

Abstract
Leishmaniasis is a parasitic disease affecting ~12 million people. Control of infection (e.g. in C57BL⁄6 mice) results from IL-12-dependent production of IFNγ by Th1⁄Tc1 cells. In contrast, BALB⁄c mice succumb to infection because of preferential Th2-type cytokine induction. Infected dendritic cells (DC) represent important sources of IL-12. Genetically determined differences in DC IL-1α⁄β production contribute to disease outcome. Whereas the course of disease was not dramatically altered in IL-1RI−⁄− mice, local administration of IL-1α to infected C57BL⁄6 mice improved disease outcome. To definitively elucidate the involvement of IL-1 in immunity against leishmaniasis, we now utilized IL-1α⁄β-double-deficient C57BL⁄6 mice. C57BL⁄6 mice are believed to be a good surrogate model for human, self limited cutaneous leishmaniasis (CL). Leishmania major-infected IL-1α⁄β−⁄− mice were resistant to experimental CL comparable to controls. In addition, DC-based vaccination against leishmaniasis in C57BL⁄6 mice was independent of IL-1. Thus, in Leishmania-resistant C57BL⁄6 mice, IL-1 signalling is dispensable for protection.
Keywords: IL-1, dendritic cells, L. major
Background
Leishmaniasis is a parasitic disease transmitted by the bite of a sand fly. The disease ranges from cutaneous leishmaniasis (CL) to visceral leishmaniasis and ~12 million people are affected worldwide (1). In murine experimental leishmaniasis, control of infection results from IL-12-dependent production of Th1⁄Tc1-derived IFNγ that activates infected macrophages (MΦ) to eliminate parasites (2–5). In disease-resistant C57BL⁄6 mice, skin DC infected with Leishmania major represent important sources of IL-12 (6). In contrast, BALB⁄c mice respond to infection with preferential Th2-type cytokine production, which is associated with disease progression. Genetically determined DC-derived factors that influence disease susceptibility of BALB⁄c mice include elevated levels of inhibitory IL-12p80 (7) and decreased release of IL-1α⁄β (8,9). Previously, we demonstrated that IL-1α⁄β facilitates Th1 induction in several inflammatory disease models (9–11). Treatment of BALB⁄c mice with IL-1 during T cell priming inhibited progressive disease by shifting the immune response towards Th1 (9). However, prolonged administration of IL-1α promoted Th2 expansion in already established infections and worsened disease outcome (11).
Question addressed
IL-1 is a key mediator of inflammation (12,13). IL-1α and IL-1β exert similar biological functions by binding to the IL-1 type I receptor (IL-1RI) (14). To definitively elucidate the involvement of IL-1 in immune responses in CL, we utilized IL-1α⁄β-double deficient C57BL⁄6 mice. Infections in mice on a C57BL⁄6 background are considered the most relevant surrogate model for human CL (3).
Experimental design and results
IL-1α⁄β−⁄− mice were infected intradermally with low dose inocula (1000 infectious-stage parasites) of L. major mimicking natural parasite transmission by sand flies. If IL-1 action contributed to the disease-resistant phenotype of C57BL⁄6 mice, IL-1α⁄β−⁄− mice should exhibit increased disease susceptibility. Surprisingly, lesion sizes in IL-1α⁄β−⁄− mice were very similar to those observed in infected wild-type mice (Fig. 1a) except for a small delay in lesion resolution between weeks 8 and 10, probably because of somewhat lower levels of antigen-specific IFNγ. In addition, even though lesional parasite burdens were reduced in IL-1α⁄β−⁄− mice in comparison with C57BL⁄6 mice in week 6 after infection, no difference between the two mouse strains was observed at week 8.
Figure 1.
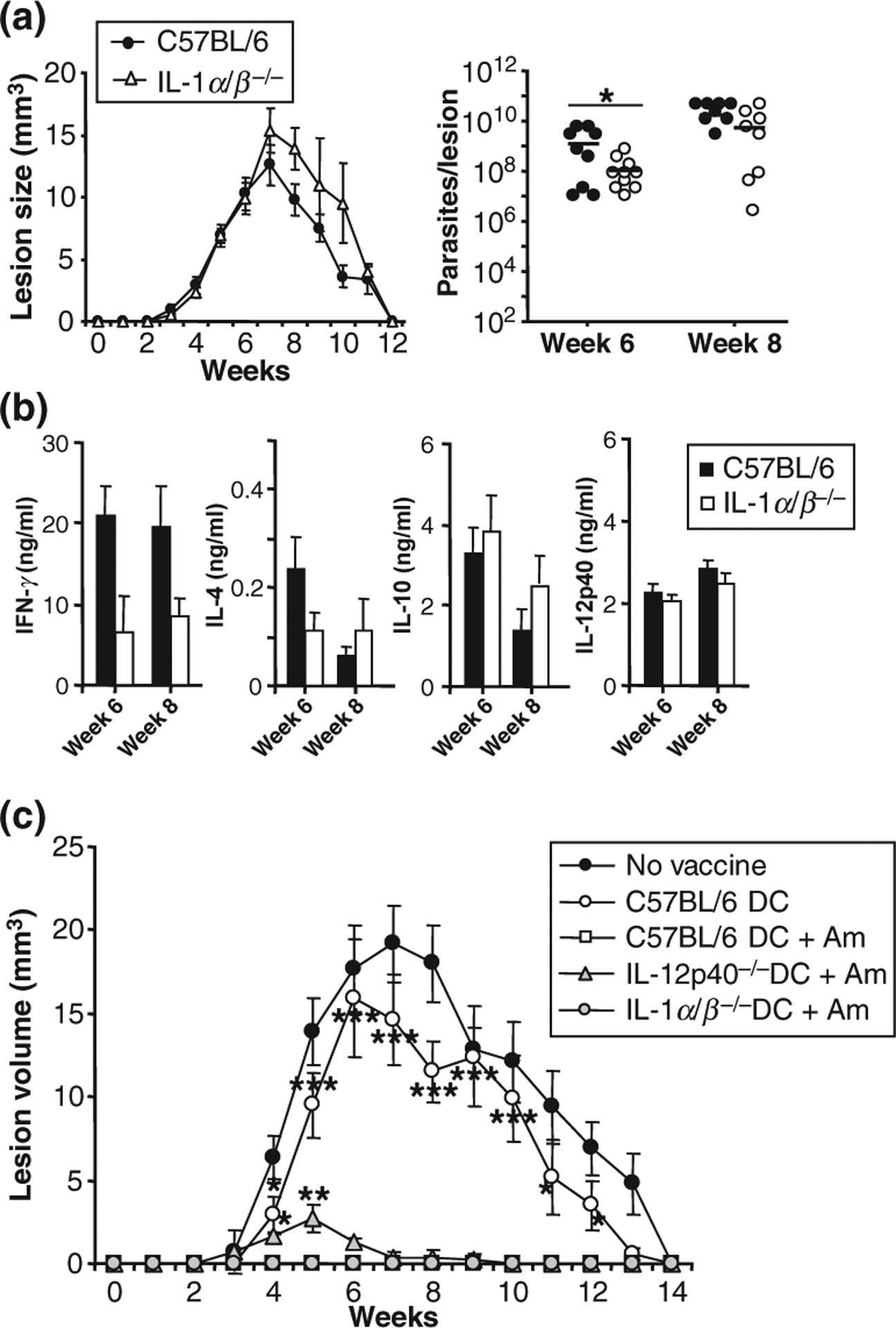
IL-1α⁄β production is dispensable for disease outcome in Leishmania major-infected, resistant C57BL⁄6 mice. Groups of ≥5 IL-1α⁄β−⁄− or C57BL⁄6 mice were infected intradermally with physiologically relevant low dose inocula of L. major (103 metacyclic promastigotes). (a) Lesion development was assessed weekly in three dimensions and calculated as ellipsoids. At weeks 6 and 8, lesional parasite burdens were determined using a limiting dilution assay. Dots represent parasite numbers in individual ears, bars indicate means. (b) Draining lymph nodes were harvested at weeks 6 and 8, plated at 1 × 106 cells⁄200 μl and restimulated with soluble Leishmania antigen (SLA, 25 μg⁄ ml). Supernatants were analysed for cytokine content by ELISA. (a + b) All data are expressed as mean ± SEM (n ≥ 9 from five independent experiments; *P ≤ 0.05). (c) Bone marrow-derived dendritic cells (DC) were generated from IL-12p40−⁄−, IL-1α⁄β−⁄−, or C57BL⁄6 mice with GM-CSF and IL-4 and harvested as immature DC on day 6. DC were plated at 2 × 105 cells⁄ml and infected overnight with L. major amastigotes (1:5). C57BL⁄6 mice were vaccinated in one ear by intradermal injection of 2 × 105 infected DC as indicated. One week later, mice were infected into the contralateral ear with physiologically relevant low dose inocula (103 parasites). Lesion development was assessed weekly in three dimensions. Lesion sizes were calculated as ellipsoids and are expressed as mean ± SEM (n ≥ 15 from three independent experiments, *P ≥ 0.05, **P ≥ 0.005, and ***P ≤ 0.002 when compared to wild-type vaccinated groups).
We also analysed the supernatants of antigen-restimulated lymph node (LN) cell cultures from Leishmania-infected C57BL⁄6 or IL-1α⁄β−⁄− mice for production of IFNγ, IL-4, IL-10 and IL-12p40 by ELISA (Fig. 1b). As expected, LN cells from C57BL⁄6 mice infected with Leishmania promastigotes produced large amounts of IFNγ upon stimulation with soluble Leishmania lysate (SLA) and very little IL-4; even though lower levels of IFNγ were found in supernatants of IL-1α⁄β−⁄− LN cells, these results were not significantly different. Additionally, the production of IL-10 and IL-12p40 was similar in both mouse strains. These results indicate that the production of IL-1α and IL-1β is dispensable for disease control in L. major-infected, resistant C57BL⁄6 mice.
Next, C57BL⁄6 wild type or IL-1α⁄β−⁄− skin-derived MΦ and immature bone marrow-derived DC (BMDC) were stimulated with amastigotes or promastigotes of L. major (5 parasites⁄cell), LPS (100 ng), or IFNγ (1000 U⁄ml). After 18 h, the supernatants were harvested and assayed for the presence of IL-12p40 and IL-10 by ELISA (Figure S1). Infection rates in cells of both mouse strains were similar, with higher infection rates in MΦ (amastigotes: 68 ± 10% vs 80 ± 11%) than in BMDC (24 ± 3 vs 32 ± 5%) as expected. Consistent with the in vivo results, MΦ and BMDC from both strains produced equal amounts of IL-12 (Figure S1). As expected, the highest level of IL-12p40 synthesis was observed in both cell types upon LPS (⁄IFNγ) stimulation, whereas L. major-induced IL-12 production was observed only in DC (6). Interestingly, weak production of IL-10 was observed from IL-1α⁄β−⁄− MΦ, while IL-10 levels from all other cells were undetectable. The significance of this finding is uncertain. Secretion of TNFα from BMDC did not differ between the two mouse strains.
Finally, we examined the potential of IL-1α⁄β−⁄−-infected DC to induce protective immunity against L. major infection. C57BL⁄6 mice were immunized intradermally in one ear with 2 × 105 infected or uninfected DC from IL-1α⁄β−⁄−, IL-12p40−⁄− or wild-type mice. Mice were challenged with 1000 L. major promastigotes injected into the contralateral ear 1 week after vaccination. Mice vaccinated with uninfected control DC revealed lesion development that was similar to unvaccinated control mice (Fig. 1c). As a positive control, L. major amastigote-infected C57BL⁄6 DC promoted full protection against infection with L. major. Importantly, this protective effect of the DC-based vaccine was also observed when infected DC from IL-1α⁄β−⁄− mice were used. On the other hand, as expected from comparable studies using IL-12p35−⁄− BALB⁄c DC, vaccination with infected DC from IL-12p40−⁄− mice did not provide complete protection when compared to wild-type DC indicated by enhanced lesion development between weeks 3 and 9 (15).
Conclusions
In summary, this study reveals that despite the fact that administration of IL-1α to infected C57BL⁄6 mice is beneficial and BALB⁄c DC produce less IL-1 than C57BL⁄6 DC (9), L. major-infected C57BL⁄6 IL-1α⁄β−⁄− mice are resistant to experimental CL. These findings are somewhat different from those obtained in IL-1RI−⁄− C57BL⁄6 mice (11, 16). In physiological low dose infections, the latter mice displayed significantly smaller lesions and decreased lesional parasite numbers together with an altered IFN-γ⁄IL-4 ratio towards Th1. However, even though effects mediated by IL-1RII (or other unidentified receptors) might contribute to this difference, in both mouse lines, absence of IL-1 signalling did not alter the overall outcome of disease. Earlier reports also demonstrated that IL-1α is necessary for Th1 development and IFNγ secretion from naïve CD4+ T cells in BALB⁄c, but not in C57BL⁄6 CD4+ T cells providing a possible explanation for our findings (17). Thus, in Leishmania-resistant C57BL⁄6 mice – a good surrogate model for human, self limited CL – IL-1 signalling is dispensable for protective immunity.
Supplementary Material
Additional Supporting Information may be found in the online version of this article:
Figure S1. Unaltered cytokine release of infected dendritic cells and macrophages from IL-1α/β−/− mice.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Acknowledgement
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG, SFB490, GK1043 and STE833⁄6-1, to E.v.S).
Abbreviations:
- CL
cutaneous leishmaniasis
- DC
dendritic cells
- MΦ
macrophages
References
- 1.Pereira Ede F, Thomaz-Soccol V, Lima HC et al. Exp Dermatol 2008: 17: 1024–1030. [DOI] [PubMed] [Google Scholar]
- 2.Reiner SL, Locksley RM. Annu Rev Immunol 1995: 13: 151–177. [DOI] [PubMed] [Google Scholar]
- 3.Sacks D, Noben-Trauth N. Nat Rev Immunol 2002: 2: 845–858. [DOI] [PubMed] [Google Scholar]
- 4.Belkaid Y, von Stebut E, Mendez S et al. J Immunol 2002: 168: 3992–4000. [DOI] [PubMed] [Google Scholar]
- 5.von Stebut E, Udey MC. Microbes Infect 2004: 6: 1102–1109. [DOI] [PubMed] [Google Scholar]
- 6.von Stebut E, Belkaid Y, Jakob T et al. J Exp Med 1998: 188: 1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nigg AP, Zahn S, Ruckerl D et al. J Immunol 2007: 178: 7251–7258. [DOI] [PubMed] [Google Scholar]
- 8.Filippi C, Hugues S, Cazareth J et al. J Exp Med 2003: 198: 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.von Stebut E, Ehrchen JM, Belkaid Y et al. J Exp Med 2003: 198: 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caucig P, Teschner D, Dinges S et al. Int Arch Allergy Immunol 2010: 152: 303–312. [DOI] [PubMed] [Google Scholar]
- 11.Kostka SL, Knop J, Konur A et al. J Invest Dermatol 2006: 126: 1582–1589. [DOI] [PubMed] [Google Scholar]
- 12.Dinarello CA Blood 1996: 87: 2095–2147. [PubMed] [Google Scholar]
- 13.Dinarello CA Annu Rev Immunol 2009: 27: 519–550. [DOI] [PubMed] [Google Scholar]
- 14.Sims JE, Gayle MA, Slack JL et al. Proc Natl Acad Sci U S A 1993: 90: 6155–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berberich C, Ramirez-Pineda JR, Hambrecht C et al. J Immunol 2003: 170: 3171–3179. [DOI] [PubMed] [Google Scholar]
- 16.Satoskar AR, Okano M, Connaughton S et al. Eur J Immunol 1998: 28: 2066–2074. [DOI] [PubMed] [Google Scholar]
- 17.Shibuya K, Robinson D, Zonin F et al. J Immunol 1998: 160: 1708–1716. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article:
Figure S1. Unaltered cytokine release of infected dendritic cells and macrophages from IL-1α/β−/− mice.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.