

Abstract
In murine leishmaniasis, healing is mediated by IFN-γ-producing CD4+ and CD8+ T cells. Thus, an efficacious vaccine should induce Th1 and Tc1 cells. Dendritic cells (DCs) pulsed with exogenous proteins primarily induce strong CD4-dependent immunity; induction of CD8 responses has proven to be difficult. We evaluated the immunogenicity of fusion proteins comprising the protein transduction domain of HIV-1 TAT and the Leishmania antigen LACK (Leishmania homolog of receptors for activated C kinase), as TAT-fusion proteins facilitate major histocompatibility complex class I-dependent antigen presentation. In vitro, TAT–LACK-pulsed DCs induced stronger proliferation of Leishmania-specific CD8+ T cells compared with DCs incubated with LACK alone. Vaccination with TAT–LACK-pulsed DCs or fusion proteins plus adjuvant in vivo significantly improved disease outcome in Leishmania major-infected mice and was superior to vaccination with DCs treated with LACK alone. Vaccination with DC + TAT–LACK resulted in stronger proliferation of CD8+ T cells when compared with immunization with DC + LACK. Upon depletion of CD4+ or CD8+ T cells, TAT–LACK-mediated protection was lost. TAT–LACK-pulsed IL-12p40-deficient DCs did not promote protection in vivo. In summary, these data show that TAT-fusion proteins are superior in activating Leishmania-specific Tc1 cells when compared with antigen alone and suggest that IL-12-dependent preferential induction of antigen-specific CD8+ cells promotes significant protection against this important human pathogen.
INTRODUCTION
Infections with Leishmania spp. are among the most frequent infectious diseases worldwide with more than 12 million people affected (Sacks and Noben-Trauth, 2002; Murray et al., 2005). Although 350 million people are at risk of infection every day, an efficacious vaccine does not exist (Murray et al., 2005). The important role of IFN-γ-producing, Leishmania-specific CD4+ T cells in protective immunity against Leishmania major in mice is well established (Reiner and Locksley, 1995; Sacks and Noben-Trauth, 2002). IFN-γ activates infected host macrophages to produce NO− and to efficiently eliminate parasites. Control of disease and long-lasting immunity in mice is thus associated with Th1-predominant immunity, whereas development of Th2 responses, as observed in BALB/c mice, leads to progressive disease (Reiner and Locksley, 1995; Sacks and Noben-Trauth, 2002). Infections of C57BL/6 mice serve as a surrogate model for self-healing cutaneous leishmaniasis of humans. Although mixed antigen-specific Th1/Th2 immune responses are observed in the peripheral blood of humans, lesional cytokine profiles clearly revealed that IFN-γ is associated with lesion resolution, whereas elevated IL-4/IL-10 levels were found in more progressive, nonhealing disease (Kharazmi et al., 1999; van Weyenbergh et al., 2004; Saha et al., 2006; Geiger et al., 2010). Several studies using the physiologically relevant murine low-dose model have indicated that IFN-γ release by CD8+ L. major-specific T cells (Tc1) also promotes the development of protective immunity (Muller et al., 1991; Belkaid et al., 2002; Uzonna et al., 2004).
Development of Th1/Tc1 immunity requires IL-12, which is released by infected (e.g., skin-derived) dendritic cells (DCs) (Flohe et al., 1998; von Stebut et al., 1998, 2000; Ahuja et al., 1999; Ritter and Osterloh, 2007; von Stebut, 2007). Upon infection, DCs become activated and efficiently present Leishmania antigen to CD4 and CD8 cells (Sacks and Noben-Trauth, 2002). Interestingly, restimulation of Leishmania-specific CD8+ T cells was possible only when L. major-containing DCs and not other antigen-presenting cells (i.e., infected macrophages) were used (Belkaid et al., 2002). In addition, infection studies with ovalbumin (OVA)-transgenic parasites showed that proteins released by L. major in infected DCs are a major source of peptides for the generation of parasite-specific CD8+ T cells (Bertholet et al., 2005). Thus, DCs infected with viable parasites present antigen in the context of both the major histocompatibility complex (MHC) class I and II and promote induction of Th1/Tc1-dependent protective immunity.
The development of an effective anti-Leishmania vaccine has yet to be realized. Vaccination approaches include whole-killed or subunit vaccines with adjuvants, genetically attenuated Leishmania parasites, Leishmania peptides expressed in attenuated organisms (e.g., Bacillus Calmette Guérin), DNA vaccines, and DC agonists (CpG motifs, Flt-3 (FMS-like tyrosine kinase 3) ligand) (Vanloubbeeck and Jones, 2004). Most of these approaches have shown some efficacy in murine models, but may be difficult to implement in humans for different reasons (ethical, non-effectiveness in humans, side effects, and so on). Utilization of recombinant peptides/proteins or DNA encoding these antigens represent promising approaches, as in the case of live parasites the risk of reactivation in immunosuppressed patients (e.g., those coinfected with strains inducing visceral disease and HIV) is problematic. Some studies have suggested that persisting viable parasites (perhaps residing in DCs) are required for long-lasting immunity (Moll et al., 1995; Belkaid et al., 2001). Previously, however, it was shown that although effector cells are lost upon complete eradication of parasites, central memory T cells mediate long-term immunity to L. major in the absence of persistent parasites (Zaph et al., 2004). These results support continued emphasis on development of a protein/peptide-based vaccine.
Several Leishmania vaccines have used DCs pulsed with parasite lysates (Flohe et al., 1998; Ahuja et al., 1999; Ramirez-Pineda et al., 2004) or recombinant parasitic proteins (Berberich et al., 2003). However, protein-pulsed DCs were not as efficient in promoting protection as were DCs infected with viable parasites (von Stebut et al., 2000). We previously purified recombinant model tumor-associated antigens containing the HIV TAT protein transduction domain (PTD) (Shibagaki and Udey, 2002, 2003). The PTD promotes accumulation of protein in the cytosol of cells independent of endocytosis (Wadia and Dowdy, 2005). This fusion protein efficiently transduced DCs and was processed by proteasomes for MHC class I-dependent presentation to cytotoxic T lymphocytes (Shibagaki and Udey, 2002, 2003). In addition, TAT-containing antigen was also presented to CD4 T cells as efficiently as native antigen. Finally, TAT-antigen-transduced DCs induced antigen-specific cytotoxic T lymphocytes in vivo and vaccinated against tumors (Shibagaki and Udey, 2002, 2003).
We have now translated this vaccination strategy into a vaccine against leishmaniasis in C57BL/6 mice. We used the Leishmania-specific antigen LACK (Leishmania homolog of receptors for activated C kinase), which is highly conserved among related Leishmania species and is expressed in both promastigote and amastigote forms of the parasite (Mougneau et al., 1995). When compared with LACK alone, TAT–LACK fusion protein was superior in inducing parasite-specific CD8+ T cells and in vaccinating against murine L. major infections.
RESULTS
Generation of fusion proteins comprising the HIV-1 TAT and LACK
Plasmids encoding TAT PTD-containing and control proteins (analogous to those generated previously using OVA; Shibagaki and Udey, 2002) were engineered, and recombinant proteins were expressed in E. coli, purified using Ni2+ affinity chromatography, dialyzed against phosphate-buffered saline (PBS), and separated using 4–12% SDS-PAGE gels (Figure 1a). The purified LACK protein was smaller than TAT–LACK as expected.
Figure 1. Dendritic cells (DCs) pulsed with TAT–LACK are superior in priming and restimulation of antigen-specific CD8+ T cells when compared with LACK DCs.
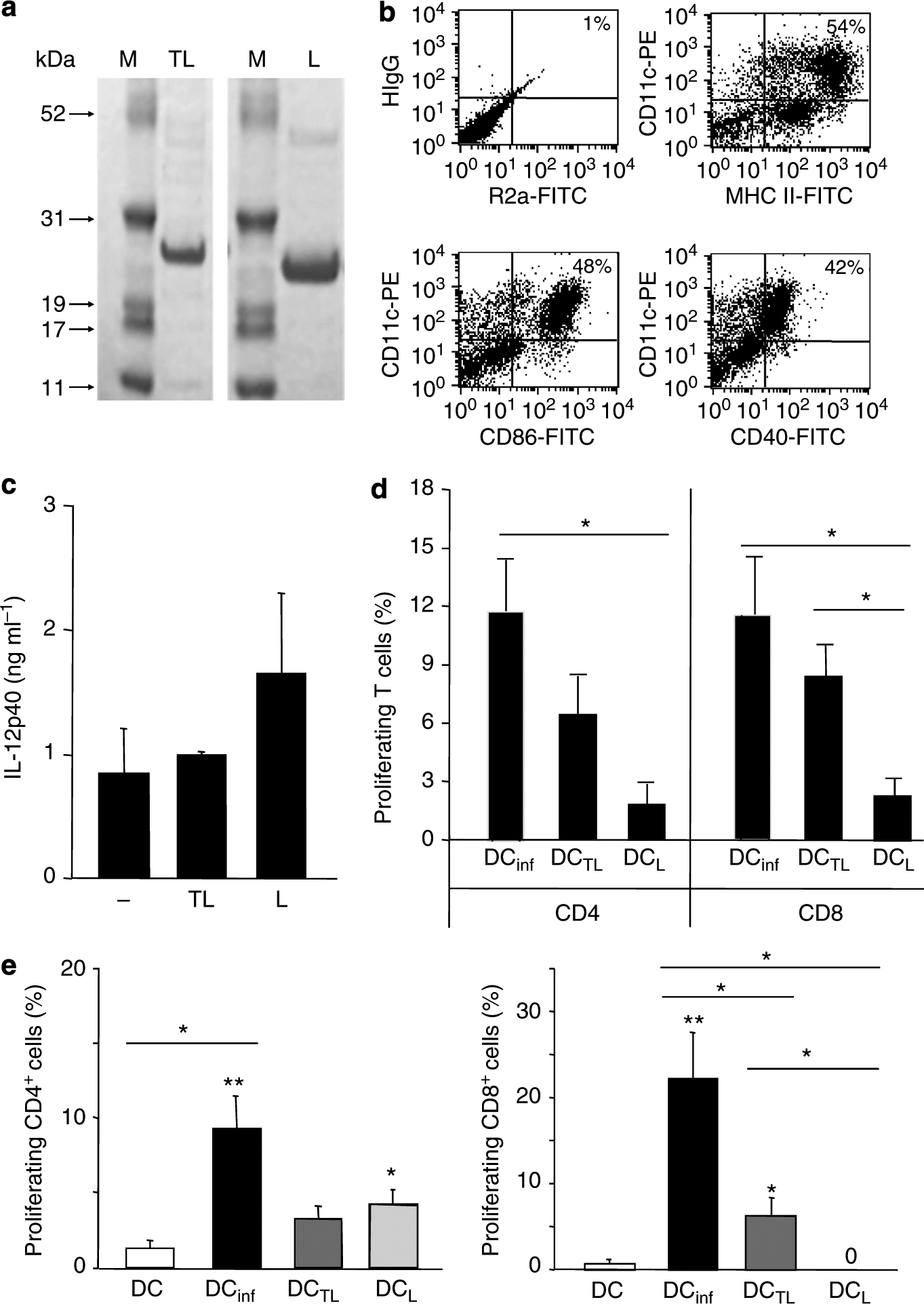
(a) Coomassie blue-stained 4–12% SDS-PAGE gel in which purified TAT–LACK and LACK were resolved. (b) Bone marrow-derived, immature DCs were generated with rIL-4 and rGM-CSF (10 ng ml−1) and harvested as immature DCs on day 6. Expression of activation markers was analyzed by flow cytometry using monoclonal antibodies against CD40, CD86, and MHC class II as well as rat anti-mouse IgG2a (R2a) and hamster anti-mouse IgG1 (HIgG) as isotype controls. One representative FACS staining is shown. (c) DCs were pulsed for 18 hours with TAT–LACK (TL) or LACK (L), or left untreated. IL-12p40 release was determined by ELISA (mean ± SEM, n⩾3). (d) TAT–LACK (DCTL)-treated and LACK (DCL)-treated DCs were obtained or DCs were infected with L. major amastigotes (DCinf, ratio 1:5). T cells were enriched from 6-week L. major-infected C57BL/6 mice using anti-Thy1.2-coated microbeads, labeled with CFSE, and restimulated with irradiated, antigen-pulsed or infected DCs (1:10 ratio of DC/T cells, 1 × 106 per 200 μl). T-cell proliferation was determined after 5 days using flow cytometry. (e) C57BL/6 mice were vaccinated with 2 × 105 untreated DCs, DCinf, DCTL, or DCL. After 1 week, lymph node (LN) cells were isolated, labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE), and antigen-specific proliferation was determined by FACS after 5 days of subculture in the presence of soluble Leishmania antigen (SLA). T cells were pregated using staining for CD4 or CD8. (d, e) For each mouse, the relative number of Leishmania-reactive cells (in percentage of total CD4+ or CD8+ T cells) was calculated when compared with untreated control cultures (mean ± SEM, n⩾3, *P⩽0.05, and **P⩽0.005 when compared with DC alone; *above lines indicate differences between stimulation groups).
Generation and protein transduction of DCs
Bone marrow-derived DCs (BMDCs) have previously been used for vaccination against Leishmania infections (Ahuja et al., 1999; Berberich et al., 2003; Ghosh et al., 2003; Ramirez-Pineda et al., 2004). We generated DCs by culturing bone marrow of C57BL/6 mice with GM-CSF and IL-4 (Inaba et al., 1992). One representative flow cytometric analysis showing the phenotype and purity of the immature DCs harvested on day 6 is provided in Figure 1b. DCs strongly expressed MHC class II and CD11c as well as intermediate and low levels of CD86 and CD40, respectively, consistent with their immature state.
Recombinant fusion proteins were treated with polymyxin B before addition to DCs. Protein transduction was performed by incubating 2 × 105 DCs with various concentrations (0.3–3 μM) of fusion or control proteins as described (Shibagaki and Udey, 2002). Cytosolic distribution of TAT–LACK when compared with LACK was confirmed using confocal microscopy (Shibagaki and Udey, 2002). Protein transduction itself did not lead to significant alterations of MHC class II/costimulatory molecules when compared with untreated DCs or induction of IL-12 release (Figure 1c).
DCs pulsed with TAT-containing fusion protein efficiently prime and restimulate antigen-specific CD8+ T cells
To assess the potential of fusion protein-pulsed DCs to restimulate antigen- specific primed T cells, we first isolated Thy1.2+ T cells (purity >84%) from mice infected for ⩾6 weeks with L. major. BMDCs were either pulsed with antigen (2 μm protein) or were infected with L. major (5:1 amastigote/DC ratio; mean infection rate 38±2%) for 18 hours. DCs were subsequently irradiated and cocultured with carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled T cells for 5 days. Antigen-specific expansion was quantified by determining the percentage of proliferating T cells after subtracting baseline proliferation induced with unpulsed DCs (Figure 1d). Results combined from two to three independent experiments are shown. TAT–LACK- and LACK-pulsed DCs induced proliferation of CD4+ T cells, but less vigorously than DCs infected with live parasites. Differences between TAT–LACK and LACK in CD4+ cells were not statistically significant. In contrast, more efficient restimulation of CD8+ T cells was achieved with TAT–LACK-pulsed DCs when compared with LACK-pulsed DCs (8.3 ± 1.6 vs. 2.3 ± 0.9%, respectively, n⩾3, P⩽0.05).
In addition to T-cell expansion, we assessed the cytokine profile of CD4 and CD8 T cells after restimulation of TAT–LACK- or LACK-pulsed DCs (Table 1). CD4 or CD8 T cells were isolated from infected mice. T cells were plated at 106 per 200 μl and restimulated with irradiated, untreated, protein-pulsed, or infected DCs (105 per 200 μl). Cytokine release from T cells was assessed after 48 hours using ELISA specific for IFN-γ and IL-4. Irradiated DCs did not release IFN-γ, IL-4, or IL-10 (data not shown). When compared with unpulsed DCs, infected DCs and DC + TAT–LACK induced significant IFN-γ release from CD4 cells. The IFN-γ/IL-4 ratios revealed that infected and TAT–LACK-pulsed DCs both induced an IFN-γ-predominant Th1 cytokine profile. Interestingly, LACK-pulsed DCs did not promote IFN-γ release from primed CD4 cells, confirming previous data that LACK-responsive CD4 cells primarily produce IL-4 (Mougneau et al., 1995). In parallel, CD8 cells restimulated with infected DCs or DC + TAT–LACK released higher amounts of IFN-γ when compared with cells stimulated with unpulsed DCs or DCs pulsed with LACK alone. Significant differences in IL-10 levels in the supernatants were not observed.
Table 1.
Cytokine responses of restimulated T cells after incubation with TAT–LACK- or LACK-pulsed DCs
| Restimulation of antigen-specific C57BL/6 T cells w/ | DC | DCinfected | DCTAT-LACK | DCLACK |
|---|---|---|---|---|
| CD4 | ||||
| IFN-γ (pg ml−1) | 20±20 | 7,236±230*** | 973±370+,* | 31±31 |
| IL-4 (pg ml−1) | 5±3 | 107±58 | 1±1 | 7±4 |
| IFN-γ/IL-4 | 7±7 | 317±163 | 681± 60+++,*** | 2±2 |
| IL-10 (pg ml−1) | 426±90 | 631±287 | 534±190 | 520±232 |
| CD8 | ||||
| IFN-γ (pg ml−1) | 777±777 | 4,914±1,754 | 1,556±1,032 | 360±360 |
| IL-4 (pg ml−1) | 2±1 | 20±7* | 28±1 | 11±9 |
| IFN-γ/IL-4 | 777±777 | 225±126 | 260±17+++ | 121±121 |
| IL-10 (pg ml−1) | 248±44 | 278±53 | 308±109 | 416±137 |
Abbreviations: DC, dendritic cell; LACK, Leishmania homolog of receptors for activated C kinase. DC were incubated with TAT–LACK, LACK, or infected (infection rate 25 ± 4%, n⩾3) as described in Materials and Methods. CD4+ or CD8+ cells were isolated from ⩾5-week infected mice, plated at 9 × 105 per 200 μl, and restimulated with DCs (1 × 105) for 48 hours. Cytokine content of supernatants was determined by ELISA (mean ± SEM; pooled data from three independent experiments; n⩾3 mice per group; statistical comparisons are provided against DC alone (*) or DCLACK (+)).
P⩽0.05, and
P⩽0.002.
We next sought to determine whether preferential induction of antigen-specific CD8+ T cells (in addition to CD4 cells) could be achieved by in vivo vaccination of mice with antigen-pulsed DCs (Figure 1e). BMDCs were pulsed overnight with 333 μm of TAT–LACK or LACK in the presence of polymyxin B or infected with amastigotes of L. major (ratio 1:5, mean infection rates after 18 hours 27 ± 3%). Cells were harvested, washed, counted, and injected intradermally into the ear skin of naive mice (2 × 105 cells per 50 μl per ear). After 1 week, lymph node (LN) cells were harvested, labeled with CFSE, and restimulated with soluble Leishmania antigen (SLA, 25 μg ml−1) or staphylococcal enterotoxin B (10 μg ml−1) as a control (data not shown). Antigen-specific expansion of CD4+ and CD8+ T cells was assessed 5 days after restimulation. Impaired proliferation of T cells was not found in any of the vaccination groups as assessed by stimulation with superantigen. Both DC + LACK and DC + TAT–LACK induced priming of antigen-specific CD4+ T cells to a similar degree, suggesting that the level of LACK uptake by DCs per se was similar. Protein-pulsed DCs did not induce responses to the extent observed with DCs infected with live parasites. DC + TAT–LACK induced clear CD8 priming as did infected DCs. Most importantly, TAT–LACK-pulsed DCs more efficiently primed CD8+ T cells in vivo when compared with DCs pulsed with LACK alone (4.5 ± 1.5 vs. 0%, respectively, n⩾4, P⩽0.05).
Vaccination with TAT–LACK-pulsed DCs is superior to DCs pulsed with LACK alone
We next investigated the vaccination potential of fusion protein-pulsed DCs in Leishmania-resistant C57BL/6 mice in vivo. LACK reactivity of naive CD4+ T cells has been detected in C57BL/6 mice (Sacks and Noben-Trauth, 2002). Data generated with Leishmania-resistant mice might be most relevant to a clinical setting because the course of disease in C57BL/6 mice most closely mimics natural L. major infections of humans.
At 1 week before infection, C57BL/6 mice were vaccinated with 2 × 105 DCs pulsed with fusion protein, LACK alone, or irrelevant controls (DC alone, DC plus TAT-β-galactosidase (βgal), or βgal; Figure 2). Mice were infected into contralateral ears with either high-dose (2 × 105 parasites, Figure 2a) or low-dose inocula (Figure 2b–f), and lesion development was assessed weekly. In both high- and low-dose infections, vaccination with TAT–LACK-pulsed DCs was more efficacious than vaccination with DCs pulsed with LACK alone. This effect was most pronounced in low-dose infections (Figure 2b). Vaccination with irrelevant controls or unpulsed DCs did not have an effect on lesion development when compared with unvaccinated mice (Figure 2c). Smaller lesion sizes in DC + TAT–LACK-vaccinated mice paralleled significantly decreased lesional parasite burdens (Figure 2d). In week 7 after infection, ears of mice vaccinated with TAT–LACK contained 4.5 ± 0.9 × 102 parasites when compared with LACK-DC-treated groups (5.1 ± 1.1 × 103 parasites, n⩾6, P⩽0.005).
Figure 2. Improved disease outcome after vaccination with fusion protein-pulsed dendritic cells (DCs).
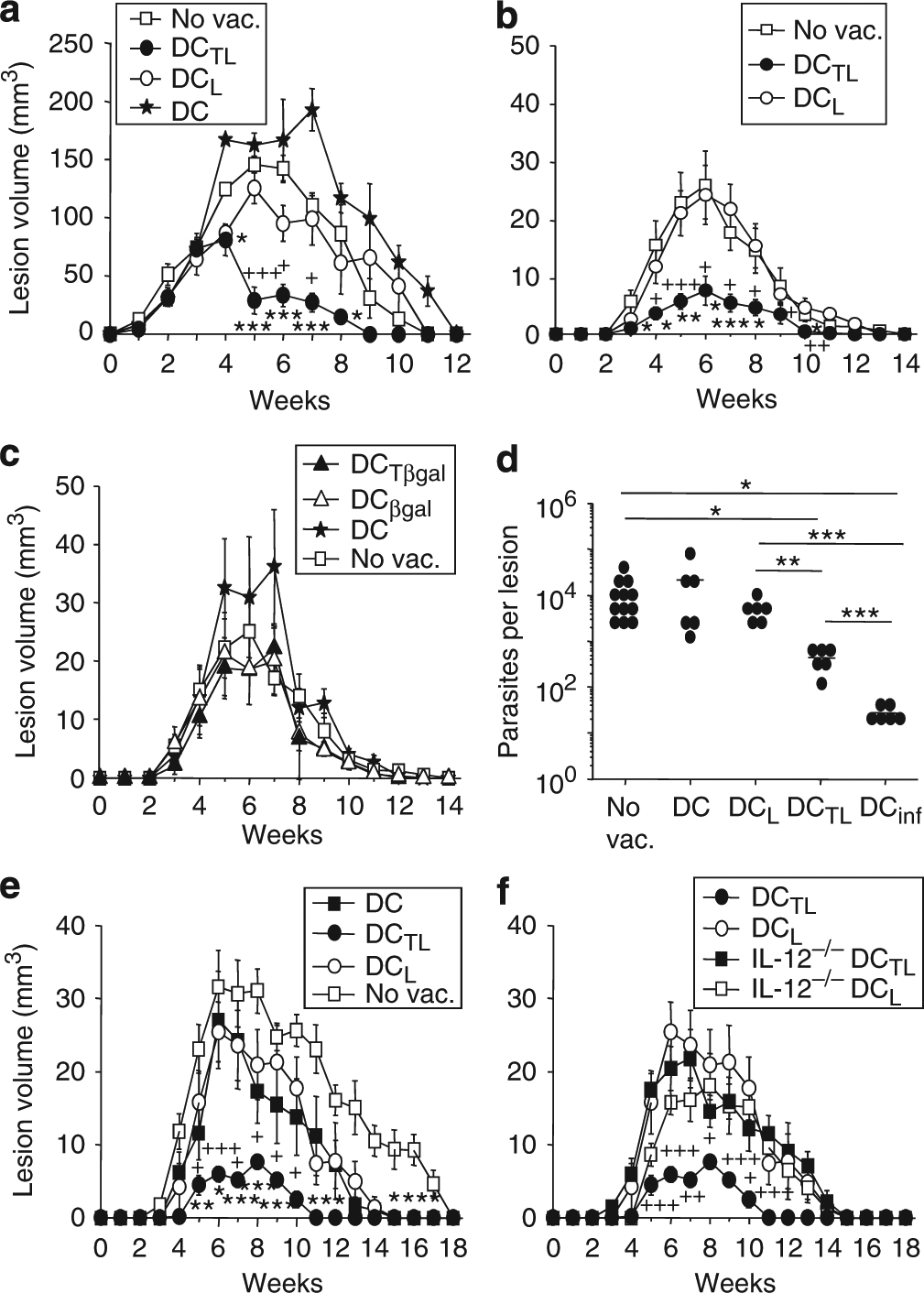
DCs were generated from IL-12p40−/− (f) or wild-type C57BL/6 (a–f) mice and pulsed overnight with TAT–LACK (DCTL), LACK alone (DCL), TAT-βgal (DCTβgal), or βgal (DCβgal). Protein-pulsed DCs (2 × 105) were injected intradermally into the ear skin of C57BL/6 mice. After 1 week, mice were infected with 2 × 105 (a) or 103 (b–f) metacyclic promastigotes of L. major. (a–c, e, f) Lesion volumes were assessed every week using a caliper (mean ± SEM, n⩾4, statistical differences with unvaccinated groups (*) and with LACK-pulsed DCs (+) are shown, *, + P⩽0.05, **, + +P⩽0.005, and ***, + + + P⩽0.002). (d) Lesional parasite loads of vaccinated C57BL/6 mice were determined by limiting dilution assays. DCs were infected with amastigotes of L. major (parasite/cell ratio 5:1). Each data point represents the number of organisms from one ear and the bars indicate arithmetic means. Pooled data of two independent experiments are shown (n⩾6; *P⩽0.05 and **P⩽0.005).
In a previous report, pretreatment of antigen-pulsed DCs with CpG oligonucleotides (ODNs) further promoted protection (independent of DC-derived IL-12 production; Ramirez-Pineda et al., 2004). In our model, cotreatment with 10 μg CpG ODN 1826 injected into vaccination sites did not have an adjuvant effect on disease outcome (data not shown).
Successful vaccination with fusion-protein-pulsed DCs is IL-12 dependent
Induction of Th1/Tc1-dependent protection against Leishmania infections is critically dependent on the release of IL-12 from infected DCs (Sacks and Noben-Trauth, 2002). We thus determined whether release of IL-12 from protein-pulsed DCs is important for the efficacy of our vaccination approach. BMDCs were generated from C57BL/6 IL-12p40−/− or wild-type controls. Characterization of IL-12p40−/− and wild-type DCs revealed no differences in expression of CD11c, MHC class II, as well as the costimulatory molecules CD40 and CD86, as determined by flow cytometry (data not shown). DCs were pulsed overnight with antigen. Similar to previous experiments, mice were vaccinated and infections were initiated 1 week later by inoculation of 103 metacyclic promastigotes into contralateral ears (Figure 2 e and f). TAT–LACK-pulsed IL-12p40-deficient DCs failed to protect vaccinated mice from L. major infection (Figure 2f). Thus, release of IL-12p40 from DCs is required for TAT–LACK-mediated immunity against L. major.
Utilization of TAT–LACK for direct in vivo vaccination
Generation/isolation of DCs ex vivo from humans is laborious and expensive and will not be feasible in Leishmania-endemic countries. We thus initiated studies analyzing the potential of a TAT–LACK fusion protein vaccine directly in vivo to circumvent ex vivo protein transduction of DCs.
First, the ability of the direct protein vaccine to induce antigen-specific T-cell responses in vivo was assessed. Mice were vaccinated with TAT–LACK, LACK, irrelevant TAT-OVA, or SLA on days −7 and −6. Subsequently, 10 μg of CpG ODN 1826 was administered into the same ears on day −5. On day 0, LN cells were isolated, labeled with CFSE, and restimulated with 25 μg ml−1 SLA. Antigen-specific expansion of CD4+ and CD8+ T cells was assessed 4 days after restimulation (Figure 3a and b). Despite the fact that the induction of antigen-specific T cells was weaker with protein when compared with the DC-based vaccine (compare Figure 1e) as expected, TAT–LACK-mediated T-cell priming was stronger than that by LACK alone for both CD4 and CD8 cells.
Figure 3. Efficacious vaccination with fusion proteins directly in vivo depends on both CD4+ and CD8+ T cells.

(a, b) C57BL/6 mice were vaccinated with 10 μg TAT–LACK (TL) or appropriate controls into ears on day −7/−6 followed by 10 μg CpG 1826 on day −5. On day 0, draining lymph node (LN) cells were harvested, single-cell suspensions were prepared, and cells were stained with carboxyfluorescein diacetate succinimidyl ester (CFSE; 1 μM). Labeled cells were plated at 1 × 106 cells per 200 μl and stimulated with antigen. After 4 days, cells were harvested and T-cell proliferation was analyzed by FACS using antibodies against CD4 and CD8. The relative number of Leishmania-reactive cells per mouse (%) was calculated when compared with untreated control cultures (mean ±SEM, n = 3). (c) C57BL/6 mice (5 mice per group) were injected with anti-CD4, anti-CD8, or irrelevant control mAb (i.p.) and vaccinated twice with 10 μg TL or LACK intradermally into the ear skin. CpG oligonucleotide (ODN) 1826 was administered the following day (10 μg per ear). After 3 weeks, after complete restoration of T cells, mice were infected with 1,000 metacyclic promastigotes of L. major. Lesion development was monitored weekly in three dimensions and expressed as mean ± SEM (pooled data from three independent experiments are shown, n⩾9, statistical differences with unvaccinated groups are shown, *P⩽0.05, and ***P⩽0.002).
We next attempted to vaccinate with TAT–LACK fusion proteins directly in vivo. TAT–LACK and LACK were administered intradermally into ear skin for two consecutive days (10 μg per ear), and infections with 103 metacyclic promastigotes were initiated 2 weeks later. When compared with PBS-treated control mice, no protective effect was observed (data not shown). Next, recombinant fusion proteins were administered for 2 days followed by a single injection of CpG ODN 1826 (10 μg per ear; Zimmermann et al., 1998; Stacey and Blackwell, 1999; Walker et al., 1999) as adjuvant on day −12 (Figure 3c). Irrelevant (fusion) proteins plus CpG ODN 1826 were included as controls (data not shown). Interestingly, only TAT–LACK + CpG ODN significantly prevented disease progression of infected C57BL/6 mice, whereas controls were ineffective. As shown for the DC-based vaccination approach, TAT–LACK-mediated protection was always superior to LACK alone (e.g., 4 ± 0.6 vs. 13 ± 2.6 mm3 in week 7 after infection, respectively, n⩾12, P⩽0.002).
The vaccination efficacy of TAT–LACK fusion protein is dependent on both CD4 and CD8 cells
To assess the dependence of the efficacy of TAT–LACK as a vaccine on CD4 and CD8 cells, we performed depletion experiments in vivo. To ensure that T cells were depleted only during vaccination, extensive dose-response experiments were performed. To achieve sufficient depletion, 125 μg of anti-CD4 mAb (clone GK1.5) or 50 μg of anti-CD8 (clone 2.43) were injected 2 days before vaccination. As a control, some mice were treated with an irrelevant isotype control antibody. The T-cell depletion efficacy was determined on the day of vaccination. The frequency of CD4+ and CD8+ cells in peripheral blood of anti-CD4- and anti-CD8-treated mice was 0.05 and 0.9%, respectively. Full restoration of the T-cell compartment was confirmed by flow cytometry from peripheral blood before mice were infected with low-dose inocula of L. major (day 21 after vaccination). Interestingly, if either CD4 or CD8 T cells were depleted during vaccination, the vaccination success of TAT–LACK was completely abrogated, as manifested by lesion volumes (Figure 3c).
DISCUSSION
Protective immunity against L. major is dependent on both Th1- and Tc1-dependent immunity. In this study we have used fusion proteins comprising the 11aa PTD from HIV-1 TAT and LACK. We showed that vaccination with fusion proteins containing TAT was consistently superior in promoting protection against cutaneous leishmaniasis when compared with proteins without TAT. Thus, in these proof-of-principle experiments, we have shown that intentional induction of antigen-specific CD8+ T cells using TAT-fusion proteins, in addition to CD4+ T cells, is beneficial for protection against this important pathogen.
Although infected (skin-derived) DCs seem to be the critical cells for the induction and maintenance of protective immunity against L. major (Flohe et al., 1998; von Stebut et al., 1998, 2000; Ahuja et al., 1999; Sato et al., 2000; Lemos et al., 2004; Iezzi et al., 2006), use of protein-loaded DCs showed only limited results when compared with cells infected with live parasites (Berberich et al., 2003; Ramirez-Pineda et al., 2004). Parasites and protein enter DCs in different ways. Parasite internalization results from FcγRI/III-mediated phagocytosis (Woelbing et al., 2006), whereas protein uptake is the result of endocytosis. Importantly, FcγR-mediated parasite uptake facilitates MHC class I and II antigen processing, whereas protein endocytosis favors MHC class II-predominant antigen presentation (Amigorena, 2002; Belkaid et al., 2002; Woelbing et al., 2006). Thus, inefficient induction of CD8 T cells (rather than suboptimal CD4 priming) may be responsible for the inability of protein-pulsed DCs to promote effective and long-term antiparasite immunity when compared with infected DCs. This concept is supported by our finding that vaccination with DC + LACK induced comparable CD4 priming as did DC + TAT–LACK, whereas CD8 priming was weaker in mice vaccinated with DCs pulsed with LACK protein alone.
The role of antigen-specific CD4+ T cells in protective immunity is well established (Reiner and Locksley, 1995; Sacks and Noben-Trauth, 2002). Additional studies have highlighted the important role that Leishmania-specific CD8+ Tc1 cells have in mediating protection, especially in the physiologically more relevant low-dose infection model mimicking natural transmission of L. major by sand flies, but not in high-dose infections (Muller et al., 1991; Erb et al., 1996; Belkaid et al., 2002; Uzonna et al., 2004). In addition, previous experiments using DNA-vaccination approaches induced CD8-dependent protection, which led to efficient protection (Gurunathan et al., 1997). In this study we showed that induction of both LACK-reactive CD4 and CD8 T cells contributed to protection against L. major infection after vaccination with TAT–LACK fusion protein. In a previous study, DCs transduced with HIV TAT-PTD-containing whole protein antigens also stimulated both antigen-specific CD8+ and CD4+ T cells (Mitsui et al., 2006). Thus, the superiority of TAT–LACK-pulsed DCs likely relates to their ability to induce CD8 priming in addition to CD4 priming, which was also achieved by LACK-treated DCs. This confirms the results of previous studies that showed that CD8+ T cells are critical for protection using LACK as the vaccinating antigen (Gurunathan et al., 1997, 2000; Shah et al., 2003; Dondji et al., 2008).
LACK has been found to be the focus of the early immune response directed against the parasites, with most LACK-reactive T cells producing IL-4, but not IFN-γ (Mougneau et al., 1995). LACK-induced preferential expansion of parasite-specific Th2 cells and a low level of IFN-γ production result in progressive infection of BALB/c mice and fatal outcome (Heinzel et al., 1989). It is accepted that C57BL/6 mice are by far the better model than BALB/c mice to investigate vaccines with potential relevance for human infections. However, as a reaction status similar to that in BALB/c mice may occur in some humans, the effects of the vaccination procedure described in this study were also assessed in susceptible BALB/c mice (data not shown). Using TAT-fusion proteins in both DC- and protein-based vaccines, TAT–LACK-treated BALB/c mice were significantly protected against progressive infection with L. major, as manifested by clearly reduced lesion volumes and parasite burdens. Similar to the findings reported in this study, TAT–LACK was superior to LACK, and TAT–LACK-mediated protection was dependent on CD4+ as well as CD8+ T cells. LACK may not be the optimal antigen choice for vaccinations. Previous experiments confirmed that LACK-pulsed DCs did not efficiently promote protection relative to that observed with other Leishmania-derived antigens (Berberich et al., 2003). Attempts to modify the immune response to the large LACK protein by using different epitopes have been successful (Jensen et al., 2009). Thus, it is reasonable to predict that if an optimized antigen combination (such as certain LACK peptides or other antigens capable of inducing more Th1/Tc1-prone immunity) was used for TAT-fusion protein-based vaccinations, the results may be even more promising.
In this study, fusion protein as well as LACK alone did not promote protection directly in vivo, confirming previous studies using proteins without adjuvant (Mougneau et al., 1995; Berberich et al., 2003; Shah et al., 2003; Ramirez-Pineda et al., 2004). Activation of skin DCs and IL-12 synthesis is required as achieved in vivo by coadministration of adjuvants such as CpG motifs or others (Zimmermann et al., 1998; Walker et al., 1999; Shah et al., 2003). In addition, we also showed that the vaccination efficacy of DCs pulsed with TAT–LACK was dependent on the production of IL-12 from the DCs, confirming previous results (Berberich et al., 2003). Independent of exogenous DCs, TAT–LACK fusion protein together with CpG efficiently induced protection against progressive leishmaniasis directly in vivo. TAT–LACK was consistently superior to LACK alone. These latter results are important, because DC-based vaccination approaches will not be widely applicable.
In summary, in this proof-of-principle study, we have shown that induction of Leishmania-specific CD8+ T cells, in addition to CD4+ cells, is beneficial for the development of protective immunity. In both DC- and protein-based vaccinations, immunization with TAT–LACK was superior to injection of LACK alone. Thus, TAT-fusion proteins represent promising tools for the induction of protective immunity against not only cancer, but also infectious diseases with intracellular pathogens. Future experiments will evaluate in more detail how this immunization strategy can be improved (e.g., by studying the durability of its efficacy) and how this vaccination approach can be translated into humans.
MATERIALS AND METHODS
Generation and characterization of fusion protein
His6-tagged TAT-hemagglutinin (HA)-OVA, HA-OVA, TAT-HA-βgal, and HA-βgal were obtained and generated as described previously (Shibagaki and Udey, 2002). Constructs encoding His6-tagged TATHA-LACK were generated by inserting LACK complementary DNA (+427 to +939, 170 aa; provided by Dr D Sacks, National Institute of Allergy and Infectious Diseases, Bethesda, MD) into multiple-cloning sites (NcoI/EcoRI) of pTAT-HA plasmid in-frame. HA-LACK constructs were generated by BamHI digestion and religation of pTAT-HA-LACK plasmids. Plasmids were then transformed into a RosettaBlue (DE3) competent bacterial strain (Novagen, Merck, Darmstadt, Germany). For protein extraction, bacterial cell pellets were sonicated in urea-containing medium. Denatured TAT-HA-LACK, HA-LACK, TAT-HA-OVA, HA-OVA, TAT-HA-βgal, and HA-βgal were then purified by sequential Ni2+ nitrilotriacetic acid-agarose chromatography and dialyzed against PBS as described (Shibagaki and Udey, 2002). Proteins were stored at −20 °C in PBS/10% glycerol and thawed immediately before use. SDS-PAGE was performed with NuPAGE 4–12% Bis-Tris gels and MOPS running buffer (Shibagaki and Udey, 2002).
Mice and parasites
C57BL/6 mice, 6–8 weeks old, were purchased from the Central Animal Facility of the University of Mainz. IL-12p40−/− C57BL/6 mice were kindly provided by Professor E Schmitt, Department of Immunology, University of Mainz. All animals were housed in accordance with institutional and federal guidelines. The experiments were undertaken with approved license from the animal care and use committee of the Region Rheinland-Pfalz, Germany. Metacyclic promastigotes or amastigotes of L. major clone VI (MHOM/IL/80/Friedlin) were prepared as described previously (Woelbing et al., 2006).
Generation and pulsing of DCs
BMDCs were generated from wild-type or IL-12p40−/− mice in GM-CSF- and IL-4-containing media (32) and harvested on day 6 of cell culture. The characteristics of the cell populations were assessed by flow cytometry using relevant surface markers. The following antibodies were used: anti-I-Ab,d/I-Ed (2G9), anti-CD11c (HL3), anti-CD40 (3/23), and anti-CD86 (GL1) (all from BD Biosciences/Becton Dickinson, Heidelberg, Germany). For protein pulsing of DCs, cells were plated at 1 × 106 per ml in RPMI/5% fetal calf serum and recombinant proteins were added at 333 nM up to 2 μM (Shibagaki and Udey, 2002). Recombinant proteins were treated with polymyxin B sulfate (50 μg ml−1, Sigma Aldrich, St Louis, MO) before addition to DCs. After 18 hours of DC pulsing, supernatants were harvested and cytokine production was analyzed using ELISA for IL12p40 (R&D Systems, Wiesbaden, Germany).
Restimulation of Leishmania-specific T cells with DCs
T cells were isolated from L. major-infected C57BL/6 mice using microbeads coated with anti-CD90 (Thy1.2) (Miltenyi, Bergisch-Gladbach, Germany). T cells (5 × 106 per ml) were labeled with 1 μm CFSE (Invitrogen, Karlsruhe, Germany) and subcultured with irradiated DCs (400 Gy) in a ratio of 10:1 for additional 5 days (106 cells per 200 μl). Antigen-specific proliferation was assessed after 5 days using flow cytometry. T cells were selected for analysis using mAbs against CD4 (L3T4, RM4–5), CD8 (Ly2, 53–6.7), or isotype control mAb (all from BD Biosciences). For each mouse, the percentage of Leishmania-reactive cells compared with nonproliferating cells was calculated as described (Woelbing et al., 2006).
To determine the T-cell cytokine profile after restimulation with TAT–LACK-transduced DCs, Leishmania-primed T cells were isolated from infected C57BL/6 mice using anti-CD4 and anti-CD8 microbeads (Miltenyi). T cells and DCs were cocultured in a ratio of 10:1 (1 × 106 cells per 200 μl). After 48 hours of incubation, supernatants were harvested and cytokine production was assessed using ELISA for IL-4 (BD Biosciences) and IFN-γ (R&D Systems).
Assessing the induction of LACK-reactive T cells in vivo
Groups of ⩾5 C57BL/6 mice were vaccinated in vivo by intradermal injection of 2 × 105 protein-pulsed DCs. For the assessment of an induction of LACK-reactive T cells, LN cells were harvested 1 week after vaccination of naive mice, labeled with CFSE as described above, and plated at 106 per 200 μl in 96-well U-bottom plates. Cells were stimulated with SLA or SEB. After 5 days, the frequency of daughter cells of proliferating antigen-reactive compared with nonproliferating LN T cells was estimated using flow cytometry (24). T cells were pregated using staining for CD4 (L3T4, RM4–5), CD8 (Ly2, 53–6.7), or isotype control mAb (all from BD Biosciences). The percentage of LACK-reactive cells was calculated separately for each mouse and the data of three independent experiments were pooled and expressed as mean ± SEM.
Vaccination schedules and L. major infection
Groups of ⩾5 mice were vaccinated with 2 × 105 per 50 μl protein-pulsed or untreated DCs injected intradermally into the ear skin. In some experiments, mice were vaccinated with fusion proteins dissolved in PBS for two consecutive days (10 μg per ear) followed by a single injection of CpG ODN 1826 (10 μg per ear). After 1 to 3 weeks, infections were initiated into contralateral ears by inoculating 2 × 105 or 103 metacyclic promastigotes as indicated (Belkaid et al., 2000). In some experiments, anti-CD4 (clone GK1.5, 125 μg per mouse) or anti-CD8 (clone 2.43, 50 μg per mouse) were injected intraperitoneally 2 days before vaccination.
Disease outcome was determined by assessing lesion volumes, the number of parasites/lesion, and cytokine profiles of draining LN cells. Lesion development was assessed weekly in three dimensions using a caliper, and lesional volumes were reported (in mm3) as ellipsoids ((a/2 × b/2 × c/2) 4/3π). Organisms present in lesional tissue were enumerated using limiting dilution assays (Woelbing et al., 2006). For measurement of cytokine production, 106 retroauricular LN cells per 200 μl were added to 96-well plates in the presence of SLA (25 μg ml−1). Antigen-specific IFN-γ (R&D Systems), IL-4, and IL-10 (BD Biosciences) production was determined after 48 hours using ELISA.
Statistics
Statistical analysis was performed using the unpaired Student’s t-test.
ACKNOWLEDGMENTS
We thank Dr Steven Dowdy for supplying the TAT-HA protein-encoding plasmids and for helpful discussions, Dr David L Sacks for providing LACK plasmids, and Drs K Steinbrink and H Jonuleit for critically reading this paper. This work was supported in part by grants from the Special Programme for Research and Training in Tropical Diseases (TDR) of the World Health Organization (WHO) (to EvS/MCU), the Deutsche Forschungsgemeinschaft (SFB 490 and 548), the MAIFOR program of the Johannes Gutenberg-University of Mainz (to EvS), the Intramural Program of the NIH, Center for Cancer Research, National Cancer Institute (to MCU), and the Boehringer Ingelheim Fonds (Bifond Travel Grant; to KK).
Abbreviations:
- BMDC
bone marrow-derived DC
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- DC
dendritic cell
- HA
hemagglutinin
- LACK
Leishmania homolog of receptors for activated C kinase
- LN
lymph node
- MHC
major histocompatibility complex
- ODN
oligonucleotide
- OVA
ovalbumin
- PBS
phosphate-buffered saline
- PTD
protein transduction domain
- SLA
soluble Leishmania antigen
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
REFERENCES
- Ahuja SS, Reddick RL, Sato N et al. (1999) Dendritic cell (DC)-based anti-infective strategies: DCs engineered to secrete IL-12 are a potent vaccine in a murine model of an intracellular infection. J Immunol 163:3890–7 [PubMed] [Google Scholar]
- Amigorena S (2002) Fcg receptors and cross-presentation in dendritic cells. J Exp Med 195:F1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Hoffmann KF, Mendez S et al. (2001) The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med 194:1497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Mendez S, Lira R et al. (2000) A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J Immunol 165:969–77 [DOI] [PubMed] [Google Scholar]
- Belkaid Y, von Stebut E, Mendez S et al. (2002) CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J Immunol 168:3992–4000 [DOI] [PubMed] [Google Scholar]
- Berberich C, Ramírez-Pineda JR, Hambrecht C et al. (2003) Dendritic cell (DC)-based protection against an intracellular pathogen is dependent upon DC derived IL-12 and can be induced by molecularly defined antigens. J Immunol 170:3171–9 [DOI] [PubMed] [Google Scholar]
- Bertholet S, Debrabant A, Afrin F et al. (2005) Antigen requirements for efficient priming of CD8+ T cells by Leishmania major-infected dendritic cells. Infect Immun 73:6620–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondji B, Deak E, Goldsmith-Pestana K et al. (2008) Intradermal NKT cell activation during DNA priming in heterologous prime-boost vaccination enhances T cell responses and protection against Leishmania. Eur J Immunol 38:706–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb K, Blank C, Ritter U et al. (1996) Leishmania major infection in major histocompatibility complex class II-deficient mice: CD8+ T cells do not mediate a protective immune response. Immunobiology 195:243–60 [DOI] [PubMed] [Google Scholar]
- Flohe SB, Bauer C, Flohe S et al. (1998) Antigen-pulsed epidermal Langerhans cells protect susceptible mice from infection with the intracellular parasite Leishmania major. Eur J Immunol 28:3800–11 [DOI] [PubMed] [Google Scholar]
- Geiger B, Wenzel J, Hantschke M et al. (2010) Resolving lesions in human cutaneous Leishmaniasis predominantly harbour chemokine receptor CXCR3-positive T helper 1/T cytotoxic type 1 cells. Br J Dermatol 162:870–4 [DOI] [PubMed] [Google Scholar]
- Ghosh M, Pal C, Ray M et al. (2003) Dendritic cell-based immunotherapy combined with antimony-based chemotherapy cures established murine visceral Leishmaniasis. J Immunol 170:5625–9 [DOI] [PubMed] [Google Scholar]
- Gurunathan S, Sacks DL, Brown DR et al. (1997) Vaccination with DNA encoding the immunodominant LACK parasite antigen confers protective immunity to mice infected with Leishmania major. J Exp Med 186:1137–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurunathan S, Stobie L, Prussin C et al. (2000) Requirements for the maintenance of Th1 immunity in vivo following DNA vaccination: a potential immunoregulatory role for CD8+ T cells. J Immunol 165:915–24 [DOI] [PubMed] [Google Scholar]
- Heinzel FP, Sadick MD, Holaday BJ et al. (1989) Reciprocal expression of interferon gamma or interleukin 4 during the resolution or progression of murine Leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J Exp Med 169:59–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iezzi G, Fröhlich A, Ernst B et al. (2006) Lymph node resident rather than skin-derived dendritic cells initiate specific T cell responses after Leishmania major infection. J Immunol 177:1250–6 [DOI] [PubMed] [Google Scholar]
- Inaba K, Inaba M, Romani N et al. (1992) Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med 176:1693–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KDC, Sercarz EE, Gabaglia CR (2009) Altered peptide ligands can modify the Th2 T cell response to the immunodominant 161–175 peptide of LACK (Leishmania homolog for the receptor of activated C kinase). Mol Immunol 46:366–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharazmi A, Kemp K, Ismail A et al. (1999) T-cell response in human Leishmaniasis. Immunol Lett 65:105–8 [DOI] [PubMed] [Google Scholar]
- Lemos MP, Esquivel F, Scott P et al. (2004) MCH class II expression restricted to CD8alpha+ and CD11b+ dendritic cells is sufficient for control of Leishmania major. J Exp Med 199:725–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui H, Inozume T, Kitamura R et al. (2006) Polyarginine-mediated protein delivery to dendritic cells presents antigen more efficiently onto MHC class I and class II and elicits superior antitumor immunity. J Invest Dermatol 126:1804–12 [DOI] [PubMed] [Google Scholar]
- Moll H, Flohe S, Rollinghoff M (1995) Dendritic cells in Leishmania major-immune mice harbor persistent parasites and mediate an antigen-specific T cell immune response. Eur J Immunol 25:693–9 [DOI] [PubMed] [Google Scholar]
- Mougneau E, Altare F, Wakil AE et al. (1995) Expression cloning of a protective Leishmania antigen. Science 268:563–6 [DOI] [PubMed] [Google Scholar]
- Muller I, Pedrazzini T, Kropf P et al. (1991) Establishment of resistance to Leishmania major infection in susceptible BALB/c mice requires parasite-specific CD8+ T cells. Int Immunol 3:587–97 [DOI] [PubMed] [Google Scholar]
- Murray HW, Berman JD, Davies CR et al. (2005) Advances in Leishmaniasis. Lancet 366:1561–77 [DOI] [PubMed] [Google Scholar]
- Ramirez-Pineda JR, Frohlich A, Berberich C et al. (2004) Dendritic cells (DC) activated by CpG DNA ex vivo are potent inducers of host resistance to an intracellular pathogen that is independent of IL-12 derived from the immunizing DC. J Immunol 172:6281–9 [DOI] [PubMed] [Google Scholar]
- Reiner SL, Locksley RM (1995) The regulation of immunity to Leishmania major. Annu Rev Immunol 13:151–77 [DOI] [PubMed] [Google Scholar]
- Ritter U, Osterloh A (2007) A new view on cutaneous dendritic cell subsets in experimental Leishmaniasis. Med Microbiol Immunol 196:51–9 [DOI] [PubMed] [Google Scholar]
- Sacks D, Noben-Trauth N (2002) The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol 2:845–58 [DOI] [PubMed] [Google Scholar]
- Saha S, Mondal S, Banerjee A et al. (2006) Immune responses in kala-azar. Indian J Med Res 123:245–66 [PubMed] [Google Scholar]
- Sato N, Ahuja SK, Quinones M et al. (2000) CC chemokine receptor (CCR)2 is required for Langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells. Absence of CCR2 shifts the Leishmania major-resistant phenotype to a susceptible state dominated by Th2 cytokines, B cell outgrowth, and sustained neutrophilic inflammation. J Exp Med 192:205–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah JA, Darrah PA, Ambrozak DR et al. (2003) Dendritic cells are responsible for the capacity of CpG oligodeoxynucleotides to act as an adjuvant for protective vaccine immunity against Leishmania major in mice. J Exp Med 198:281–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibagaki N, Udey MC (2002) Dendritic cells transduced with protein antigens induce cytotoxic lymphocytes and elicit antitumor immunity. J Immunol 168:2393–401 [DOI] [PubMed] [Google Scholar]
- Shibagaki N, Udey MC (2003) Dendritic cells transduced with TAT protein transduction domain-containing tyrosinase-related protein 2 vaccinate against murine melanoma. Eur J Immunol 33:850–60 [DOI] [PubMed] [Google Scholar]
- Stacey KJ, Blackwell JM (1999) Immunostimulatory DNA as an adjuvant in vaccination against Leishmania major. Infect Immun 67:3719–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzonna JE, Joyce KL, Scott P (2004) Low dose Leishmania major promotes a transient T helper cell type 2 response that is down-regulated by interferon gamma-producing CD8+ T cells. J Exp Med 199:1559–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Weyenbergh J, Santana G, D’Oliveira A Jr et al. (2004) Zinc/copper imbalance reflects immune dysfunction in human Leishmaniasis: an ex vivo and in vitro study. BMC Infect Dis 17:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanloubbeeck Y, Jones DE (2004) The immunology of Leishmania infection and the implications for vaccine development. Ann NY Acad Sci 1026:267–72 [DOI] [PubMed] [Google Scholar]
- von Stebut E (2007) Immunology of cutaneous Leishmaniasis: the role of mast cells, phagocytes and dendritic cells for protective immunity. Eur J Dermatol 17:115–22 [DOI] [PubMed] [Google Scholar]
- von Stebut E, Belkaid Y, Jakob T et al. (1998) Uptake of Leishmania major amastigotes results in activation and interleukin 12 release from murine skin-derived dendritic cells: implications for the initiation of anti-Leishmania immunity. J Exp Med 188:1547–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Stebut E, Belkaid Y, Nguyen BV et al. (2000) Leishmania major-infected murine Langerhans cell-like dendritic cells from susceptible mice release IL-12 after infection and vaccinate against experimental cutaneous Leishmaniasis. Eur J Immunol 30:3498–506 [DOI] [PubMed] [Google Scholar]
- Wadia JS, Dowdy SF (2005) Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Adv Drug Deliv Rev 57:579–96 [DOI] [PubMed] [Google Scholar]
- Walker PS, Scharton-Kersten T, Krieg AM et al. (1999) Immunostimulatory oligodeoxynucleotides promote protective immunity and provide systemic therapy for Leishmaniasis via IL-12- and IFN-gamma-dependent mechanisms. Proc Natl Acad Sci USA 96:6970–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woelbing F, Lopez Kostka S, Moelle K et al. (2006) Uptake of Leishmania major by dendritic cells is mediated by Fcgamma receptors and facilitates acquisition of protective immunity. J Exp Med 203:177–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaph C, Uzonna J, Beverley SM et al. (2004) Central memory T cells mediate long-term immunity to Leishmania major in the absence of persistent parasites. Nat Med 10:1104–10 [DOI] [PubMed] [Google Scholar]
- Zimmermann S, Egeter O, Hausmann S et al. (1998) CpG oligodeoxynucleotides trigger protective and curative Th1 responses in lethal murine Leishmaniasis. J Immunol 160:3627–30 [PubMed] [Google Scholar]