reference [18], our sequencing data were analysed with Hisat/StringTie/Ballgown, using those transcripts built with ‘anchored’ method as reference, and all tissues-derived annotation provided at the portal for CLS data (https://public-docs.crg.es/rguigo/CLS/). We associated the IDs and biotypes from ref [18] to those equivalent from GENCODE, by means of ‘Transcript-to-biotype’ file that is available at the web portal of CLS annotation. We particularly worked with those transcripts defined as ‘lncRNAs’, and considering those transcripts with variance ≥1 and FPKM ≥ 0.1. For the DE transcripts, we considered those with log2 (FC)≥|2|. We took p-values ≤ 0.01 for the LZ vs 2C and PS vs LZ transitions, and FDR ≤ 0.05 for the RS vs PS transition as statistical values. We then compared the obtained data with ours (analysed with the same pipeline but using Ensembl v93 as reference, and only considering lincRNAs). Pearson’s correlation coefficients with their statistical significance were calculated for the coincidences.
RT-qPCR validation
For confirmative RT-qPCR, 3,000-cell fractions from 2C, LZ, PS and RS populations were sorted as explained above, but using a MoFlo Astrios EQ (Beckman Coulter) in Purify mode. Generation of cell lysates, reverse transcription and qPCR were performed using Power SYBR Green Cells-to-Ct kit (Ambion, Life Technologies) following the instructions of the manufacturer. For qPCR step, 2 µL cDNA in 20 µL final volume reaction mix was used. All the reactions were made in a CFX96 Touch Real-Time PCR Detection System 1 (BioRad, Hercules, CA), with three biological replicas each.
As the expression levels of commonly used control genes such as Gapdh and Actb significantly vary across the different testicular cell populations (e.g. see Fig. 5I), we chose Surf4 (Surfeit gene 4) as normalizing gene because it exhibited similar expression levels in the four cell populations in our RNAseq data (55.99 ± 2.91 FPKM in 2C; 59.91 ± 10.31 FPKM in LZ; 59.36 ± 5.76 FPKM in PS; 45.09 ± 4.38 FPKM in RS). The coding genes and their AS lncRNAs or neighbour lincRNAs selected for confirmation by RT-qPCR are shown in Fig. 5, and all especially designed primers are listed in Supplementary Table S6.
Amplification efficiency of the primers was >93%. We made relative expression quantification using the 2−ΔΔCt method, and 2C as calibrator condition.
Stellaris® RNA fluorescence in situ hybridization
We designed probe sets using the Stellaris® FISH probe designer (https://www.biosearchtech.com/support/tools/design-software/stellaris-probe-designer), and had them synthesized (Biosearch Technologies) as follows: probes against Kcnmb4 were conjugated to Quasar570, while probes against Kcnmb4os1 were conjugated to Quasar670. Ready-to-use Malat1 probes conjugated to Quasar570 (SMF-3008-19) were purchased from the same company.
Testes of male mice at 25 dpp were cut in halves, fixed in 4% paraformaldehyde in PHEM buffer (60 mM PIPES pH 7.4, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2) for 1 h at room temperature, and cryoprotected as previously described [87]. Sections of 10 µm in thickness were obtained, transferred to poly-L-lysine-coated slides, and kept at −20°C.
In situ hybridization was performed following the protocols as indicated by the manufacturer for ‘frozen tissues’ [65], and using the commercial buffers from the company.
For those cases where FISH and co-immunostaining were performed on the same sections, immunodetection was done before the in situ hybridization. Anti-MVH antibody (Abcam, ab13840) was used at 1:2,000 dilution, followed by incubation with secondary anti-rabbit Alexa 488 antibody (A-11034, Invitrogen) 1:1,000. We followed the protocol as suggested by Biosearch Technologies for ‘sequential IF’. All the protocols are in Biosearch Technologies web page (https://www.biosearchtech.com/support/resources/stellaris-protocols).
The sections were examined under a Zeiss LSM 800 confocal microscope, and photographed with Axiocam 506 colour digital camera (Carl Zeiss Microscopy, Germany). Overlapping between sense and AS probes was analysed by using Colocalization Finder plugin in Fiji ImageJ.
Funding Statement
This work was supported by Agencia Nacional de Investigación e Innovación (ANII, Uruguay) under grant FCE-1-2014-1-104251 to AG (including a PhD scholarship to MFT), and Comisión Sectorial de Investigación Científica (CSIC), UDELAR (Uruguay), under an I+D Groups grant to AG and RB. MFT was awarded with a Boehringer Ingelheim Fonds travel grant, and a short-term research grant from DAAD. Flow Cytometry purifications with Astrios EQ were carried out under ANII grant PEC_1_2016_1_133123.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Romrell LJ, Bellvé AR, Fawcet DW.. Separation of mouse spermatogenic cells by sedimentation velocity. Dev Biol. 1976;19:119–131. [DOI] [PubMed] [Google Scholar]
- [2].Meistrich ML. Separation of spermatogenic cells and nuclei from rodent testes. Methods Cell Biol. 1977;15:15–54. [DOI] [PubMed] [Google Scholar]
- [3].Gaysinskaya V, Bortvin A. Flow cytometry of murine spermatocytes. Curr Protoc Cytom. 2015;72:7.44.1–7.44.24. [DOI] [PubMed] [Google Scholar]
- [4].Da Cruz I, Rodríguez-Casuriaga R, Santiñaque FF, et al. Transcriptome analysis of highly purified mouse spermatogenic cell populations: gene expression signatures switch from meiotic-to-postmeiotic-related processes at pachytene stage. BMC Genomics. 2016;17(294). DOI: 10.1186/s12864-016-2618-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bolcun-Filas E, Handel MA. Meiosis: the chromosomal foundation of reproduction. Biol Reprod. 2018;99:112–126. [DOI] [PubMed] [Google Scholar]
- [6].Kleene KC. A possible meiotic function of the peculiar patterns of gene expression in mammalian spermatogenic cells. Mech Dev. 2001;106:3–23. [DOI] [PubMed] [Google Scholar]
- [7].Geisinger A. Spermatogenesis in mammals: a very peculiar cell differentiation process. In Cell Differentiation Research Developments. Ivanova LB, ed. New York, NY: Nova Publishers; 2008. p. 97–123. [Google Scholar]
- [8].Gan H, Cai T, Lin X, et al. Integrative proteomic and transcriptomic analyses reveal multiple post-transcriptional regulatory mechanisms of mouse spermatogenesis. Mol Cell Proteomics. 2013;12:1144–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Meikar O, Vagin VV, Chalmel F, et al. An atlas of chromatoid body components. RNA. 2014;20:483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Soumillon M, Necsulea A, Weier M, et al. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep. 2013;3:2179–2190. [DOI] [PubMed] [Google Scholar]
- [11].Atkinson SR, Marguerat S, Bähler J. Exploring long non-coding RNAs through sequencing. Semin Cell Dev Biol. 2012;23:200–205. [DOI] [PubMed] [Google Scholar]
- [12].Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;8:145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Amaral PP, Clark MB, Gascoigne DK, et al. LncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 2011;39:D146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li LJ, Leng RX, Fan YG, et al. Translation of noncoding RNAs: focus on lncRNAs, pri-miRNAs, and circRNAs. Exp Cell Res. 2017;361:1–8. [DOI] [PubMed] [Google Scholar]
- [15].Ginger MR, Shore AN, Contreras A, et al. A noncoding RNA is a potential marker of cell fate during mammary gland development. Proc Natl Acad Sci. 2006;103:5781–5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mehler MF, Mattick JS. Noncoding RNAs and RNA editing in brain development, functional diversification, and neurological disease. Physiol Rev. 2007;87:799–823. [DOI] [PubMed] [Google Scholar]
- [17].Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lagarde J, Uszczynska-Ratajczak B, Carbonell S, et al. High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing. Nat Genet. 2017;49:1731–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wilhelm BT, Marguerat S, Watt S, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–1243. [DOI] [PubMed] [Google Scholar]
- [20].Cabili MN, Trapnell C, Goff L, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Necsulea A, Soumillon M, Warnefors M, et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014;505:635–640. [DOI] [PubMed] [Google Scholar]
- [22].Ma L, Bajic VB, Zhang Z. On the classification of long non-coding RNAs. RNA Biol. 2013;10:925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Guenzl PM, Barlow DP. Macro lncRNAs: a new layer of cis-regulatory information in the mammalian genome. RNA Biol. 2012;9:731–741. [DOI] [PubMed] [Google Scholar]
- [24].Carninci P. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. [DOI] [PubMed] [Google Scholar]
- [25].Hon CC, Ramilowski JA, Harshbarger J, et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature. 2017;543:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wu T, Du Y. LncRNAs: from basic research to medical application. Int J Biol Sci. 2017;13:295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Barman P, Reddy D, Bhaumik SR. Mechanisms of antisense transcription initiation with implications in gene expression, genomic integrity and disease pathogenesis. Noncoding RNA. 2019;5:E11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hong SH, Kwon JT, Kim J, et al. Profiling of testis-specific long noncoding RNAs in mice. BMC Genomics. 2018;19(1). DOI: 10.1186/s12864-018-4931-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chalmel F, Lardenois A, Evrard B, et al. High-resolution profiling of novel transcribed regions during rat spermatogenesis. Biol Reprod. 2014;91:5. [DOI] [PubMed] [Google Scholar]
- [31].Ran M, Chen B, Li Z, et al. Systematic identification of long noncoding RNAs in immature and mature porcine testes. Biol Reprod. 2016;94:77. [DOI] [PubMed] [Google Scholar]
- [32].Jan SZ, Vormer TL, Jongejan A, et al. Unraveling transcriptome dynamics in human spermatogenesis. Development. 2017;144:3659–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Luk AC, Chan WY, Rennert OM, et al. Long noncoding RNAs in spermatogenesis: insights from recent high-throughput transcriptome studies. Reproduction. 2014;147:R131–41. [DOI] [PubMed] [Google Scholar]
- [34].Nakajima R, Sato T, Ogawa T, et al. A noncoding RNA containing a SINE-B1 motif associates with meiotic metaphase chromatin and has an indispensable function during spermatogenesis. PLoS One. 2017;12:e0179585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bao J, Wu J, Schuster AS, et al. Expression profiling reveals developmentally regulated lncRNA repertoire in the mouse male germline. Biol Reprod. 2013;89:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liang M, Li W, Tian H, et al. Sequential expression of long noncoding RNA as mRNA gene expression in specific stages of mouse spermatogenesis. Sci Rep. 2014;4:5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Laiho A, Kotaja N, Gyenesei A, et al. Transcriptome profiling of the murine testis during the first wave of spermatogenesis. PLoS One. 2013;8:e61558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lin X, Han M, Cheng L, et al. Expression dynamics, relationships, and transcriptional regulations of diverse transcripts in mouse spermatogenic cells. RNA Biol. 2016;13:1011–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wichman L, Somasundaram S, Breindel C, et al. Dynamic expression of long noncoding RNAs reveals their potential roles in spermatogenesis and fertility. Biol Reprod. 2017;97:313–323. [DOI] [PubMed] [Google Scholar]
- [40].Pertea M, Kim D, Pertea GM, et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11:1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen Y, Zheng Y, Gao Y, et al. Single-cell RNA-seq uncovers dynamic processes and critical regulators in mouse spermatogenesis. Cell Res. 2018;28:879–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rodríguez-Casuriaga R, Santiñaque FF, Folle GA, et al. Rapid preparation of rodent testicular cell suspensions and spermatogenic stages purification by flow cytometry using a novel blue-laser-excitable vital dye. MethodsX. 2014;1:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Geisinger A, Rodríguez-Casuriaga R. Flow cytometry for the isolation and characterization of rodent meiocytes. Methods Molec Biol. 2017;1471:217–230. [DOI] [PubMed] [Google Scholar]
- [44].Ilott NE, Ponting CP. Predicting long non-coding RNAs using RNA sequencing. Methods. 2013;63:50–59. [DOI] [PubMed] [Google Scholar]
- [45].Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet. 2013;14:880–893. [DOI] [PubMed] [Google Scholar]
- [46].Khalil AM, Guttman M, Huarte M, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009;106:11667–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ørom UA, Derrien T, Beringer M, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang Y, Xue S, Liu X, et al. Analyses of long non-coding RNA and mRNA profiling using RNA sequencing during the pre-implantation phases in pig endometrium. Sci Rep. 2016;6:20238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liu Y, Sun Y, Li Y, et al. Analyses of long non-coding RNA and mRNA profiling using RNA sequencing in chicken testis with extreme sperm motility. Sci Rep. 2017;22:9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ulitsky I, Bartel DP. LincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Breschi A, Gingeras TR, Guigó R. Comparative transcriptomics in human and mouse. Nat Rev Genet. 2017;18:425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Roux BT, Heward JA, Donnelly LE, et al. Catalog of differentially expressed long non-coding RNA following activation of human and mouse innate immune response. Front Immunol. 2017;8:1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hezroni H, Koppstein D, Schwartz M, et al. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015;11:1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sun R, Shen R, Li J, et al. Lyzl4, a novel mouse sperm-related protein, is involved in fertilization. Acta Biochim Biophys Sin (Shanghai). 2011;43:346–353. [DOI] [PubMed] [Google Scholar]
- [55].Zhao J, Zhao J, Xu G, et al. Deletion of Spata2 by CRISPR/Cas9n causes increased inhibin alpha expression and attenuated fertility in male mice. Biol Reprod. 2017;97:497–513. [DOI] [PubMed] [Google Scholar]
- [56].Lin RY, Moss SB, Rubin CS. Characterization of S-AKAP84, a novel developmentally regulated A kinase anchor protein of male germ cells. J Biol Chem. 1995;270:27804–27811. [DOI] [PubMed] [Google Scholar]
- [57].Andersen OM, Yeung CH, Vorum H, et al. Essential role of the apolipoprotein E receptor-2 in sperm development. J Biol Chem. 2003;278:23989–23995. [DOI] [PubMed] [Google Scholar]
- [58].Liang AJ, Wang GS, Ping P, et al. The expression of the new epididymal luminal protein of PDZ domain containing 1 is decreased in asthenozoospermia. Asian J Androl. 2018;20:154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yue F, Cheng Y, Breschi A, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang CC, Brodnicki T, Copeland NG, et al. Conserved linkage of NK-2 homeobox gene pairs Nkx2-2/2-4 and Nkx2-1/2-9 in mammals. Mamm Genome. 2000;11:466–468. [DOI] [PubMed] [Google Scholar]
- [61].Iwamori T, Lin YN, Ma L, et al. Identification and characterization of RBM44 as a novel intercellular bridge protein. PLoS One. 2011;6:e17066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fagerberg L, Hallström BM, Oksvold P, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Miki K, Qu W, Goulding EH, et al. Glyceraldehyde 3-phosphate dehydrogenase-S, a sperm-specific glycolytic enzyme, is required for sperm motility and male fertility. Proc Natl Acad Sci USA. 2004;101:16501–16506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Nayeri S, Sargolzaei M, Abo-Ismail MK, et al. Genome-wide association for milk production and female fertility traits in Canadian dairy Holstein cattle. BMC Genet. 2016;17:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Orjalo AV Jr, Johansson HE. Stellaris® RNA fluorescence in situ hybridization for the simultaneous detection of immature and mature long noncoding RNAs in adherent cells. Methods Mol Biol. 2016;1402:119–134. [DOI] [PubMed] [Google Scholar]
- [66].Weiger TM, Holmqvist MH, Levitan IB, et al. A novel nervous system beta subunit that downregulates human large conductance calcium-dependent potassium channels. J Neurosci. 2000;20:3563–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].West JA, Davis C, Sunwoo H, et al. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell. 2014;55:791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Conesa A. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Guil S, Esteller M. Cis-acting noncoding RNAs: friends and foes. Nat Struct Mol Biol. 2012;19:1068–1075. [DOI] [PubMed] [Google Scholar]
- [71].Liu Z, Zhao P, Han Y, et al. lncRNA FEZF1-AS1 is associated with prognosis in lung adenocarcinoma and promotes cell proliferation, migration, and invasion. Oncol Res. 2018;27:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Yung Y, Ophir L, Yerushalmi GM, et al. HAS2-AS1 is a novel LH/hCG target gene regulating HAS2 expression and enhancing cumulus cells migration. J Ovarian Res. 2019;12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chowdhury TA, Kleene KC. Identification of potential regulatory elements in the 5ʹ and 3ʹ UTRs of 12 translationally regulated mRNAs in mammalian spermatids by comparative genomics. J Androl. 2012;33:244–256. [DOI] [PubMed] [Google Scholar]
- [74].Lee K, Haugen HS, Clegg CH, et al. Premature translation of protamine 1 mRNA causes precocious nuclear condensation and arrests spermatid differentiation in mice. Proc Natl Acad Sci USA. 1995;92(26):12451–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tseden K, Topaloglu Ö, Meinhardt A, et al. Premature translation of transition protein 2 mRNA causes sperm abnormalities and male infertility. Mol Reprod Dev. 2007;74:273–279. [DOI] [PubMed] [Google Scholar]
- [76].Faghihi MA. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010;11:R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Lehtiniemi T, Kotaja N. Germ granule-mediated RNA regulation in male germ cells. Reproduction. 2018;155:R77–R91. [DOI] [PubMed] [Google Scholar]
- [78].Shukla KK, Mahdi AA, Rajender S. Ion channels in sperm physiology and male fertility and infertility. J Androl. 2012;33:777–788. [DOI] [PubMed] [Google Scholar]
- [79].Yang CT, Zeng XH, Xia XM, et al. Interactions between beta subunits of the KCNMB family and Slo3: beta4 selectively modulates Slo3 expression and function. PLoS One. 2009;4:e6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. [DOI] [PubMed] [Google Scholar]
- [81].Rodríguez-Casuriaga R, Folle GA, Santiñaque F, et al. Simple and efficient technique for the preparation of testicular cell suspensions. J Vis Exp. 2013;78. DOI: 10.3791/50102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Rodríguez-Casuriaga R, Geisinger A, Santiñaque FF, et al. High-purity flow sorting of early meiocytes based on DNA analysis of guinea pig spermatogenic cells. Cytometry A. 2011;79:625–634. [DOI] [PubMed] [Google Scholar]
- [83].Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Pertea M, Pertea GM, Antonescu CM, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Frazee AC, Pertea G, Jaffe AE, et al. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol. 2015;33:243–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Pertea M, Shumate A, Pertea G, et al. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 2018;19:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Capoano CA, Wettstein R, Kun A, et al. Spats 1 (Srsp1) is differentially expressed during testis development of the rat. Gene Expr Patterns. 2010;10:1–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.