

ABSTRACT
Optic perineuritis can be a manifestation of infectious and systemic inflammatory disorders, but the majority of cases are idiopathic. Myelin oligodendrocyte glycoprotein (MOG)-IgG-positive optic neuritis has been reported to be associated with optic nerve sheath enhancement. This report describes two MOG-IgG patients with clinical, radiological and therapeutic response consistent with optic perineuritis. MOG-IgG may account for many cases of previously described idiopathic optic perineuritis. Vision loss with optic nerve sheath enhancement on MRI should prompt testing for MOG-IgG.
KEYWORDS: Optic perineuritis, myelin oligodendrocyte glycoprotein (MOG), aquaporin-4 (AQP4), optic neuritis, optic nerve sheath
Introduction
Optic perineuritis (OPN) is an uncommon inflammatory disorder affecting the optic nerve sheath.1 OPN can be a manifestation of infectious and systemic inflammatory disorders, such as syphilis and sarcoidosis, but in most cases remains idiopathic.1,2 The clinical presentation is similar to demyelinating optic neuritis and characterised by vision loss and pain on eye movements, but patients with OPN more commonly have optic disc oedema, sparing of their central vision and peripheral visual field loss. The diagnosis is established by magnetic resonance imaging (MRI) with the demonstration of optic nerve sheath enhancement with sparing of the optic nerve itself.1,2 Differentiating OPN from demyelinating optic neuritis is important as it has therapeutic and prognostic implications. OPN is not associated with multiple sclerosis, is often exquisitely sensitive to corticosteroids, and can be steroid dependent.1
Autoantibodies targeting neural proteins such as aquaporin 4 (AQP4) and myelin oligodendrocyte glycoprotein (MOG) are established biomarkers of autoimmune optic neuritis.3 Most reports of optic perineuritis precede the contemporary antibody testing practice; thus, the frequency of detection of AQP4 IgG and MOG-IgG in “idiopathic” OPN cases is unknown. In this case series, we describe two patients with MOG-IgG positive OPN.
Methods
The study protocol was approved by the Mayo Clinic IRB (08–006647). AQP4-IgG and MOG-IgG1 serostatus were determined using a validated flow cytometry assay utilizing live M1-AQP4-transfected and full-length MOG-transfected HEK293 cells as previously described.4 The clinical manifestations and paraclinical findings (MRI and optical coherence tomography [OCT]) were reviewed.
Results
Patient 1
A 49-year-old male suffered an episode of acute disseminated encephalomyelitis (ADEM). Workup revealed that he was MOG-IgG positive at the onset with a titre of 1:100. AQP4-IgG was negative. He was treated with intravenous methylprednisolone (IVMP) and plasma exchange (PLEX) with full recovery, then started on a prednisone taper. Four months later, while on 15 mg/day of prednisone, he developed prominent bilateral pain on eye movements. Visual acuity was 20/20 OU with normal colour vision. There was subtle optic disc elevation without pallor in both eyes. Automated perimetry showed mild enlargement of the blind spots bilaterally with mild peripheral depression in the right eye (Figure 1c). OCT showed a mildly thickened retinal nerve fibre layer (RNFL) of 133 µm OD and 113 µm OS. Visual evoked potentials showed mild delayed latencies in both eyes. MRI demonstrated optic nerve sheath enhancement bilaterally with sparing of the optic nerve consistent with bilateral OPN (Figure 1a,b). Extensive serological testing for infectious, rheumatological and other inflammatory disorders was negative.
Figure 1.
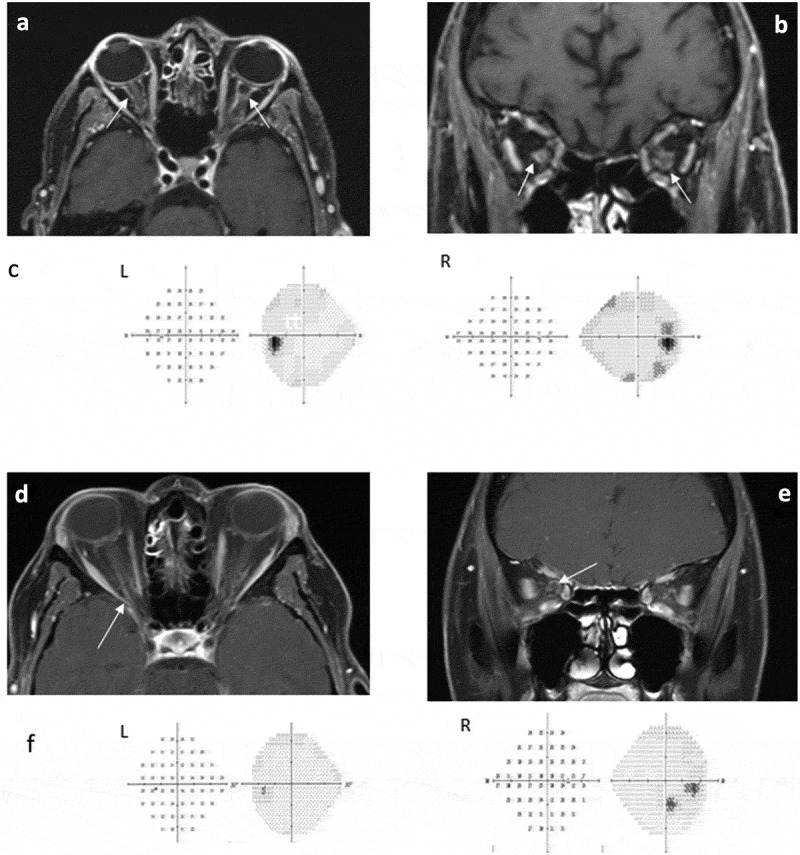
Patient 1: A, Axial and B, Coronal T1-weighted magnetic resonance imaging showing bilateral optic nerve sheath enhancement (arrows); C, Visual fields demonstrating mild enlargement of the blind spots OU with mild peripheral depression OD. Patient 2. D, Axial and E, Coronal T1-weighted images show optic nerve sheath enhancement in the right eye (arrow); F, Visual fields show a paracentral scotoma OD, and normal OS.
His visual function and optic disc oedema resolved with IVMP followed by oral prednisone. Nine months later, there was a recurrence of the peripheral visual loss that resolved with an increase in the dose of the oral steroids and initiation of azathioprine (200mg/day). Another nine months later, a second recurrence occurred leading to an increase of the azathioprine to 300mg/day. The patient has remained stable without recurrence over the past 18 months and remains with a visual acuity of 20/20 OU with normal automated perimetry.
Patient 2
A 53-year-old woman presented with a two-day history of visual loss in the right eye described as a “spot” in her vision. Her visual acuity was 20/25 OD and 20/20 OS with normal colour vision. Dilated fundus examination showed grade 2 optic disc swelling OD and a normal appearing optic disc OS. Automated visual fields showed a paracentral scotoma OD but were full OS (Figure 1f). On OCT the RNFL was slightly thickened at 131 µm OD with a normal RNFL OS at 80 µm. Orbital MRI showed perineural enhancement of the right optic nerve with some loss of signal within the nerve sheath on T2 weighted images (Figure 1d,e). She was started on high dose oral prednisone with a taper over the next 6–8 weeks with complete resolution of symptoms. MOG-IgG was positive with a titre of 1:100 and AQP4-IgG was negative. Similar to patient 1, an extensive serological testing for infectious, rheumatological and other inflammatory disorders was negative.
She has been stable after 12 months, with no recurrent attacks. There is a small, residual paracentral scotoma OD with mild optic disc pallor. OCT performed 11 months after presentation showed mild, diffuse RNFL thinning OD, at 72 µm, with focal supero-temporal thinning. The OCT was a normal OS. Ganglion cell layer thickness was mildly decreased OD at 71 µm but the normal OS. Repeat MOG antibody testing performed 11 months after initial testing showed persistently positive titres at 1:40. Since she has been relapsed free, she is being observed without treatment.
Discussion
This report describes two patients with MOG-IgG positive OPN. Although MOG-IgG seropositive patients typically experience longitudinal involvement of the optic nerve, often associated with severe vision loss, our two patients demonstrated optic nerve sheath enhancement with sparing of the optic nerve itself. There are features of MOG-IgG involvement of the optic nerve that parallel some of the features of previously described idiopathic OPN. First, MOG-IgG associated ON is often very corticosteroid responsive and can be corticosteroid dependent, which is a common feature of idiopathic OPN. Second, MOG-IgG positive optic neuritis is often associated with optic disc oedema with up to 86% of cases having visible optic disc oedema, which is usually present in idiopathic OPN.1 Third, optic nerve sheath enhancement can be a prominent feature of MOG-IgG associated ON and is present in up to 50% of patients with MOG-IgG positive ON.5,6
Patients with OPN have been traditionally evaluated for infectious and inflammatory aetiologies, such as polyangiitis with granulomatosis, syphilis, and sarcoidosis. MOG-IgG may account for many cases of previously described idiopathic OPN given the shared characteristics between the two entities and therefore we propose that MOG-IgG should also be tested in patients with OPN. MOG-IgG testing in OPN could be helpful to confirm the diagnosis of an immune-mediated aetiology and can assist in decision-making regarding treatment and prognostication.
Funding Statement
Mayo Clinic foundation, Department of Laboratory Medicine and Pathology and Center for MS and Autoimmune Neurology.
Acknowledgements
The authors thank John Schmeling for technical support, Mary Curtis for secretarial assistance, and Jessica Sagan for study coordination.
Author contributions
Dr Lopez Chiriboga, Dr Van Stavern, and Dr Chen were involved in drafting and revising the manuscript for content, including medical writing for content, study concept and design, analysis and interpretation of data, acquisition of data and study supervision.
Revising the manuscript for content and analysis and interpretation of data: all authors.
Declaration of interest
S. Lopez, J. Fryer, G. Van Stavern and J.Chen have no disclosures.
E. P. Flanagan receives research support from Medimmune but has not received personal compensation.
M. T. Bhatti is a consultant for Celgene.
S.J. Pittock holds patents that relate to functional AQP4/NMO-IgG assays and NMO-IgG as a cancer marker; has a patent pending for GFAP as markers of neurological autoimmunity and paraneoplastic disorders; consulted for Alexion and Medimmune; and received research support from Grifols, Medimmune, and Alexion. All compensation for consulting activities is paid directly to Mayo Clinic.
References
- 1.Purvin V, Kawasaki A, Jacobson DM.. Optic perineuritis: clinical and radiographic features. Arch Ophthalmol. 2001;119(9):1299–1306. doi:. [DOI] [PubMed] [Google Scholar]
- 2.Bergman O, Andersson T, Zetterberg M. Optic perineuritis: a retrospective case series. Int Med Case Rep J. 2017;10:181–188. doi: 10.2147/IMCRJ.S125972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jitprapaikulsan J, Chen JJ, Flanagan EP, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein autoantibody status predict outcome of recurrent optic neuritis. Ophthalmology. 2018;125(10):1628–1637. doi: 10.1016/j.ophtha.2018.03.041. [DOI] [PubMed] [Google Scholar]
- 4.López-Chiriboga AS, Majed M, Fryer J, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG–associated disorders. JAMA Neurol. July 2018;75:1355. doi: 10.1001/jamaneurol.2018.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. Myelin oligodendrocyte glycoprotein antibody–positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8–15. doi: 10.1016/j.ajo.2018.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramanathan S, Reddel SW, Henderson A, et al. Dale and Fabienne Brilot. Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol - Neuroimmunol Neuroinflammation. 2014;1(4):e40. doi: 10.1212/NXI.0000000000000040 [DOI] [PMC free article] [PubMed] [Google Scholar]