

ABSTRACT
Particulate matter (PM) is able to induce airway epithelial injury, while the detailed mechanisms remain unclear. Here we demonstrated that PM exposure inactivated MTOR (mechanistic target of rapamycin kinase), enhanced macroautophagy/autophagy, and impaired lysosomal activity in HBE (human bronchial epithelial) cells and in mouse airway epithelium. Genetic or pharmaceutical inhibition of MTOR significantly enhanced, while inhibition of autophagy attenuated, PM-induced IL6 expression in HBE cells. Consistently, club-cell-specific deletion of Mtor aggravated, whereas loss of Atg5 in bronchial epithelium reduced, PM-induced airway inflammation. Interestingly, the augmented inflammatory responses caused by MTOR deficiency were markedly attenuated by blockage of downstream autophagy both in vitro and in vivo. Mechanistically, the dysregulation of MTOR-autophagy signaling was partially dependent on activation of upstream TSC2, and interacted with the TLR4-MYD88 to orchestrate the downstream NFKB activity and to regulate the production of inflammatory cytokines in airway epithelium. Moreover, inhibition of autophagy reduced the expression of EPS15 and the subsequent endocytosis of PM. Taken together, the present study provides a mechanistic explanation for how airway epithelium localized MTOR-autophagy axis regulates PM-induced airway injury, suggesting that activation of MTOR and/or suppression of autophagy in local airway might be effective therapeutic strategies for PM-related airway disorders.
Abbreviations: ACTB: actin beta; AKT: AKT serine/threonine kinase; ALI: air liquid interface; AP2: adaptor related protein complex 2; ATG: autophagy related; BALF: bronchoalveolar lavage fluid; COPD: chronic obstructive pulmonary disease; CXCL: C-X-C motif chemokine ligand; DOX: doxycycline; EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; EPS15: epidermal growth factor receptor pathway substrate 15; HBE: human bronchial epithelial; H&E: hematoxylin & eosin; IKK: IKB kinase; IL: interleukin; LAMP2: lysosomal-associated membrane protein 2; LPS: lipopolysaccharide; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; MTEC: mouse tracheal epithelial cells; MTOR: mechanistic target of rapamycin kinase; MYD88: MYD88 innate immune signal transduction adaptor; NFKB: nuclear factor of kappa B; NFKBIA: NFKB inhibitor alpha; PM: particulate matter; PtdIns3K: phosphatidylinositol 3-kinase; Rapa: rapamycin; RELA: RELA proto-oncogene, NFKB subunit; SCGB1A1: secretoglobin family 1A member 1; siRNA: small interfering RNAs; SQSTM1: sequestosome 1; TEM: transmission electronic microscopy; TLR4: toll like receptor 4; TSC2: TSC complex subunit 2.
KEYWORDS: Airway epithelial injury, airway inflammation, autophagy, MTOR, particulate matter
Introduction
Air pollution is a worldwide problem affecting human health. Accumulating epidemiological and clinical studies show that exposure to air pollution, in particular airborne particulate matter (PM), increases morbidity and mortality for respiratory diseases, such as asthma, chronic obstructive pulmonary disease (COPD), and lung cancer [1,2]. Thus, it is of great importance to explore the potential molecular mechanisms by which air pollutants trigger airway injury, in order to propose new effective therapeutic strategies. Recently, several studies have already demonstrated that PM is able to deposit in the respiratory tract or come into alveoli, consequently induces impairment of airway epithelial barrier, oxidative stress, autophagy, DNA damage, and genomic instability [3,4]. However, the detailed molecular mechanisms mediating the adverse effects of PM remain to be further investigated.
MTOR (mechanistic target of rapamycin kinase) is a serine-threonine protein kinase of the phosphatidylinositol 3-kinase (PtdIns3K)-related family. Extensive studies have established a dominant role for MTOR in regulating cellular growth and metabolism in response to growth factors and nutrients, and reveal that MTOR signaling pathway is implicated in the progression of cancer, obesity, type 2 diabetes, as well as the aging process [5]. Due to different sensitivities to rapamycin, MTOR functions in two distinct complexes named MTOR complex 1 and MTOR complex 2. Generally, MTOR complex 1 is considered as the master regulator of autophagy [5,6]. Accumulating evidence suggests that MTOR and autophagy play critical roles in pulmonary diseases [7–10]. Our previous study has demonstrated that autophagy is essential for environmental ultrafine PM-induced inflammation and mucus hyperproduction in airway epithelial cells [11], and emerging studies further suggest that PM induces autophagy via inhibition of PtdIns3K-AKT (AKT serine/threonine kinase)-MTOR signaling pathway in human bronchial epithelial cells [12], macrophages [13], or endothelial cells [14]. However, to the best of our knowledge, several critical issues remain unclear. What is the main function of MTOR in PM-induced damage in vitro and in vivo? Whether the function of MTOR in PM-induced airway inflammation is dependent on the downstream autophagy? What are the underlying mechanisms by which MTOR-autophagy interrelate in PM-induced airway inflammation?
Here, through genetic and pharmaceutical approaches to inhibit MTOR in vitro, and using mice with specific knockdown of Mtor in club cells, we demonstrate that Mtor deletion exacerbates airway inflammation induced by intratracheal PM instillation, and the suppressive effect of MTOR is autophagy-dependent. We also clarify that PM inactivates MTOR and induces autophagy in airway epithelial cells via TSC2 (TSC complex subunit 2) pathway. The MTOR-autophagy axis and TLR4 (toll like receptor 4)-MYD88 (MYD88 innate immune signal transduction adaptor) pathway interacts with each other to orchestrate the PM-induced inflammatory responses via NFKB (nuclear factor of kappa B) signaling, and autophagy modulates the PM endocytosis likely via EPS15 (epidermal growth factor receptor pathway substrate 15).
Results
PM exposure inactivates MTOR, enhances autophagy, and impairs lysosomal activity in human bronchial epithelial (HBE) cells and in mouse airway epithelium
In our previous study [11], we have demonstrated that ultrafine PM triggered a typical convergence of endocytosis and autophagy in HBE cells. Since MTOR is the major negative regulator of autophagy, we examined whether the expression of MTOR is modulated by PM in vivo and in vitro. As expected, treatment of HBE cells with PM time- and dose-dependently decreased the levels of p-MTOR and p-RPS6, while induced the MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta)-II expression (Figure 1A to D). In the primary mouse tracheal epithelial cells cultured in an air-liquid interface (ALI) system, PM also significantly inactivated MTOR, as revealed by the level of p-RPS6 (Figure 1E and F). Further analysis demonstrated that the MTOR was decreased in both SCGB1A1 (secretoglobin family 1A member 1)-positive and -negative cells (Figure 1G–I). In a mouse model of PM-induced airway injury, immunofluorescence analysis demonstrated that the MTOR activity was declined predominantly in airway epithelium, especially in the SCGB1A1-positive cells (Figure 1J and K). Furthermore, we found an elevation of autophagic vacuoles in epithelial cells of relatively large airways by transmission electron microscopy (TEM) (Figure 1L and M). Taken together, these results indicated that PM suppressed the MTOR activity and induced autophagy in HBE cells and in mouse airway epithelial cells, including both SCGB1A1-positive and -negative cells.
Figure 1.
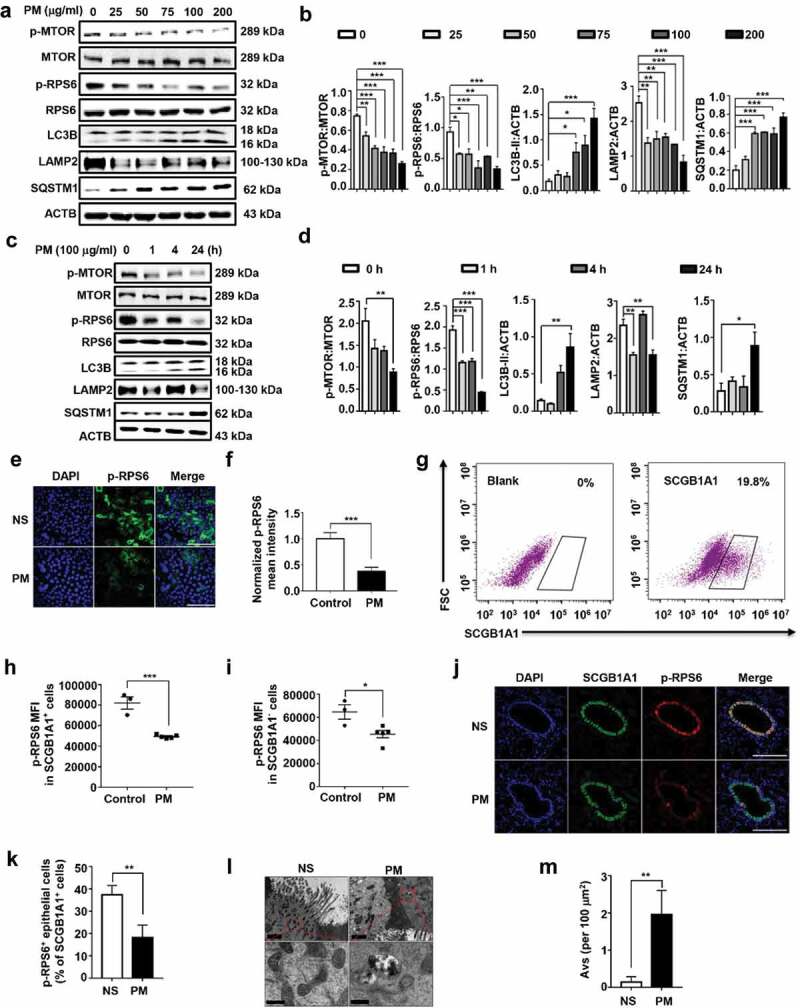
PM exposure causes MTOR inactivation and induces autophagy in HBE cells and in mouse airway epithelium. (A and B) Representative immunoblots (A) and semi-quantification (B) of dose-dependent levels of p-MTOR, MTOR, p-RPS6, RPS6, LC3B, LAMP2, and SQSTM1 in HBE cells exposed to PM for 24 h. (C and D) Representative immunoblots (C) and semi-quantification (D) of time-dependent expression of indicated proteins in HBE cells exposed to PM at 100 μg/ml. Blots from 3–5 independent experiments were quantified by ImageJ software. (E to I) MTECs were differentiated in an air-liquid interface culture system. After well differentiation, cells were treated with PM at 100 μg/ml for 24 h. (E and F) Representative images (n = 10–14 images for each group) of p-RPS6 staining (E) and semi-quantification (F) in PM-treated MTECs. Scale bar: 60 μm. (G) Representative images of SCGB1A1 staining in MTECs. (H and I) PM exposure decreased the levels of p-RPS6 in both SCGB1A1-positive (H) and -negative (I) MTECs. (J and K) Representative images (n = 10–14 images for each group) (J) and semi-quantification (K) of p-RPS6 in mouse airway epithelial cells exposed to PM. Lung sections stained for SCGB1A1 or p-RPS6 24 h after last PM exposure. Scale bar: 150 μm. (L and M) TEM images (n = 5–8 images for each group) (L) and semi-quantified level (M) of autophagic vacuoles in isolated mouse bronchus. Scale bar: upper panels, 2 μm; lower panels, 0.4 μm. Error bars, mean ± SEM. Differences between two groups were identified using the Student t-test (F, H, I, K, and M) and multiple groups using one-way ANOVA (B and D). *P < 0.05, **P < 0.01, ***P < 0.001.
Notably, PM treatment also decreased the expression of LAMP2 (lysosomal-associated membrane protein 2) while increased the levels of SQSTM1 (sequestosome 1) (Figure 1A–D), suggesting that PM also impaired the lysosomal activity. Degradation of EGFR (epidermal growth factor receptor) has been proved to proceed specifically in lysosomes [15]. In HBE cells, EGFR localized on the surface of cells at basal conditions and EGF treatment induced EGFR internalization and degradation. Interestingly, EGFR degradation was suppressed in PM-treated cells (Figure S1A and B). Western blot analysis further demonstrated that the degradation of EGFR was blocked in PM-treated cells (Figure S1C). Moreover, we monitored the autophagy flux by using RFP-GFP-LC3 plasmid and found that there were more yellow dots (autophagosomes) than red dots (autolysosomes) in PM-treated cells (Figure S1D). Taken together, these data suggested that PM impaired lysosomal activity.
Blockade of MTOR signaling significantly augments PM-induced production of inflammatory cytokines in airway epithelial cells
We have demonstrated that autophagy is required for PM-induced expression of inflammatory cytokines in HBE cells and is essential for PM-induced airway inflammation in vivo [11]. To determine whether reduced MTOR activity was associated with PM-induced inflammatory responses, we used MTOR small interfering RNAs (siRNA). The knockdown effects of all siRNA used in this study were displayed in Figure S2. As shown in Figure 2A and D, PM exposure induced a notable increase of IL6 (Interleukin 6) expression in HBE cells, and MTOR knockdown further enhanced the IL6 production. However, knockdown of MTOR failed to affect the PM-induced IL8 expression (Figure S3A). To further confirm the function of MTOR on PM-induced expression of IL6 and IL8, two widely used MTOR inhibitors, rapamycin (Rapa) and Torin 1, were used. Consistently, these compounds also significantly enhanced the PM-induced IL6 (Figure 2B,C,D,E, and F), while again exerted no considerable effect on IL8 production (Figure S3B and C). Interestingly, in the ALI culture of primary mouse tracheal epithelial cells, Torin1 also remarkably augmented the PM-induced expression of IL6, CXCL1 (C-X-C motif ligand 1), and CXCL2 (Figure 2G–K).
Figure 2.
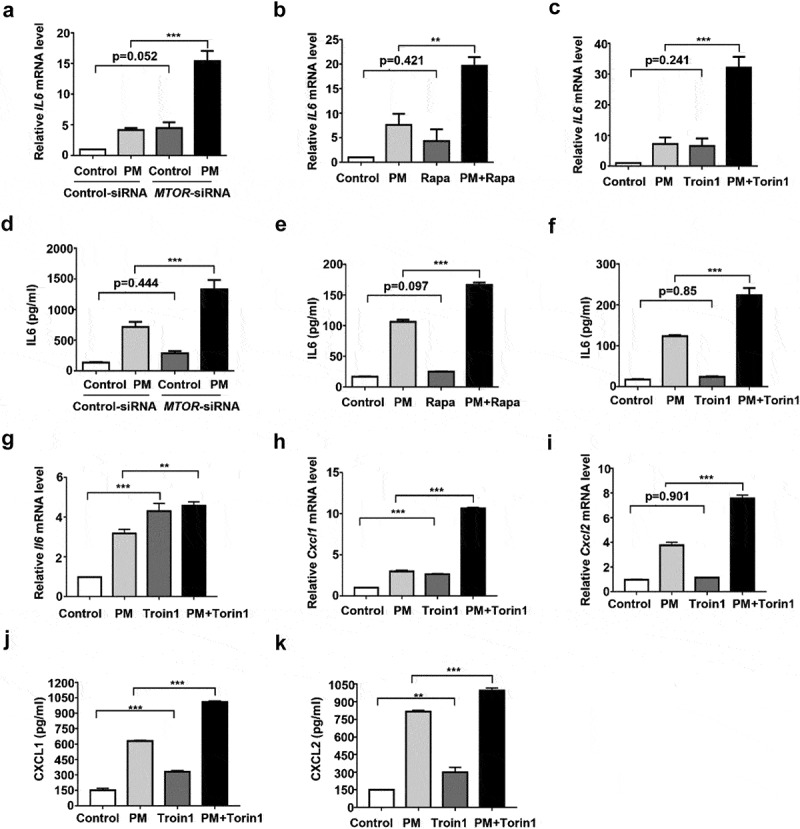
MTOR impairment enhances PM-induced IL6 expression in HBE cells. HBE cells were transfected with MTOR-siRNA (A and D) for 24 h, and then stimulated with PM (100 μg/ml) for an addition 24 h, or treated with Rapa (5 nM) (B and E) or Torin1 (250 nM) (C and F) together with PM (100 μg/ml) for 24 h. The relative mRNA levels of IL6 (A to C) were measured by quantitative real-time PCR, and the secretion of IL6 (D to F) in cell culture supernatants was determined by ELISA. (G to K) MTECs were differentiated in an air-liquid interface culture system. After well differentiation, cells were treated with Torin1 (250 nM) together with PM (100 μg/ml) for 24 h. The relative mRNA levels of Il6 (G), Cxcl1 (H), and Cxcl2 (I) were measured by quantitative real-time PCR, while the protein levels of CXCL1 (J) and CXCL2 (K) were detected by ELISA. Data are representative of 3–5 independent experiments. Error bars, mean ± SEM. Differences were identified using one-way ANOVA. **P < 0.01, ***P < 0.001.
Club-cell-specific deletion of MTOR aggravates PM-induced airway inflammation
Next, we sought to examine the effect of airway epithelial cell-localized MTOR in regulating PM-induced airway inflammation in vivo. To conditionally knockout the Mtor gene in club cells, Scgb1a1-rtTA/(tetO)7-Cre/mtorflox/flox mice were generated as described before [10], and were given doxycycline to induce Cre expression and Mtor deletion (mtor∆/∆). Knockdown efficiency of Mtor was determined by the decreased p-RPS6 phosphorylation in airway epithelial cells in mtor∆/∆ mice (Figure S4A to C).
To establish a mouse model of airway inflammation, mice were treated with 100 μg PM per day by intratracheal instillation for 4 days. Inflammation responses were revealed by remarkable accumulation of total inflammatory cells and neutrophils in BALF (bronchoalveolar lavage fluid), increased expression of inflammatory cytokines such as IL6, CXCL1, and CXCL2 both in lung and BALF supernatant, and elevated inflammation score by using immunohistological analysis. All of these were significantly aggravated in mtor∆/∆ mice in response to PM exposure (Figure 3A–J), demonstrating that club cell-localized MTOR suppresses PM-induced airway inflammation in vivo.
Figure 3.
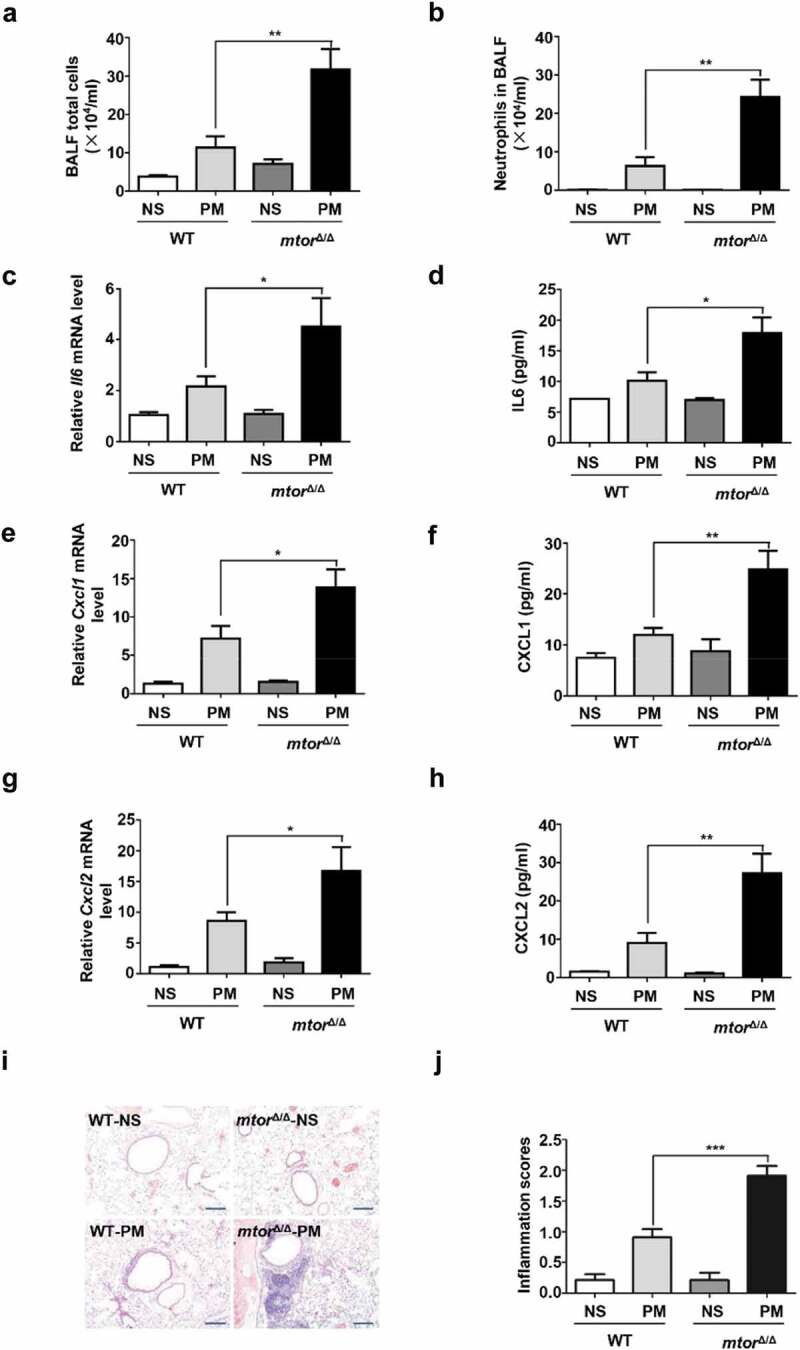
Selective disruption of MTOR in airway epithelial cells exacerbates PM-induced airway inflammation. mtor∆/∆ mice (defined as mice with specific knockdown of MTOR in club cells) and WT (control) littermates (n = 4 to 13 for each group) were exposed to PM at 100 μg/d for 4 days. 24 h after last PM exposure, the total inflammatory cells (A) and the number of neutrophils (B) in the BALF were assessed. The relative mRNA levels of Il6 (C) Cxcl1 (E), and Cxcl2 (G) in lung tissue were measured by quantitative PCR. Protein levels of IL6 (D), CXCL1 (F), and CXCL2 (H) in the BALF were detected by ELISA. (I and J) Representative images (I) and semi-quantification (J) of lung sections stained with H&E (n = 19–22 images for each group). Scale bar: 200 μm. Data are presented as mean ± SEM. Differences were identified using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001.
Inhibition of autophagy attenuated the IL6 production induced by PM exposure and MTOR deficiency in HBE cells
Next, we sought to determine whether the effect of MTOR on PM-induced IL6 production was dependent on the downstream autophagy. As expected, inhibition of MTOR by Rapa enhanced the PM-induced LC3B-II (Figure 4A and B). To examine the involvement of autophagy in PM-induced inflammatory responses, we pretreated HBE cells with ATG5 (autophagy related 5)-siRNA or spautin1 prior to PM stimulation. Consistently, ATG5-siRNA or spautin1 markedly reduced the expression of IL6 induced by PM exposure (Figure 4C–F). Moreover, spautin1 was able to rescue the exacerbated production of IL6 caused by MTOR knockdown in HBE cells (Figure 4G and H). These results supported the conclusion that autophagy mediated the suppressive effect of MTOR in PM-induced epithelial injury in HBE cells.
Figure 4.
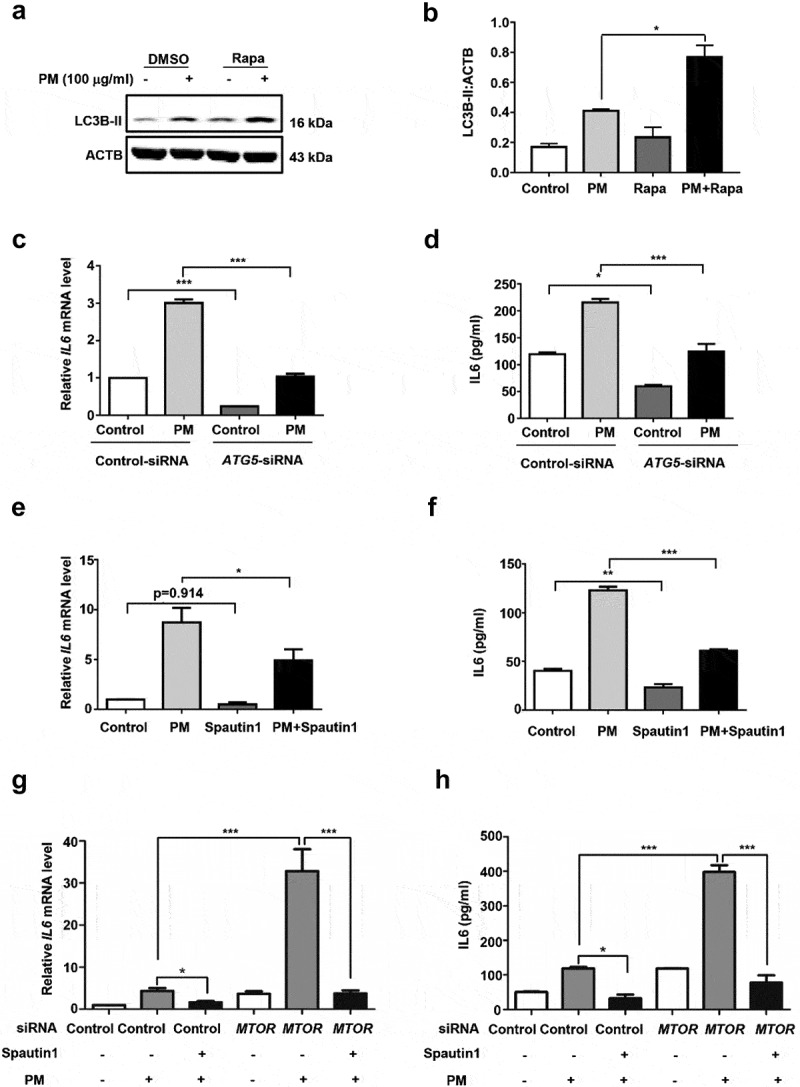
The role of MTOR in PM-induced IL6 production is dependent on the downstream signaling autophagy in HBE cells. (A) Cells were treated with Rapa, together with PM (100 μg/ml) for 10 h, then harvested for western blot analysis of LC3B. (B) Quantification of the immunoblots presented in Figure 4A, and blots from 3 independent experiments were quantified by ImageJ software. Cells were infected with indicated Control- or ATG5-siRNA (C and D) for 24 h, and then stimulated with PM (100 μg/ml) for an addition 24 h, or treated with or without spautin1 (E and F) together with PM (100 μg/ml) for 24 h. The relative mRNA level of IL6 (C and E) were measured by quantitative real-time PCR, and IL6 (D and F) in cell culture supernatants was detected by ELISA. (G and H) Cells were transfected with indicated Control- or MTOR-siRNA for 24 h, and then exposed to PM (100 μg/ml), co-treated with or without spautin1 for an addition 24 h to measure the mRNA levels (G) and protein levels (H) of IL6. Data are representative of 3–7 independent experiments. Error bars, mean ± SEM. Differences were identified using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001.
Club-cell-specific deletion of atg5 attenuates PM-induced airway inflammation
To investigate the function of club-cell-specific autophagy in PM-induced airway inflammation, Scgb1a1-rtTA/(tetO)7-Cre/atg5flox/flox mice were generated (Figure 5A). Knockdown efficiency of ATG5 was confirmed by the decreased immunofluorescence in airway epithelial cells and by declined protein expression in the lung tissues of atg5∆/∆ mice (Figure 5B–D). In contrast to the effects of Mtor deletion, club-cell-specific knockdown of Atg5 notably decreased the PM-induced inflammatory responses, including the inflammatory cells in BALF and around the airways, and the production of Il6 (Figure 5E–I).
Figure 5.
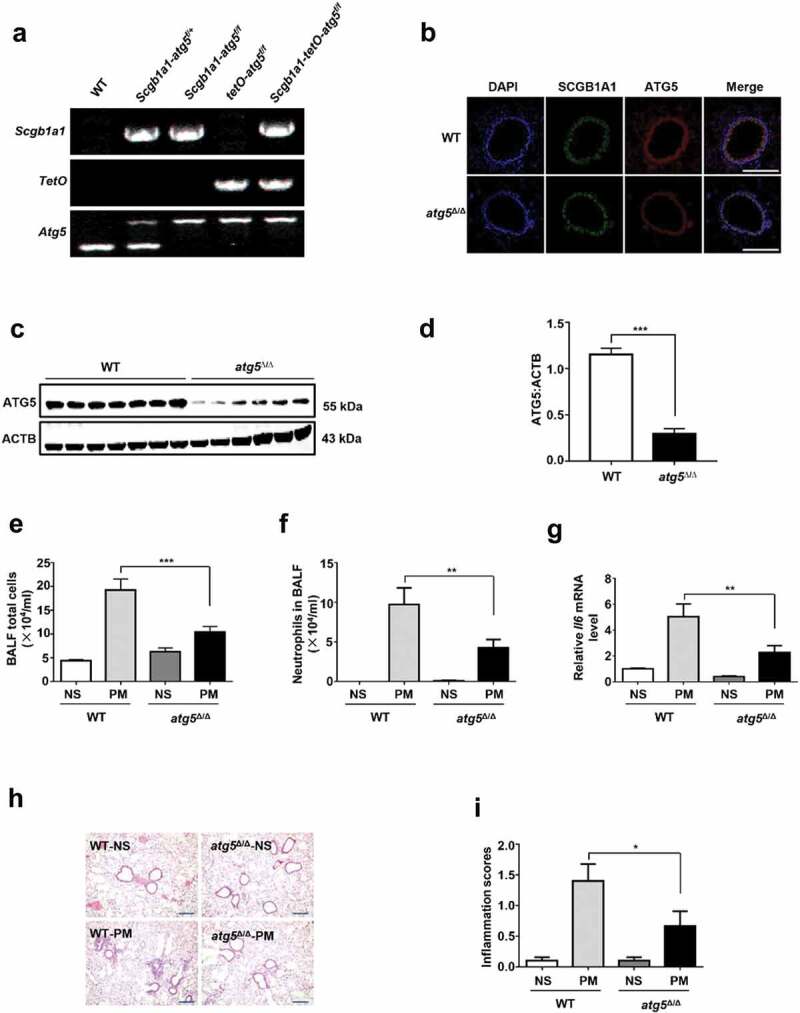
Club-cell-specific knockdown of ATG5 attenuates PM-induced airway inflammation. (A) Genotyping of atg5∆/∆ mice by PCR using genomic DNA from mouse tails. (B) Representative immunofluorescence images of mouse lung sections stained with SCGB1A1 (Green), ATG5 (red), and nuclei (DAPI, blue). Scale bar: 150 μm. (C and D) Representative immunoblots (C) and semi-quantification (D) of ATG5 in mouse lung tissues. (E–I) atg5∆/∆ mice (defined as mice with specific knockdown of ATG5 in club cells) and WT (control) littermates (n = 5 to 8 for each group) were exposed to PM at 100 μg/d for 4 days. 24 h after last PM exposure, the total inflammatory cells (E) and the number of neutrophils (F) in the BALF were assessed. The relative mRNA levels of Il6 (G) in lung tissue were measured by quantitative PCR. (H and I) Representative images (H) and semi-quantification (I) of lung sections stained with H&E (n = 20–30 images for each group). Scale bar: 200 μm. Data are presented as mean ± SEM of 3 independent experiments. Differences between two groups were identified using the Student t-test (D) and multiple groups using one-way ANOVA (E, F, G, and I). *P < 0.05, **P < 0.01, ***P < 0.001.
Lc3b deletion attenuates the aggravated inflammation caused by MTOR disruption in vivo
To explore whether the role of MTOR in regulation of airway inflammation is dependent on the downstream autophagy in vivo, lc3b−/− mice, which have been displayed impaired autophagy in lungs and other organs [16–18], were crossed with mtor∆/∆ to generate mtor∆/∆ lc3b−/− mice. Similar to our observations in HBE cells, when upon PM exposure, the aggravated inflammation caused by specific Mtor deletion in club cells was also remarkably attenuated in the mtor∆/∆lc3b−/− mice (Figure 6A to J).
Figure 6.
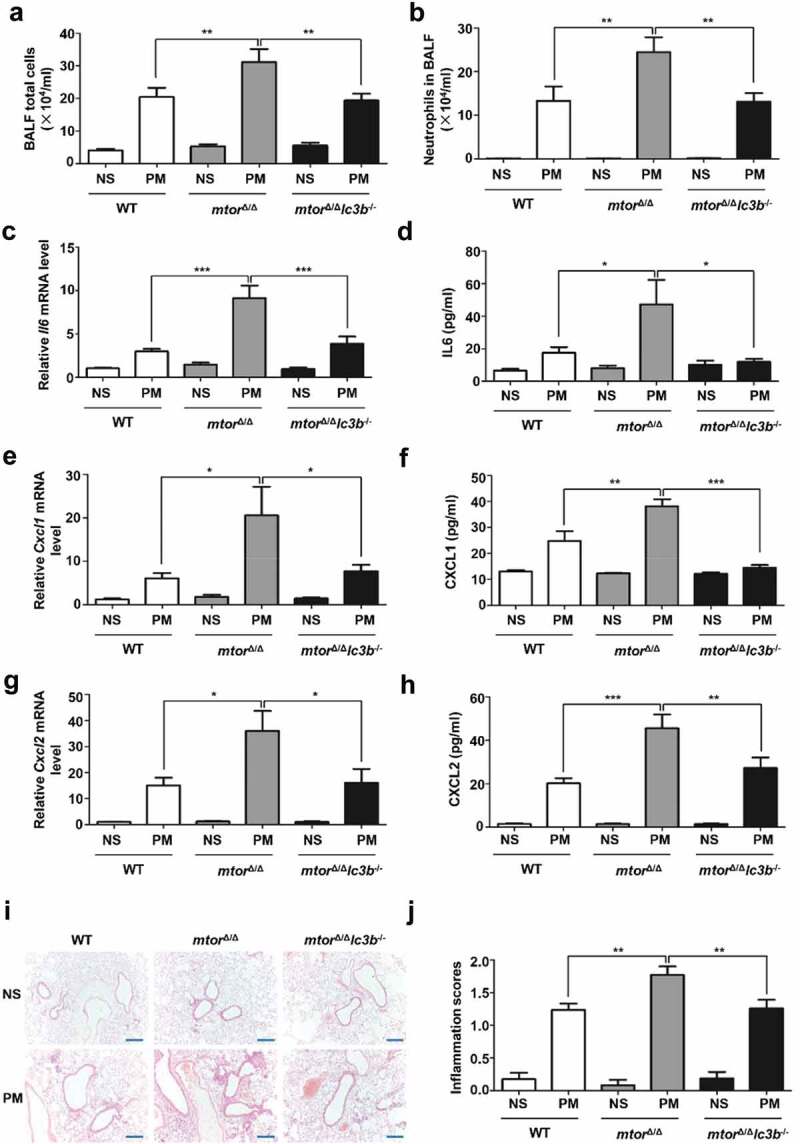
LC3B deletion ameliorates the exacerbated PM-induced inflammation caused by MTOR deficiency. Mice (n = 7 to 19 for each group) were exposed to PM at 100 μg/d for 4 days. 24 h after last PM exposure, the total inflammatory cells (A) and the number of neutrophils (B) in the BALF were assessed. The relative mRNA levels of Il6 (C) Cxcl1 (E), and Cxcl2 (G) in lung tissue were measured by quantitative PCR. Protein levels of IL6 (D), CXCL1 (F), and CXCL2 (H) in the BALF were detected by ELISA. (I and J) Representative images (I) and semi-quantification (J) of lung sections stained with H&E (n = 12–22 images for each group). Scale bar: 200 μm. Data are presented as mean ± SEM of 3 independent experiments. Differences were identified using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001.
PM inactivates MTOR and induces autophagy via upstream TSC2 in airway epithelial cells
Tuberous sclerosis complex (TSC1/2) has been reported as a crucial negative regulator of MTOR activation in response to cellular energy, growth factors, and nutrient [19]. We therefore assessed the role of TSC2 in modulating MTOR activity after PM treatment. As expected, exposure of HBE cells to PM time- and dose-dependently caused an increase in p-TSC2 phosphorylation (Figure 7A–D). Immunohistochemical staining for p-TSC2 further revealed that PM upregulated the activation of TSC2 in both airway and alveolar epithelial cells (Figure 7E). Moreover, knockdown of TSC2 by siRNA notably rescued the PM-induced decrease of MTOR activity (Figure 7F and G). We then further studied the function of TSC2 in PM-induced IL6 expression in HBE cells. As expected, TSC2 deficiency caused by two different siRNAs both significantly decreased the mRNA level of IL6 (Figure 7H), as well as the secretion of IL6 in culture supernatant after PM exposure in HBE cells (Figure 7I). Furthermore, rapamycin could partially reverse the protective effect of TSC2 knockdown on PM-induced IL6 production (Figure 7J and K). Taken together, these data suggested that PM inactivated MTOR and induced autophagy through upstream TSC2 in airway epithelial cells.
Figure 7.

Upstream TSC2 mediates PM-induced dysregulation of MTOR-autophagy in airway epithelial cells. (A) Dose-dependent levels of p-TSC2 and TSC2 in HBE cells exposed to PM for 24 h. (B) HBE cells, exposed to PM at 100 μg/ml, were harvested 0, 1, 4, and 24 h after exposure and analyzed for protein levels of p-TSC2 and TSC2 by western blot analysis. (C and D) Quantification of the immunoblots presented in Figure 6A (C) and 6B (D), and blots from 3–4 independent experiments were quantified by ImageJ software. (E) Representative images of lung sections stained for p-TSC2. Scale bar: 50 μm. (F) Cells were transfected with indicated Control- or TSC2-siRNA for 24 h, and then stimulated with PM (100 μg/ml) for an addition 24 h to analyze the levels of MTOR and p-MTOR by western blot. (G) Quantification of the immunoblots presented in Figure 6F. (H and I) Cells were infected with indicated TSC2-siRNA-1 or TSC2-siRNA-2 for 24 h, and then stimulated with PM (100 μg/ml) for an addition 24 h to measure the IL6 mRNA levels by Q-PCR (H). Cell culture supernatants were examined for IL6 by ELISA (I). (J and K) Cells were transfected with indicated Control- or TSC2-siRNA for 24 h, and then exposed to PM (100 μg/ml), co-treated with or without Rapa for an addition 24 h to measure the mRNA levels (J) and protein levels (K) of IL6. Data are representative of 3–7 independent experiments. Error bars, mean ± SEM. Differences were identified using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001.
The TLR4-MYD88 and MTOR-autophagy pathways interact with each other to regulate PM-induced inflammatory response in HBE cells
We also examined the possible involvement of other pathways in PM-modulated MTOR signaling and inflammatory response. We found that PM treatment rapidly activated TLR4-MYD88-IKK-NFKB signaling (Figure S5A and B). TLR4 knockdown by siRNA decreased the MTOR activity and increased the levels of LC3B-II (Figure S5C and D), and subsequently ameliorated the production of IL6 (Figure S5E to F). The TLR4 signaling also consistently regulated the LAMP2 expression, while it inconsistently modulated the levels of p-TSC2 (Figure S5C and D). On the other side, we observed autophagy deficiency led to decreased activation of TLR4-MYD88 signaling (Figure S6A and C). Thus, the TLR4-MYD88 and MTOR-autophagy pathways might regulate each other in the context of PM-induced epithelial injury.
MTOR suppresses PM-induced inflammation through NFKB pathway in airway epithelium
Next, we attempted to figure out the underlying mechanisms by which MTOR-autophagy axis regulated IL6 production after PM treatment in HBE cells. NFKB pathway is well recognized to regulate the production of inflammatory cytokines. Not surprisingly, genetic knockdown of RELA (RELA proto-oncogene, NFKB subunit) by siRNA diminished IL6 production in HBE cells (Figure S7A–D). Moreover, inhibition of MTOR by Rapa strongly increased (Figure 8A and B), whereas inhibition of autophagy by spautin1 or ATG5-siRNA decreased the activation of NFKB caused by PM exposure (Figure 8C–F), as determined by the altered levels of p-RELA. We further clarified the effect of MTOR deficiency on NFKB activity in mouse lung. Consistently, the expression of p-RELA, which reflected the activation of NFKB, was elevated in mouse lung after PM exposure and was further enhanced by Mtor deficiency (Figure 8G and H). These data altogether indicated that MTOR suppressed PM-induced inflammation through the NFKB pathway in airway epithelium.
Figure 8.

MTOR suppresses PM-induced inflammation via the NFKB pathway in airway epithelium. (A to F) Cells were treated with Rapa (A), spautin1 (C), or ATG5-siRNA (E) together with PM (100 μg/ml) for 24 h, then harvested for western blot analysis of p-RELA, RELA, and NFKBIA. Quantification of the immunoblots presented in Figure 8A (B), 8C (D) or 8E (F). (G) mtor∆/∆ mice and WT littermates were exposed to PM at 100 μg/d for 4 days, 24 h after last PM exposure, the protein levels of p-RELA and RELA in lung tissue were examined by western blot. (H) Quantification of the immunoblots presented in Figure 8G. (I and J) Flow cytometric analysis of NFKBIA in HBE cells treated with PM for 24 h. (K and L) Representative images (n = 8–11 images for each group) and semi-quantification of NFKBIA in HBE cells exposed to PM for 24 h. Scale bar: 100 μm. (M) Co-localization of NFKBIA (red) and autophagic vesicles (GFP-LC3 punctations) in PM-treated HBE cells. HBE cells were transfected with GFP-LC3 for 24 h, and were treated with PM at 100 μg/d for 4 h. Scale bar: 25 μm. (N and P) Cells were treated with spautin1 (N) or ATG5-siRNA (P), together with PM (100 μg/ml) for 24 h, then harvested for western blot analysis of p-CHUK/p-IKBKB (p-IKK) and p-NFKBIA. (O and Q) Quantification of the immunoblots presented in Figure 8N (O) and 8P (Q). Data are representative of 3–6 independent experiments. Error bars, mean ± SEM. Differences were identified using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001.
To clarify how autophagy activated NFKB, we examined the levels of its molecule inhibitor, NFKBIA (NFKB inhibitor alpha). We found that PM decreased NFKBIA (Figure 8I–L), and Rapa augmented, while spautin1 or ATG5-siRNA reduced the degradation of NFKBIA (Figure 8A–F). Further investigation demonstrated that part of the NFKBIA was localized with the GFP-LC3 punctations (Figure 8M), indicating PM-induced autophagy might directly degrade NFKBIA. Moreover, we also observed that PM increased the level of NFKBIA phosphorylation, and spautin1 or ATG5 deficiency evidently restrained PM-induced NFKBIA phosphorylation (Figure 8N–Q). Taken together, the decrease of NFKBIA by PM might due to the cooperation of direct degradation by autophagy with classical NFKBIA phosphorylation signaling.
Blocking autophagy decreases the basal expression of EPS15 and reduces PM endocytosis in HBE cells
As we have clearly shown that PM was endocytosed into HBE cells and some of the PM-containing endosomes fused with autophagosomes to form amphisomes [11], we then examined the possible crosstalk between autophagy and endocytosis. EPS15 is known to critically regulate endocytosis [20]. Interestingly, although PM treatment failed to modulate the expression of EPS15 in HBE cells (Figure 9A and B), knockdown of EPS15 showed significantly decreased endocytosed PM (Figure 9C and D). In addition, EPS15 efficiency exerted inconsiderable effects on the expression of p-MTOR and LC3B, though it modulated the LAMP2 expression (Figure 9E and F). However, inhibition of EPS15 significantly reduced the PM-induced production of IL6 (Figure 9G and H). These results were further confirmed by knockdown of AP2 (Figure 9I and J), another important protein involved in endocytosis. On the other side, blocking autophagy by BECN1/Beclin 1- or ATG5-siRNA remarkably decreased the basal expression of EPS15 (Figure 9K–M). Moreover, these autophagy-related siRNAs apparently diminished the levels of endocytosed PM in HBE cells (Figure 9N and O).
Figure 9.
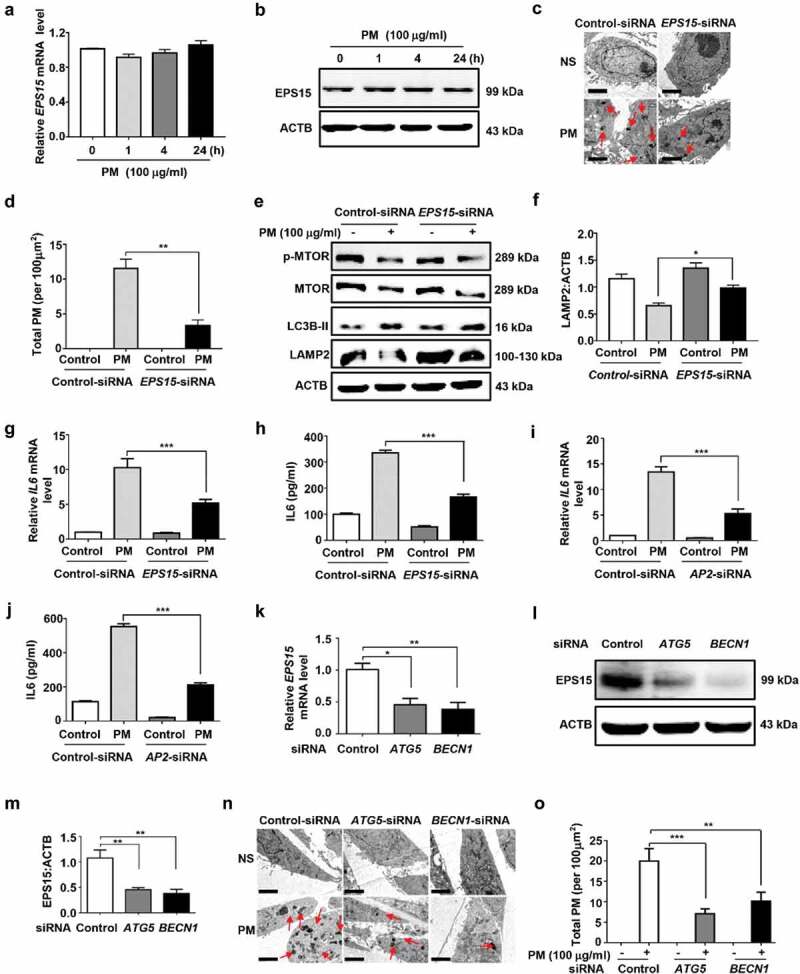
Autophagy modulates basal expression of EPS15 and regulates PM endocytosis in HBE cells. (A and B) PM treatment exerted no effects on EPS15 expression, as determined by mRNA (A) or protein (B) expression. (C and D) Effects of EPS15-siRNA on PM endocytosis. Cells were transfected with indicated Control- or EPS15-siRNA for 24 h, and then treated with PM (100 μg/ml) for an addition 24 h. Total PM in cells were observed by electron microscopy. Representative images (C) and the semi-quantification results (D) were shown. Scale bar: 2 μm. (E to H) Effects of EPS15-siRNA on PM-induced expression of p-MTOR, MTOR, LC3B, and LAMP2, and production of IL6 in HBE cells. The protein expression (E and F) was determined by western blotting analysis, the relative mRNA level of IL6 (G) were measured by Q-PCR, and IL6 (H) in cell culture supernatants was detected by ELISA. (I and J) Effects of AP2-siRNA on PM-induced expression of IL6 in HBE cells. (K to O) Effects of autophagy in regulation of the expression of EPS15 and PM endocytosis. (K to M) Cells were transfected with siRNA for 24 h, and were harvested for analysis of mRNA (K), and protein levels (L) with semi-quantification (M). (N and O) After siRNA transfection, cells were treated with PM (100 μg/ml) for an addition 24 h. Representative images of the incidence of black particles in cells (N) and the semi-quantification results (O) were shown. Scale bar: 2 μm. Data are representative of 3–6 independent experiments. Error bars, mean ± SEM. Differences were identified using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
In this study we demonstrate that PM inactivates MTOR and induces autophagy in HBE cells and in mouse airway epithelium, and club-cell-specific deletion of MTOR aggravates PM-induced airway inflammation. We further identify that the role of Mtor deficiency is dependent on the downstream autophagy in vitro and in vivo. The activation of upstream TSC2 directly contributes to downstream dysregulated MTOR-autophagy in response to PM treatment. Moreover, the MTOR-autophagy axis cross-regulates with TLR4-MYD88 signaling to orchestrate PM-induced airway inflammation through activation of NFKB. Autophagy also modulates the endocytosis of PM, and the endocytosis and TLR signaling contribute the PM-induced lysosomal impairment (Figure 10).
Figure 10.

Summarization of the role of MTOR-autophagy axis in PM-induced airway inflammation. PM exposure inactivates MTOR and induces autophagy through activation of upstream TSC2 in airway epithelium. The dysregulated MTOR-autophagy axis cross-regulates with TLR4-MYD88 signaling to orchestrate PM-induced NFKB activation and subsequent airway inflammation likely through NFKBIA direct degradation by autophagy or its phosphorylation. Autophagy also modulates PM endocytosis, and both endocytosis and TLR4 pathways contribute to the impairment of lysosomal functions.
The function of autophagy in lung diseases is either protective or deleterious, dependent on stimuli and cell type [7,21]. As for the role of autophagy in PM-induced airway inflammation, it has been indicated to be deleterious in epithelial cells according to multiple studies [11,12,22,23]. Consistently, we have recently found that PM triggers a typical convergence of endocytosis and autophagy, and autophagy is required for PM-induced inflammation and mucus hyperproduction in airway epithelium [11]. Furthermore, a recent study [22] has reveals that the PtdIns3K-AKT-MTOR pathway may be a key modulator of PM-induced autophagy in Beas-2B cells. In consistent with this research, our present results also showed that PM exposure inactivated MTOR and induced autophagy not only in HBE cells but also in mouse airway epithelium. Generally, autophagy is associated with upstream signaling MTOR, plays an important role in regulating airway epithelial inflammatory responses, such as acute lung injury [10], COPD [24] and asthma (paper under review). However, an abundance of evidence [25–28] reveals the fact that MTOR is not the unique regulator of autophagy. For instance, Henson et al [27] have demonstrated that fundamental metabolic requirements of senescent primary human CD8+ T cells and MAPK14/p38 MAPK (mitogen-activated protein kinase) blockade reverses senescence via autophagy in an MTOR-independent manner. Another study [25] has also shown that L-type Ca2+ channel antagonists, the K+ATP channel opener minoxidil, and the Gi signaling activator clonidine induce autophagy in an MTOR-independent manner. In the present study, it was the first time we clearly elaborated the physiological relevance of airway epithelium localized MTOR in regulating PM-induced airway inflammation in vitro and in vivo, and we further confirmed the role of MTOR deficiency was dependent on the downstream autophagy and LC3B.
It should be noted in our present study that PM exposure induced a notable increase of IL6 and IL8 expression in HBE cells, and MTOR knockdown further enhanced the induced production of IL6, but not IL8. Currently we have demonstrated that MTOR-autophagy axis orchestrates PM-induced NFKB activation likely through degradation of NFKBIA in airway epithelial cells, and we have previously demonstrated that NFKB regulates PM-induced IL8 production in HBE cells [11]. Thus, it is likely that MTOR knockdown increases IL8 expression though NFKB, while it decreases IL8 expression via some unknown pathways. Another issue worthy of attention was that although impaired MTOR showed no considerable effect on IL8 production, mice of club-cell-specific deletion of Mtor displayed aggravated airway inflammation, including increased expression of inflammatory cytokines such as IL6, CXCL1 and CXCL2 both in lung and BALF supernatant. One plausible explanation is that airway epithelium deletion of MTOR results in elevated IL6 production in epithelial cells, subsequently contributes to aggravated inflammatory response in lung, including but not limit to enhanced recruitment of inflammatory cells and expression of IL6, CXCL1, CXCL2. To support this, several studies [29–32] have shown that the secretion of IL6 causes secondary production of chemokines such as IL8 and CD46 molecule by mononuclear cells, macrophages as well as expression of ICAM (intercellular adhesion molecule)-1, and other adhesion molecules on endothelial cells, eventually leading to enhanced neutrophil migration. Moreover, IL6 alone is able to promote neutrophilia [33]. Additionally, IL6 induces the production of proinflammatory cytokines such as IL17 and IL23 via IL6RA (interleukin 6 receptor, alpha) in mature neutrophils to establish a Th17 cells-polarizing positive feedback loop [32,34].
TSC1 and TSC2 are usually considered as a complex (TSC1/2), and inactivation of TSC1 and TSC2 results in a similar phenotype, implying that they have similar functions in cell growth and proliferation [35–37]. Recently, breakthroughs in TSC1/2 researches connect the TSC1/2 with the MTOR signaling, showing that TSC2 acts as a RHEB-GAP to suppress MTOR signaling [19]. Therefore, TSC2 may function as a master regulator of MTOR in response to nutrient, growth factors and energy signals. For example, Ng and colleagues [38] demonstrate that tsc2-null mouse embryonic fibroblasts are sensitive to apoptosis in response to amino acid starvation and hypoxia, and further confirm that constitutive activation of MTOR in tsc2−/− cells results in suppression of autophagy and subsequently enhances susceptibility to stress-mediated cell death. However, some other studies [39,40] have also reported that the effect of TSC2 could be MTOR-independent. In consistent with these former studies, our current data clearly demonstrated that the activation of upstream TSC2 pathway directly contributed to downstream dysregulated MTOR-autophagy in response to PM treatment.
NFKB is widely involved in the regulation of inflammation and tumor development, and NFKB is generally pro-inflammatory in both acute lung injury and COPD. Additionally, we observe that activation of NFKB in airway epithelium mediates PM-induced airway inflammation in a recent study [11]. Autophagy have been implicated in the process of NFKB activation, whereas the role of autophagy in NFKB activation is complex and likely varies depending on cell type and stimuli [10,11,41–45]. For example, we report that autophagy in airway epithelium is essential for PM-induced activation of NFKB and subsequent airway inflammation [11,41]. Conversely, in lipopolysaccharide (LPS)-induced acute lung injury, autophagy is proved to act negatively in regulation of NFKB activation [10]. The detailed mechanisms that NFKB activation can be either positively or negatively regulated by MTOR-autophagy in airway epithelial cells remain unknown and need further investigation. Of note, NFKB signaling, showed in some case [46], can in turn regulate autophagy. Although there are numerous studies looking into the crosstalk between autophagy and NFKB, little is known about the relationship between MTOR and NFKB. Similarly, the functions of MTOR in lung diseases also appear to be cell type and stimuli-specific. In fact, we have recently found that inactivation of MTOR leads to enhanced cigarette smoke-induced production of inflammatory cytokines in airway epithelial cells [24], showing MTOR acts negatively in the context of COPD pathogenesis. On the contrary, recent studies have demonstrated that Rapa suppresses the activation of NFKB induced by LPS [47,48], and we further confirm that activation of MTOR promotes LPS-induced inflammation cytokines through elevation of NFKB activation in HBE cells [10]. In our study, MTOR functioned negatively, while autophagy acted positively in modulation of NFKB activation in PM-induced inflammation.
The crosstalk between endocytosis/phagocytosis and autophagy is complex. The xeno-material containing endosomes are readily to fuse with autophagosomes to form amphisomes [49]. However, whether endocytosis is required for autophagy induction remains unclear. In our study, knockdown of EPS15 exerted inconsiderable effects on PM-induced dysregulation of MTOR-autophagy, suggesting that endocytosis was not necessary for PM-induced autophagy. On the contrary, blocking autophagy reduced the expression of EPS15 and eventually decreased the endocytosed PM in HBE cells, indicating that autophagy positively regulated PM endocytosis. However, it has been shown that atg7−/− macrophages exhibit increased phagocytosis of Mycobacterium tuberculosis [50]. Thus, the eventual functions of autophagy in regulation of endocytosis/phagocytosis appear to be cell- and pathogen -dependent.
It is worth noting that the protective role of MTOR is observed not only in the PM-induced airway epithelial injury, but also in asthma (paper under review) and COPD [24]. Thus, MTOR activation could be an effective therapeutic approach being extensively used for various chronic airway diseases. Also, the present study proposes a mechanistic explanation that MTOR inhibition could cause airway epithelial injury, implying that MTOR inhibitors should be used with caution, particularly for patients with such chronic airway diseases or airway disorders induced by air-borne particulate pollution.
It should be pointed out that one weakness of the current study is that PM was suspended in saline and was instilled into mouse airway intratracheally. As human exposure to PM is predominantly inhaled, a nebulization approach would be more ideal for animal study. Nonetheless, the present study demonstrates that PM inactivates MTOR and induces autophagy through upstream TSC2 signaling, and the MTOR-autophagy axis cross-regulates with TLR4-MYD88 to activate NFKB, leading to subsequent inflammatory response in airway epithelial cells. The massive induction of autophagy is required for PM-induced airway inflammation. Activation of MTOR and/or suppression of autophagy in airway epithelial cells might be effective therapeutic strategies for preventing airway inflammation induced by air-borne particulate pollution.
Materials and methods
Reagents
Antibodies against ACTB (Santa Cruz Biotechnology, sc-130300), MTOR (Cell Signaling Technology, 2972), p-MTOR (Cell Signaling Technology, 5536), p-RPS6 (Cell Signaling Technology, 4858), p-RPS6 (Cell Signaling Technology, 4803), RPS6 (Cell Signaling Technology, 2217), LC3B (Sigma-Aldrich, L7543), LAMP2 (Cell Signaling Technology, 49067), SQSTM1 (Cell Signaling Technology, 39749), EGFR (Abcam, ab76153), TSC2 (Cell Signaling Technology, 3990), p-TSC2 (Cell Signaling Technology, 3617), p-TSC2 (Abcam, ab109403), p-CHUK/p-IKBKB (p-IKK; Cell Signaling Technology, 2697), NFKBIA (Cell Signaling Technology, 4814), p-NFKBIA (Cell Signaling Technology, 2859), RELA (Cell Signaling Technology, 8242), p-RELA (Cell Signaling Technology, 3039), SCGB1A1 (Santa Cruz Biotechnology, sc-365992), EPS15 (Abcam, ab174291), TLR4 (Santa Cruz Biotechnology, sc-293072), MYD88 (Santa Cruz Biotechnology, sc-74532), and ATG5 (Abcam, ab108327) were used. A set of siRNAs of Control (sc-37007), MTOR (sc-35409), TSC2 (sc-36762), ATG5(sc-41445), RELA (sc-29410), EPS15 (sc-35321), AP2 (sc-29610), TLR4 (sc-40260), and BECN1/Beclin 1 (sc-29797) were purchased from Santa Cruz Biotechnology. Another siRNA of TSC2 (TSC2-siRNA-2) was designed and synthesized by Shanghai Gene Pharma for verifying the effects. The siRNA transfection reagent (SL100568) were obtained from SignaGen Laboratories. RNAiso plus (9109), Reverse Transcription Reagents (DRR037A), and SYBR Green Master Mix (DRR041A) were from Takara Biotechnology. All primers used in this study were synthesized by Sangon Biotech, Shanghai. Doxycycline (D9891) was purchased from Sigma-Aldrich. Rapamycin (S1039), Torin1 (S2827), and spautin1 (S7888) were purchased from Selleck, and the final concentrations in the culture medium were 5 nM, 250 nM, and 10 nM, respectively. EGF was purchased from Invitrogen (E3480).
Cell culture and PM treatment
We obtained HBE cells from American Type Culture Collection (CRL-2741). HBE cells were cultured in RPMI 1640 (Gibco, C11875500BT) supplemented with 10% FBS (Gibco, 10082147). PM, purchased from National Institute of Standards and Technology (NIST, 1649b), was suspended and sonicated in sterile saline to a final concentration at 2 mg/ml, and HBE cells were exposure to PM at 100 μg/ml.
Primary mouse tracheal epithelial cells (MTEC) were isolated from the respiratory tract of mice and cultured at air liquid interface (ALI) as previously described [51]. MTEC were maintained in proliferation medium for 14 days to attained 100% confluence. Then ALI was created for MTEC differentiation for an additional 14 days. Culture medium was renewed every other day. At day 29, after PM treatment at 100 μg/ml for 24h, CXCL1 and CXCL2 expression in basolateral supernatants were measured by ELISA, and cells were collected to evaluate Cxcl1, Cxcl2, and Il6 mRNA expression by Q-PCR, or to evaluate p-RPS6 expression by flow cytometry.
Transfection
Each well of 6-well plates were seeded with 5 × 104 cells and grown overnight. Then the siRNA transfection was performed with the transfection reagent (SignaGen Laboratories, SL100568), while the GFP-LC3 or mRFP-GFP-LC3 plasmid was transfected into cells by using PloyJet in vitro DNA Transfection reagent (SignaGen Laboratories, SL100688) according to the manufacturer’s protocol.
Animals and mouse model of PM-induced airway inflammation
Scgb1a1-rtTA/(tetO)7-Cre/mtorflox/flox mice were generated by crossing the mtorflox/flox mice (C57BL/6 background; obtained from the Jackson Laboratory, 011009) with Scgb1a1-rtTA/(tetO)7-Cre transgenic mice (C57BL/6 background). Age- and sex-matched Scgb1a1-rtTA/(tetO)7-Cre/Mtor+/+ animals were used as controls in the experiments. In order to induce the expression of Cre, 6-wk-old mice were fed with doxycycline in drinking water (2 mg/ml) for 20 days before establishing the model of PM-induced airway inflammation, and the mice were kept with doxycycline at all the time until they were sacrificed. The Scgb1a1-rtTA/(tetO)7-Cre/mtorflox/flox and Scgb1a1-rtTA/(tetO)7-Cre/Mtor+/+ mice after doxycycline-induced Cre activation were designated as mtor∆/∆ and WT mice, respectively.
Club-cell-specific atg5∆/∆ mice were generated the same as above.
lc3b−/− mice were obtained from Jackson laboratory. Furthermore, the mtor∆/∆ mice were mated with lc3b−/− mice to generate Scgb1a1-rtTA/(tetO)7-Cre/mtorflox/flox- lc3b−/− (mtor∆/∆lc3b−/−) mice, which were deleted both Mtor and Lc3b alleles in airway epithelial cells. We used genomic DNA extracted from tails of mice for genotyping. All primers used for genotyping were listed in Table 1. Animal care and experimental protocols were approved by the Ethical Committee for Animal Studies at Zhejiang University.
Table 1.
Primers used for genotyping.
| Genes | Primer sequence (5ʹ-3ʹ) |
|---|---|
| Scgb1a1 | Forward AAA ATCTTGCCAGCTTTCCCC |
| Reverse ACTGCCCATTGCCCAAACAC | |
| TetO | Forward TGCCACGACCAAGTGACAGCAATG |
| Reverse AGAGACGGA AATCCATCGCTCG | |
| Mtor | Forward TTATGTTTGATAATTGCAGTTTTGGCTAGC AGT |
| Reverse TTTAGGACTCCTTCTGTGACATAC ATTTCCT | |
| Lc3b | Lc3b-1 GACACCTGTACACTCTGATGCACT |
| Lc3b-2 CCTGCCGTCTGCTCTAAGCTG | |
| Lc3b-3 CCACTCCCACTGTCCTTTCCTAAT | |
| Atg5 | Atg5-1 GAATATGAAGGCACACCCCTGAAATG |
| Atg5-2 ACAACGTCGAGCACAGCTGCGCAAGG | |
| Atg5-3 GTACTGCATAATGGTTTAACTCTTGC |
To establish a mouse model of PM-induced airway inflammation, PM was prepared as described detailed in vitro, and we exposure of mice to 100 μg PM (in 50 μl saline) per day by intratracheal instillation for 4 days. Meanwhile, control mice were treated with the same volume of saline.
Quantitative real-time polymerase chain reaction (PCR)
HBE cells and lung homogenates were lysed with RNAiso plus (Takara Biotechnology, 9109), and total RNA was extracted. By using reverse transcription reagents (Takara Biotechnology, DRR037A), RNAs were reverse-transcribed. Then, the expression of genes was measured by quantitative real-time PCR, which performed on a StepOne real-time PCR system (Applied Biosystems, Foster City, CA) using SYBR Green Master Mix (Takara Biotechnology, DRR041A). All protocols were performed following the manufacturer’s instructions. All primers used for quantitative real time PCR analysis are listed in Table 2.
Table 2.
Primers used for quantitative real time PCR analysis.
| Species | Genes | Primer sequence (5ʹ-3ʹ) |
|---|---|---|
| Human | ACTB | Forward: CATGTACGTTGCTATCCAGGC |
| Reverse: CTCCTTAATGTCACGCACGAT | ||
| Human | IL6 | Forward: ACTCACCTCTTCAGAACGAATTG |
| Reverse: CCATCTTTGGAAGGTTCAGGTTG | ||
| Mouse | Actb | Forward: GGCTGTATTCCCCTCCATCG |
| Reverse: CCAGTTGGTAACAATGCCATGT | ||
| Mouse | Cxcl1 | Forward: CTGGGATTCACCTCAAGAACATC |
| Reverse: CAGGGTCAAGGCAAGCCTC | ||
| Mouse | Cxcl2 | Forward: TGTCCCTCAACGGAAGAACC |
| Reverse: CTCAGACAGCGAGGCACATC | ||
| Mouse | Il6 | Forward: CTGCAAGAGACTTCCATCCAG |
| Reverse: AGTGGTATAGACAGGTCTGTTGG |
Western blot assay
The lysates of PM-treated HBE cells and lung tissue were prepared with RIPA buffer (Beyotime, P0013B) containing protease (Roche Diagnostics GmbH, 04-693-116-001) and phosphatase inhibitors (Roche Diagnostics GmbH, 04-906-837-001). The supernatants of cell lysates were run on gels and incubated with relevant antibodies using standard methods. ACTB was used as a loading control. Quantification was performed by densitometry and analyzed using ImageJ software.
ELISA
The concentration of IL6 in culture supernatants and mouse cytokines such as CXCL1, CXCL2 and IL6 in BALF supernatants, were measured by ELISA kits following the manufacturer’s protocol. The concentration of IL6 in culture supernatants and mouse cytokines such as CXCL1, CXCL2 and IL6 in BALF supernatants, were measured by ELISA kits following the manufacturer’s protocol. ELISA kits for human IL6 (D6050), mouse CXCL1 (MKC00B), mouse CXCL2 (MM200) and mouse IL6 (M6000B) were purchased from R&D systems.
BALF collection and analysis
Mice were sacrificed 24 h after the last exposure to PM, and lavaged with 0.4 ml PBS (Solarbio,NO. P1010) by injecting into the lungs and drawing to collect cells for 3 times. The total number of BALF cells was counted, then the remaining BALF was centrifuged (400 g for 10min at 4°C). The supernatant was retained for further analysis, while the cell pellet was resuspended in PBS moderately and centrifuged on glass slides. Then cells on glass slides were stained with Wright–Giemsa stain (Baso, BA-4017), and differential counts were assessed by counting 200 total cells.
Histological analysis
After exposure to PM, the lungs were removed and fixed in 4% paraformaldehyde at 4°C for 24 h. After fixation, the lungs were embedded in paraffin for hematoxylin & eosin (H&E) analysis and inflammation score was determined according to published guidelines [52].
Immunohistochemistry and immunofluorescence staining
Paraformaldehyde-fixed and paraffin-embedded lung sections were prepared and immunostained for p-TSC2 following standard methods. Images were photographed using Olympus BX53 inverted microscope (Olympus, Melville, NY). Quantitative analysis was performed as described before [53]. p-TSC2-positive bronchial epithelial cells were revealed as a percentage of total epithelial cells.
The expression of p-RPS6 and ATG5 were measured by immunofluorescence staining. Lung sections were stained with anti-p-RPS6 and anti-SCGB1A1, or with anti-ATG5 and anti-SCGB1A1 as published previously [10]. Fluorescence images were captured with confocal microscope. p-RPS6-positive bronchial epithelial cells were revealed as a percentage of SCGB1A1-positive epithelial cells.
HBE cells were fixed and stained with anti-NFKBIA antibody (Cell Signaling Technology, 4814; 1:1600) at 4°C for 4 h. MTEC were stained with anti-p-RPS6 on the transwell membrane as described previously [54]. The relative fluorescence intensity was measured with ImageJ software, and the mean relative fluorescence intensities were normalized to the levels of control.
Flow cytometry
MTEC cells were harvested, fixed, and permeabilized, then intracellularly stained with SCGB1A1 antibody (Santa Cruz Biotechnology, sc-365992-PE; 1:200) and p-RPS6 antibody (Cell Signaling Technology, 4803; 1:400) at 4°C for 30 min. For analysis of NFKBIA expression, HBE cells were staining with NFKBIA (Cell Signaling Technology, 4814; 1:1600) for 30 min, followed by 1 μg of PE goat anti-mouse IgG antibody (Biolegend, 405307) for 20 min at 4°C. The corresponding isotype control antibodies were used. Flow cytometric analysis were performed on CytoFlex flow cytometry system (Beckman Coulter, Brea, California). FlowJo software (Tree Star, Ashland, OR) was used for analysis and graphical output.
Transmission electron microscopy (TEM)
For TEM examination, the isolated bronchus samples from mouse after the last PM exposure in vivo or HBE cells after experimental manipulations were fixed in 2.5% glutaraldehyde in PBS for 24 h. Then the samples were prepared according to standard methods. Images were scanned with TECNA1 10 transmission electron microscope (FEI, Hillsboro, Oregon, USA).
Semi-quantification was performed by observers blinded to group and treatment as before [11].
Statistics
Data are presented as means ± SEM. Statistical analysis was performed with GraphPad Prism (GraphPad software, San Diego, California, USA). Differences between two groups were identified using the Student t-test and multiple groups using one-way ANOVA. Values of P less than 0.05 was considered statistically significant.
Funding Statement
This work was supported by the National Key R&D Program of China (Grant 2016YFA0501602 to Z.-H. C.), Key Project of Chinese National Programs for Fundamental Research and Development (973 program, 2015CB553405 to Z.-H. C.), the Major Project (81490532 to H.-H. S.) and the General Projects (81670031 to Z.-H. C., 81370126 to W. L., and 81570021 to H.-Q. H.) from the National Natural Science Foundation of China, and the Precision Medicine Research of the National Key Research and Development Plan of China (2016YFC0905800).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Materials
Supplemental Materials data for this article can be accessed here.
References
- [1].Guan WJ, Zheng XY, Chung KF, et al. Impact of air pollution on the burden of chronic respiratory diseases in China: time for urgent action. Lancet. 2016;388(10054):1939–1951. [DOI] [PubMed] [Google Scholar]
- [2].Gordon SB, Bruce NG, Grigg J, et al. Respiratory risks from household air pollution in low and middle income countries. Lancet Respir Med. 2014;2(10):823–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Santibanez-Andrade M, Quezada-Maldonado EM, Osornio-Vargas A, et al. Air pollution and genomic instability: the role of particulate matter in lung carcinogenesis. Environ Pollut. 2017;229:412–422. [DOI] [PubMed] [Google Scholar]
- [4].Oberdörster G, Oberdörster E, Oberdörster J.. Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ Health Perspect. 2005;113(7):823–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Saxton RA, Sabatini DM.. MTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ryter SW, Choi AM. Autophagy in lung disease pathogenesis and therapeutics. Redox Biol. 2015;4:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li ZY, Wu YF, Xu XC, et al. Autophagy as a double-edged sword in pulmonary epithelial injury: a review and perspective. Am J Physiol Lung Cell Mol Physiol. 2017;313(2):L207–L217. [DOI] [PubMed] [Google Scholar]
- [8].Nakahira K, Pabon Porras MA, Choi AM. Autophagy in plumonary disease. Am J Respir Cirt Care Med . 2016;194(10):1196––1207.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hu Y, Liu J, Wu YF, et al. mtor and autophagy in regulation of acute lung injury: a review and perspective. Microbes Infect. 2014;16(9):727–734. [DOI] [PubMed] [Google Scholar]
- [10].Hu Y, Lou J, Mao YY, et al. Activation of MTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Autophagy. 2016;12(12):2286–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen ZH, Wu YF, Wang PL, et al. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy. 2016;12(2):297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu T, Wu B, Wang Y, et al. Particulate matter 2.5 induces autophagy via inhibition of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin kinase signaling pathway in human bronchial epithelial cells. Mol Med Rep. 2015;12(2):1914–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Su R, Jin X, Zhang W, et al. Particulate matter exposure induces the autophagy of macrophages via oxidative stress-mediated PI3K/AKT/mtor pathway. Chemosphere. 2017;167:444–453. [DOI] [PubMed] [Google Scholar]
- [14].Ding R, Zhang C, Zhu X, et al. ROS-AKT-mtor axis mediates autophagy of human umbilical vein endothelial cells induced by cooking oil fumes-derived fine particulate matters in vitro. Free Radic Biol Med. 2017;113:452–460. [DOI] [PubMed] [Google Scholar]
- [15].Liu B, Fang M, Hu Y, et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy. 2014;10(3):416–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen ZH, Lam HC, Jin Y, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010;107(44):18880–18885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lam HC, Cloonan SM, Bhashyam AR, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2013;123(12):5212–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li Y, Corradetti MN, Inoki K, et al. TSC2: filling the GAP in the mtor signaling pathway. Trends Biochem Sci. 2004;29(1):32–38. [DOI] [PubMed] [Google Scholar]
- [20].van Bergen En Henegouwen PM. Eps15: a multifunctional adaptor protein regulating intracellular trafficking. Cell Commun Signaling. 2009;7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Racanelli AC, Kikkers SA, Choi AMK, et al. Autophagy and inflammation in chronic respiratory disease. Autophagy. 2017;13:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Deng X, Zhang F, Rui W, et al. PM2.5-induced oxidative stress triggers autophagy in human lung epithelial A549 cells. Toxicol In Vitro. 2013;27(6):1762–1770. [DOI] [PubMed] [Google Scholar]
- [23].Deng X, Zhang F, Wang L, et al. Airborne fine particulate matter induces multiple cell death pathways in human lung epithelial cells. Apoptosis. 2014;19(7):1099–1112. [DOI] [PubMed] [Google Scholar]
- [24].Wang Y, Liu J, Zhou JS, et al. MTOR suppresses cigarette smoke-induced epithelial cell death and airway inflammation in chronic obstructive pulmonary disease. J Immunol. 2018;200(8):2571–2580. [DOI] [PubMed] [Google Scholar]
- [25].Williams A, Sarkar S, Cuddon P, et al. Novel targets for Huntington’s disease in an mtor-independent autophagy pathway. Nat Chem Biol. 2008;4(5):295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Harder LM, Bunkenborg J, Andersen JS. Inducing autophagy: a comparative phosphoproteomic study of the cellular response to ammonia and rapamycin. Autophagy. 2014;10(2):339–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Henson SM, Lanna A, Riddell NE, et al. p38 signaling inhibits mtorC1-independent autophagy in senescent human CD8(+) T cells. J Clin Invest. 2014;124(9):4004–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang X, Chen S, Song L, et al. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy. 2014;10(4):588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Saggini A, Chimenti S, Chiricozzi A. IL-6 as a druggable target in psoriasis: focus on pustular variants. J Immunol Res. 2014;2014:964069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bartoccioni E, Scuderi F, Marino M, et al. IL-6, monocyte infiltration and parenchymal cells. Trends Immunol. 2003;24(6):298–299. [DOI] [PubMed] [Google Scholar]
- [31].Ishihara K, Hirano T. IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev. 2002;13(4–5):357–368. [DOI] [PubMed] [Google Scholar]
- [32].Romano M, Sironi M, Toniatti C, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6(3):315–325. [DOI] [PubMed] [Google Scholar]
- [33].Kaplanski G, Marin V, Montero-Julian F, et al. IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003;24(1):25–29. [DOI] [PubMed] [Google Scholar]
- [34].Ogura H, Murakami M, Okuyama Y, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008;29(4):628–636. [DOI] [PubMed] [Google Scholar]
- [35].Hodges AK, Li SW, Maynard J, et al. Pathological mutations in TSC1 and TSC2 disrupt the interaction between hamartin and tuberin. Hum Mol Genet. 2001;10(25):2899–2905. [DOI] [PubMed] [Google Scholar]
- [36].Tapon N, Ito N, Dickson BJ, et al. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell. 2001;105(3):345–355. [DOI] [PubMed] [Google Scholar]
- [37].Potter CJ, Huang H, Xu T. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 2001;105(3):357–368. [DOI] [PubMed] [Google Scholar]
- [38].Ng S, Wu YT, Chen B, et al. Impaired autophagy due to constitutive mtor activation sensitizes TSC2-null cells to cell death under stress. Autophagy. 2011;7(10):1173–1186. [DOI] [PubMed] [Google Scholar]
- [39].Agarwal S, Bell CM, Rothbart SB, et al. AMP-activated Protein Kinase (AMPK) control of mtorC1 Is p53- and TSC2-independent in pemetrexed-treated carcinoma cells. J Biol Chem. 2015;290(46):27473–27486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rozas NS, Redell JB, McKenna J 3rd, et al. Prolonging the survival of Tsc2 conditional knockout mice by glutamine supplementation. Biochem Biophys Res Commun. 2015;457(4):635–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xu XC, Wu YF, Zhou JS, et al. Autophagy inhibitors suppress environmental particulate matter-induced airway inflammation. Toxicol Lett. 2017;280:206–212. [DOI] [PubMed] [Google Scholar]
- [42].Chang CP, Su YC, Hu CW, et al. TLR2-dependent selective autophagy regulates NF-kappaB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ. 2013;20(3):515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Criollo A, Chereau F, Malik SA, et al. Autophagy is required for the activation of NFkappaB. Cell Cycle. 2012;11(1):194–199. [DOI] [PubMed] [Google Scholar]
- [44].Lopez-Alonso I, Aguirre A, Gonzalez-Lopez A, et al. Impairment of autophagy decreases ventilator-induced lung injury by blockade of the NF-kappaB pathway. Am J Physiol Lung Cell Mol Physiol. 2013;304(12):L844–52. [DOI] [PubMed] [Google Scholar]
- [45].Colleran A, Ryan A, O’Gorman A, et al. Autophagosomal IkappaB alpha degradation plays a role in the long term control of tumor necrosis factor-alpha-induced nuclear factor-kappaB (NF-kappaB) activity. J Biol Chem. 2011;286(26):22886–22893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pan H, Zhang Y, Luo Z, et al. Autophagy mediates avian influenza H5N1 pseudotyped particle-induced lung inflammation through NF-kappaB and p38 MAPK signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2014;306(2):L183–95. [DOI] [PubMed] [Google Scholar]
- [47].Fielhaber JA, Carroll SF, Dydensborg AB, et al. Inhibition of mammalian target of rapamycin augments lipopolysaccharide-induced lung injury and apoptosis. J Immunol. 2012;188(9):4535–4542. [DOI] [PubMed] [Google Scholar]
- [48].Lorne E, Zhao X, Zmijewski JW, et al. Participation of mammalian target of rapamycin complex 1 in Toll-like receptor 2- and 4-induced neutrophil activation and acute lung injury. Am J Respir Cell Mol Biol. 2009;41(2):237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Klionsky DJ, Eskelinen EL, Deretic V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes … wait, I’m confused. Autophagy. 2014;10:549–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bonilla DL, Bhattacharya A, Sha Y, et al. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity. 2013;39:537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lam HC, Choi AM, Ryter SW. Isolation of mouse respiratory epithelial cells and exposure to experimental cigarette smoke at air liquid interface. J Vis Exp. 2011;21:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lee KS, Lee HK, Hayflick JS, et al. Inhibition of phosphoinositide 3-kinase delta attenuates allergic airway inflammation and hyperresponsiveness in murine asthma model. Faseb J. 2006;20(3):455–465. [DOI] [PubMed] [Google Scholar]
- [53].Xanthou G, Alissafi T, Semitekolou M, et al. Osteopontin has a crucial role in allergic airway disease through regulation of dendritic cell subsets. Nat Med. 2007;13(5):570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Dickinson JD, Alevy Y, Malvin NP, et al. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy. 2016;12(2):397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.