

Abstract
Milk fat globule-epidermal growth factor 8 (MFG-E8) is an anti-inflammatory glycoprotein that mediates the clearance of apoptotic cells and is implicated in the pathogenesis of autoimmune and inflammatory diseases. Because MFG-E8 also controls bone metabolism, we investigated its role in rheumatoid arthritis (RA), focusing on inflammation and joint destruction. The regulation of MFG-E8 by inflammation was assessed in vitro using osteoblasts, in arthritic mice and in patients with RA. K/BxN serum transfer arthritis (STA) was applied to MFG-E8 knock-out mice to assess its role in the pathogenesis of arthritis. Stimulation of osteoblasts with lipopolysaccharide (LPS) and tumor necrosis factor (TNF)-α downregulated the expression of MFG-E8 by 30% to 35%. MFG-E8-deficient osteoblasts responded to LPS with a stronger production of pro-inflammatory cytokines. In vivo, MFG-E8 mRNA levels were 52% lower in the paws of collagen-induced arthritic (CIA) mice and 24% to 42% lower in the serum of arthritic mice using two different arthritis models (CIA and STA). Similarly, patients with RA (n = 93) had lower serum concentrations of MFG-E8 (–17%) compared with healthy controls (n = 140). In a subgroup of patients who had a moderate to high disease activity (n = 21), serum concentrations of MFG-E8 rose after complete or partial remission had been achieved (+67%). Finally, MFG-E8-deficient mice subjected to STA exhibited a stronger disease burden, an increased number of neutrophils in the joints, and a more extensive local and systemic bone loss. This was accompanied by an increased activation of osteoclasts and a suppression of osteoblast function in MFG-E8-deficient mice. Thus, MFG-E8 is a protective factor in the pathogenesis of RA and subsequent bone loss. Whether MFG-E8 qualifies as a novel biomarker or therapeutic target for the treatment of RA is worth addressing in further studies.
Keywords: MFG-E8, RHEUMATOID ARTHRITIS, INFLAMMATION, NEUTROPHILS, BONE LOSS
Introduction
Rheumatoid arthritis (RA) is a systemic inflammatory disorder that affects ~ 1% of the population. Characteristic clinical features include swollen joints and severe cartilage and bone destruction. In addition, internal organs, the skin, eyes, and the nervous system can be involved in the systemic inflammatory process. Although the pathogenesis of RA is not fully understood, the imbalance between pro- and anti-inflammatory cytokines is critical in the development of autoimmune processes and the consecutive uncontrolled inflammatory response. The increased knowledge on the underlying cytokine perturbations in RA has already led to a remarkable improvement of RA therapy by targeting specific cytokines such as TNF-α and IL-6.(1) Nevertheless, further research is required to unravel the underlying mechanisms that lead to the cytokine imbalance and clinical manifestation of RA.
Milk fat globule-epidermal growth factor 8 (MFG-E8) was originally identified in the lactating mammary glands in mice and, meanwhile, has been found in a variety of cell types.(2,3) One of the main functions of MFG-E8 is mediating the clearance of apoptotic cells via phagocytes.(2,4,5) The lack of apoptotic cell clearance is considered one of the main mechanisms leading to the development of the autoimmune lupus-like disease in MFG-E8 knock-out (KO) mice.(6) However, besides its scavenging role, MFG-E8 has been increasingly appreciated for its anti-inflammatory properties. Using mouse models of inflammation-induced tissue injury in the colon, lungs, and periodontium, MFG-E8 was identified as an anti-inflammatory factor by suppressing NF-κB-induced transcription of pro-inflammatory cytokines and inhibiting neutrophil infiltration of inflamed tissues.(7–11) In line, low expression levels of MFG-E8 were found in inflamed lung and colon tissue,(10,11) suggesting a prominent role of MFG-E8 in the pathogenesis of inflammatory diseases.
Recent studies further identified MFG-E8 in osteoblasts and osteoclasts being a critical negative regulator of osteoclast differentiation and thus bone homeostasis.(8,12–14) The absence of MFG-E8 was associated with a higher number of osteoclasts, suppressed bone formation, and an increased bone loss in mouse models of periodontitis and ovariectomy-induced bone loss.(8,12) Treatment of murine or human osteoclasts with recombinant MFG-E8 reduces their differentiation capacity and inhibits their differentiation.(8,12) Thus, the aim of this study was to clarify the role of MFG-E8 in the pathogenesis of RA, in particular focusing on the progress of inflammation and bone loss.
Materials and Methods
Arthritis models and arthritis assessment
All animal procedures were approved by the institutional animal care committee and the Landesdirektion Sachsen. All mice were fed a standard diet with water ad libitum and were kept in groups of 5 animals per cage. Mice were exposed to a 12-hour light/dark cycle and an air-conditioned room at 23°C (no specific pathogen-free room). Enrichment was provided in forms of cardboard houses and bedding material.
Collagen-induced arthritis (CIA) is a well-established mouse model for human RA. Arthritic mice develop swollen joints, chronic inflammation, as well as joint destruction over a period of 3 to 4 weeks. CIA was induced using a type II collagen/complete Freud’s adjuvant (Sigma-Aldrich, Mannheim, Germany) emulsion injected at the base of the tail in 20 10-week-old male DBA/1 mice (purchased from Janvier, Le Genest Saint Isle, France) as previously published.(15) From day 14 on, mice were monitored every second day for symptoms of arthritis. 80% of the animals developed arthritis. Serum was collected 10 days after arthritis onset. Ten randomly chosen arthritic animals were additionally treated with dexamethasone (DEX, 100 mg/mouse/intraperitoneal injection) 3 times per week to suppress inflammation. Five additional animals did not receive any treatment and served as healthy controls. All 25 animals were included in the analysis.
To assess the role of MFG-E8 in arthritis, we used the K/BxN serum transfer arthritis (STA) model, which is a well-established model for RA in humans because it displays major features including joint swelling, systemic inflammation, and subchondral bone damage.(16,17) Arthritis was induced in 8-week-old MFG-E8 KO male mice and their age- and sex-matched WT littermate controls (C57BL/6 background)(18,19) by injecting 150 μL K/BxN serum intraperitoneally on day 0 and day 2. At this age, MFG-E8 KO mice did not display any lupus-like features. Mice were monitored daily for arthritis in a blinded fashion. All mice developed arthritis. Serum was collected 4 days after arthritis onset. This experiment was conducted two independent times with each 7 to 10 mice per group. All animals that entered the study were included in the data analysis.
The disease activity score represents the sum of all four paws graded as 0, normal paw to 3, severe paw swelling. In addition, thermal imaging was performed using a high-resolution thermal imaging camera (VarioCAM high resolution; Carl Zeiss, Jena, Germany). Photographs were taken before the induction of arthritis and 4 days after arthritis onset. The paw temperature was measured at the hind paw using the supplied software.(15)
Structural bone analyses
Bone mineral density (BMD) was assessed by peripheral quantitative computed tomography (pQCT; Stratec, Birkenfeld, Germany) as reported previously.(20) The measurements were performed at a voxel size of 70 μm at the fourth vertebral body. Contour mode 1 and peel mode 20 were used to calculate the trabecular BMD.
Micro-CT was performed on the entire paw up to the ankle ex vivo using the vivaCT40 (Scanco Medical, Brüttisellen, Switzerland) with an X-ray energy of 70 kVp, 114 mA, 200 msec integration time, and an isotropic voxel size of 20 μm. Predefined scripts from Scanco were used for the reconstruction and evaluation of the metatarsophalangeal joint area.
Bone histomorphometry
Dynamic bone histomorphometry was performed as described previously.(15,20) Briefly, mice received two intraperitoneal injections of calcein (20 mg/kg, Sigma-Aldrich) on days 5 and 2 before euthanization. Bones from the third lumbar vertebra and proximal tibia were fixed in 4% PBS-buffered paraformaldehyde and dehydrated in an ascending ethanol series. Subsequently, bones were embedded in methacrylate and cut into 4-μm sections for staining and 7-μm sections to assess fluorescence labels. The sections were stained with von Kossa and toluidine blue to analyze bone volume/total volume (BV/TV), trabecular number (Tb.N), trabecular separation (Tb.Sp), and trabecular thickness (Tb.Th). Unstained sections were analyzed using fluorescence microscopy to determine the mineralized surface/bone surface (MS/BS), the mineral apposition rate (MAR), and the bone formation rate/bone surface (BFR/BS).
Tartrate-resistant acid phosphatase (TRAP) staining was used to assess the osteoclast surface per bone surface (Oc.S/BS) and number of osteoclasts per bone perimeter (N.Oc/B.Pm) on paraffin sections of the spine and paws. Histomorphometric analyses was performed with the Osteomeasure software (OsteoMetrics, Decatur, GA, USA) according to international standards. The bone erosion score and cartilage integrity score were determined by grading the integrity of the subchondral bone and cartilage surface at three independent locations (0, smooth, to 3, rough surface).
Primary murine bone marrow stromal cell culture
Primary murine bone marrow stromal cells (BMSC) were differentiated to osteogenic cells using standard osteogenic medium in DMEM (Invitrogen, Darmstadt, Germany) for 7 days.(8,12–14) Before treatment, cells were switched to DMEM containing 1% FCS overnight and then treated with 1 μg/mL LPS (Sigma-Aldrich) or 50 ng/mL TNF-α (R&D Systems, Frankfurt, Germany) for 48 hours. In some experiments, BMSC were isolated from TLR2/4 KO mice (C57BL/6 backgroud). To block TNF-α actions, cells were pretreated for 2 hours with 1 μg/mL of soluble TNF receptors (TNFR1 and TNFR2, both from R&D Systems).
RNA isolation, RT, and real-time RT-PCR
RNA was extracted from the hind paws by crushing them in liquid nitrogen and collecting the bone powder in Trifast (Peqlab, Erlangen, Germany). Total RNA from cell culture was isolated using the High Pure RNA Isolation Kit (Roche, Mannheim, Germany) according to the manufacturer’s protocol. Five hundred nanograms RNA were reverse transcribed using Superscript II (Invitrogen, Darmstadt, Germany) and subsequently used for SYBR Green-based real-time PCRs using a standard protocol (Applied Biosystems, Carlsbad, CA, USA). The primer sequences were: β-actin s: ATCTGGCACCACACCTTCT, β-actin as: GGGGTGTTGAAGGTCTCAAA; CCL2 s: GTTGGCTCAGCCAGATGCA, CCL2 as: AGCCTACTCATTGGGATCATCTTG; CTSK s: AAGTGGTTCAGAAGATGACGGGAC, CTSK as: TCTTCAGAGTCAATGCCTCCGTTC; IL-1β s: ACAAGGAGAACCAAGCAACG, IL-1β as: GCCGTCTTTCATTACACAGG; IL-6 s: GGA TAC CAC TCC CAA CAG ACC, IL-6 as: TCC AGT TTG GTA GCA TCC ATC; MFG-E8 s: CTACTGCCTCTGCCCTGAAG, MFG-E8 as: CCAGACATTTGGCATCATTG; OPG s: CCTTGCCCTGACCACTCTTA, OPG as: ACACTGGGC TGCAATACACA; OSCAR s: TGGCGGTTTGCACTCTTCA, OSCAR as: GATCCGTTACCAGCAGTTCCAGA; PGRP s: GGAACTTCATGGACCG GGTA, PGRP as: TTCCCAGCTTTGGATGACCT; RANKL s: GCAGAAG GAACTGCAACACA, RANKL as: GATGGTGAGGTGTGCAAATG; TNF-α s: CCTCTTCTCATTCCTGCTTGTG, TNF-α as: CACTTGGTGGT TTGCTACGAC; TRAP s: ACTTGCGACCATTGTTAGCC, TRAP as: AGAGGGATCCATGAAGTTGC. The results were calculated using the ΔΔCT method and are presented in x-fold increase relative to β-actin mRNA levels.
Western blot analysis
Osteogenic cells were lysed in lysis buffer containing 20 mM Tris/HCl pH 7.4, 1% SDS, a protease inhibitor (complete mini, Roche), and a phosphatase inhibitor (phosSTOP, Roche). Lysates were processed through a 24-gauge needle and centrifuged at 20,000g for 30 minutes. For electrophoresis, 20 μg of heatdenatured and reduced proteins were loaded on a 10% SDS-PAGE and transferred onto a 0.2-mm nitrocellulose membrane (Whatman, Sigma-Aldrich). After blocking for 1 hour with 5% non-fat dry milk in Tris-buffered saline with 1% Tween-20 (TBS-T), membranes were incubated with an anti-MFG-E8 antibody (R&D Systems, 1:500) overnight and washed three times with TBS-T. Thereafter, incubation with an appropriate HRP-conjugated secondary antibody for 1 hour at RT followed. Finally, membranes were washed with TBS-T and incubated with an ECL substrate (Pierce, Thermo Fisher Scientific, Waltham, MA, USA). The proteins were then visualized using the MF-ChemiBIS 3.2 bioimaging system (Biostep, Jahnsdorf, Germany).
Flow cytometric analyses of cells in the knee joints
Both knee joints from arthritic mice were dissected carefully without disintegrating the femur or tibia. Muscle tissue was removed from the femur and tibia. Afterwards, knee joints were cracked open and digested using a collagenase (1.2 mg/mL, C8051)/dispase II (25 U/mL, D4693) (both from Sigma-Aldrich) solution for 30 minutes at 37°C. The digestion was stopped by adding 10% FCS and the solution was passed through a 50-μm mesh. Synovial cells were washed twice with PBS/2% FCS, blocked with anti-rat IgG and CD16/CD32, and subsequently stained with anti-CD11b-FITC (1:800), anti-CD45-PE (1:200), anti-B220-PE-Cy7 (1:500), anti-CD3-APC (1:200), and an anti-Gr1-A700 (1:100) antibody for 30 minutes at 4°C in the dark (all antibodies were purchased from eBioscience, Mannheim, Germany). Thereafter, cells were washed twice with PBS and analyzed with the BD LSR II flow cytometer (BD Biosciences, San Jose, CA, USA) and the FlowJo vX software (Tree Star Inc., Ashland, OR, USA).
Study populations
Serum samples from 93 patients with RA were collected at the Department of Rheumatology at the Technische Universität Dresden. RA was defined according to the 2010 ACR/EULAR classification criteria. Patient characteristics are given in Table 1. Eighty-four patients (90%) were taking disease-modifying antirheumatic drugs (DMARDs). Forty-seven patients (51%) received conventional DMARDs, 28 patients (30%) a DMARD combination (conventional and biologic), 6 patients (6.5%) biologic DMARDs, and 1 patient (1%) glucocorticoid therapy alone. Seven patients were not yet treated with glucocorticoids or DMARDs (8%). Twenty-one patients showed a moderate to high disease activity (CDAI > 10) at the time of measurement (19 females, 2 males). After having adjusted therapy, all patients achieved a low disease activity, and a follow-up measurement was performed (median follow-up time: 202 days [1st and 3rd quartiles: 78 to 309 days]).
Table 1.
Patient Characteristics
| SHIP-1 (n = 140) | RA (n = 93) | p Value | |
|---|---|---|---|
| Male (n [%]) | 45 (32.1) | 27 (29.0) | 0.61 |
| Age (years) | 59.5 (51.0–70.5) | 61.0 (52.0–71.0) | 0.33 |
| BMI (kg/m2) | 26.7 (23.9–29.8) | 26.1 (23.1–30.3) | 0.66 |
| P1NP (ng/mL) | 40.0 (30.5–51.5) | 34.4 (26.1–46.0) | 0.01 |
| OCN (ng/mL) | 16.8 (12.8–22.4) | 10.3 (8.2–15.5) | <0.01 |
| CTX (ng/mL) | 0.30 (0.20–0.48) | 0.19 (0.11–0.30) | <0.01 |
| MFG-E8 (ng/mL) | 4.04 (2.94–5.54) | 3.34 (2.53–4.38) | <0.01 |
BMI = body mass index; P1NP = intact amino-terminal propeptide of type I procollagen; OCN = osteocalcin; CTX = carboxy-terminal telopeptide of type I collagen; MFG-E8 = milk fat-globule protein E8.
Data are number (proportion) or median (1st–3rd quartile). Group differences were tested with chi-square or Kruskal-Wallis tests.
Two missing values in P1NP and OCN; 3 missing in CTX in RA patients.
Control samples (n = 140) were obtained from the Study of Health in Pomerania (SHIP)-1 cohort, which includes 3300 men and women aged 25 to 86 years. Details on sampling methods and study protocols of the study have been reported previously.(21) Control subjects were selected to match patients with RA with regard to age, sex, and BMI. Subjects were not eligible as controls if they had a condition affecting bone metabolism (ie, self-reported osteoporosis, rheumatoid arthritis, spondylarthropathies, malignant disease, hyper- or hypothyroidism defined as thyroid-stimulating hormone <0.25 or >2.12 mU/L, primary and secondary hyperparathyroidism defined as parathyroid hormone >120 pg/mL, hypogonadism in men defined as testosterone <10.4 mmol/L, chronic kidney disease defined as eGFR <30 mL/min/1.73m2, risky alcohol consumption defined as ≥30 g/d in men and ≥20 g/d in women, acute inflammation defined as serum high-sensitivity C-reactive protein concentration >10 mg/L, or pregnancy) or were taking medications affecting bone metabolism (heparin, calcineurin inhibitors, anti-epileptic medication, bisphosphonates, parathyroid hormone, selective estrogen receptor modulators).
The investigations in SHIP-1 and collecting RA patient samples were carried out in accordance with the Declaration of Helsinki, including written informed consent of all participants. The study methods were approved by the Institutional Review Board of the University of Greifswald and the Technische Universität Dresden, respectively.
Serum analysis
Serum intact amino-terminal propeptide of type I procollagen (P1NP, IDS), osteocalcin (IDS), and carboxy-terminal telopeptide of type I collagen (CTX, IDS) concentrations were measured on the IDS-iSYS Multi-Discipline Automated Analyzer (Immunodiagnostic Systems Limited, Frankfurt am Main, Germany). Human and mouse MFG-E8 was measured manually using ELISAs from R&D Systems (Minneapolis, MN, USA).
Statistical analysis
Data are presented as mean ± standard deviation (SD). Statistical evaluations of two group comparisons were performed using a two-sided Student’s t test. Paired Student’s t tests were used for analyses of Fig. 3E–G. One-way analyses of variance (ANOVA) was used for experiments with more than two groups. Two-way ANOVA was used for analyzing genotype and arthritis effects. Group differences for human samples were tested with chi-square or Kruskal-Wallis tests. Correlations were analyzed using the Spearman correlation coefficient. Any p values < 0.05 were considered statistically significant.
Fig. 3.
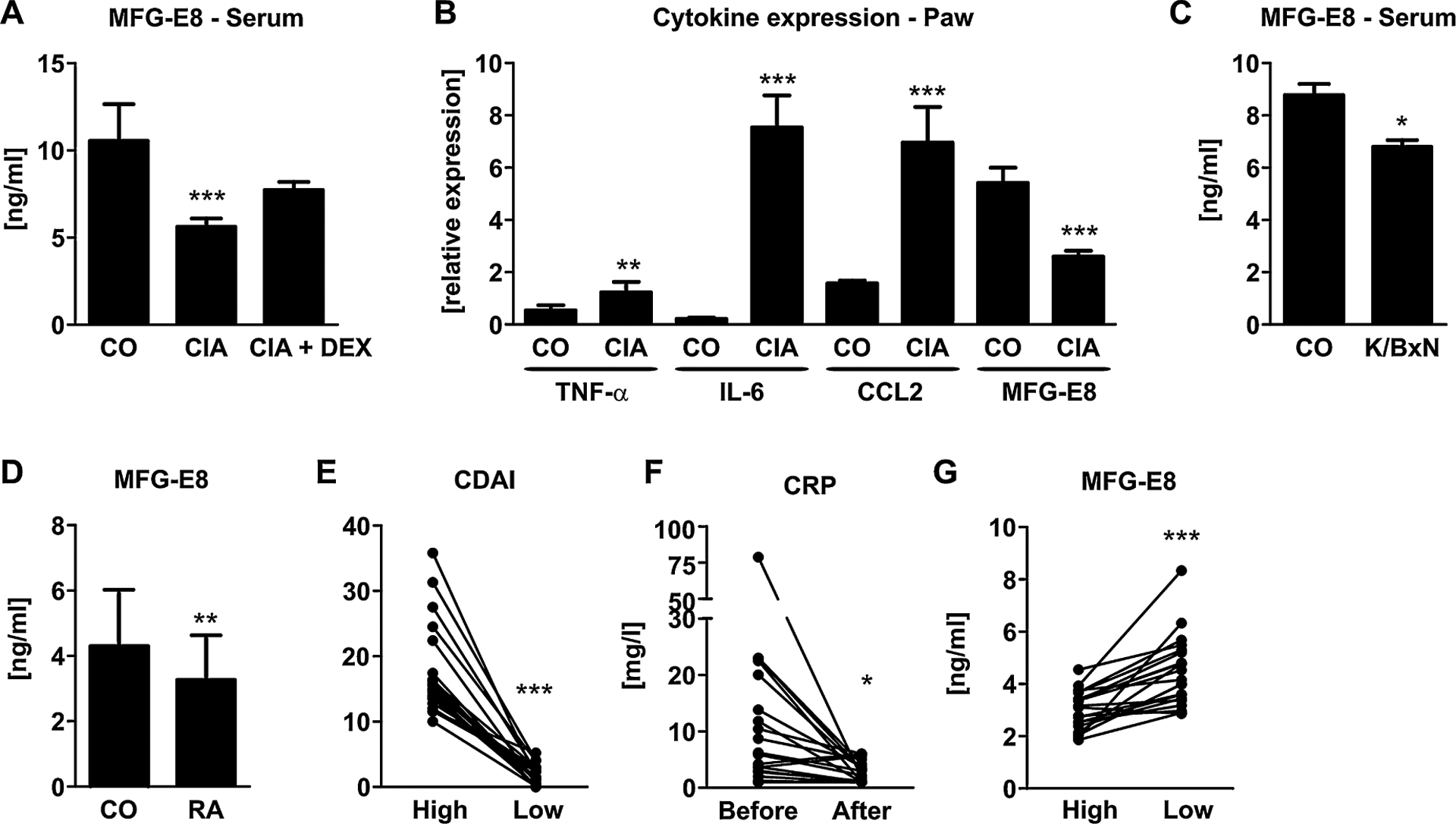
MFG-E8 is decreased in inflammatory arthritis in mice and men. (A) MFG-E8 serum concentrations were measured in healthy mice (CO) and mice with collagen-induced arthritis (CIA) 10 days after arthritis onset using an ELISA. Ten arthritic mice were treated with dexamethasone (CIAþDEX) starting at disease onset to treat inflammation. n = 7–10. (B) mRNA was isolated from the paws of CO and CIA mice, reverse transcribed, and subjected to qPCR analyses for tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), chemokine (C-C motif) ligand 2 (CCL2), and MFG-E8. Expression was normalized to β-actin. n = 6–8. (C) MFG-E8 serum concentrations were measured in healthy mice (CO) and mice with K/BxN serum transfer arthritis (STA) 4 days after arthritis onset using an ELISA. (D) Gene expression analyses of MFG-E8 in the paws of healthy and STA mice. n = 7–10. (E) MFG-E8 serum concentrations were measured in patients with rheumatoid arthritis (RA, n = 93) and 140 age-, sex-, and BMI-matched healthy controls (CO). (F–H) Follow-up analyses from 15 patients who initially had a high disease burden (first visit) and after successful treatment had a low disease activity at the second visit. (E) Disease activity as reflected by the clinical disease activity index (CDAI), (F) serum levels of C-reactive protein (CRP), (G) serum levels of MFG-E8. *p < 0.05; **p < 0.01; ***p < 0.001.
Results
MFG-E8 is downregulated by LPS and TNF-α
To investigate the regulation of MFG-E8 under inflammatory conditions in osteogenic cells in vitro, we stimulated murine osteoblasts from WT mice with LPS and TNF-α for 24 hours, respectively. Both stimuli downregulated the mRNA expression of MFG-E8 by 35% and 30%, respectively (Fig. 1A). Suppression of MFG-E8 was confirmed at protein level using Western blot analyses (Fig. 1B). To assess whether LPS regulates MFG-E8 expression through its cognate receptor Toll-like receptor 4 (TLR4), we used osteoblasts from TLR2/4 KO mice. TLR2/4-deficient osteoblasts exhibited a much lower MFG-E8 expression compared with osteoblasts from WT animals and as expected were not sensitive to LPS stimulation (Fig. 1C). Additionally, blocking TNF-α with the soluble receptors TNFR1 and TNFR2, which in high concentrations serve as decoys sequestering TNF-α,(22,23) abrogated MFG-E8 downregulation (Fig. 1D). Hence, under the influence of inflammatory mediators such as LPS and TNF-α, MFG-E8 expression is suppressed.
Fig. 1.
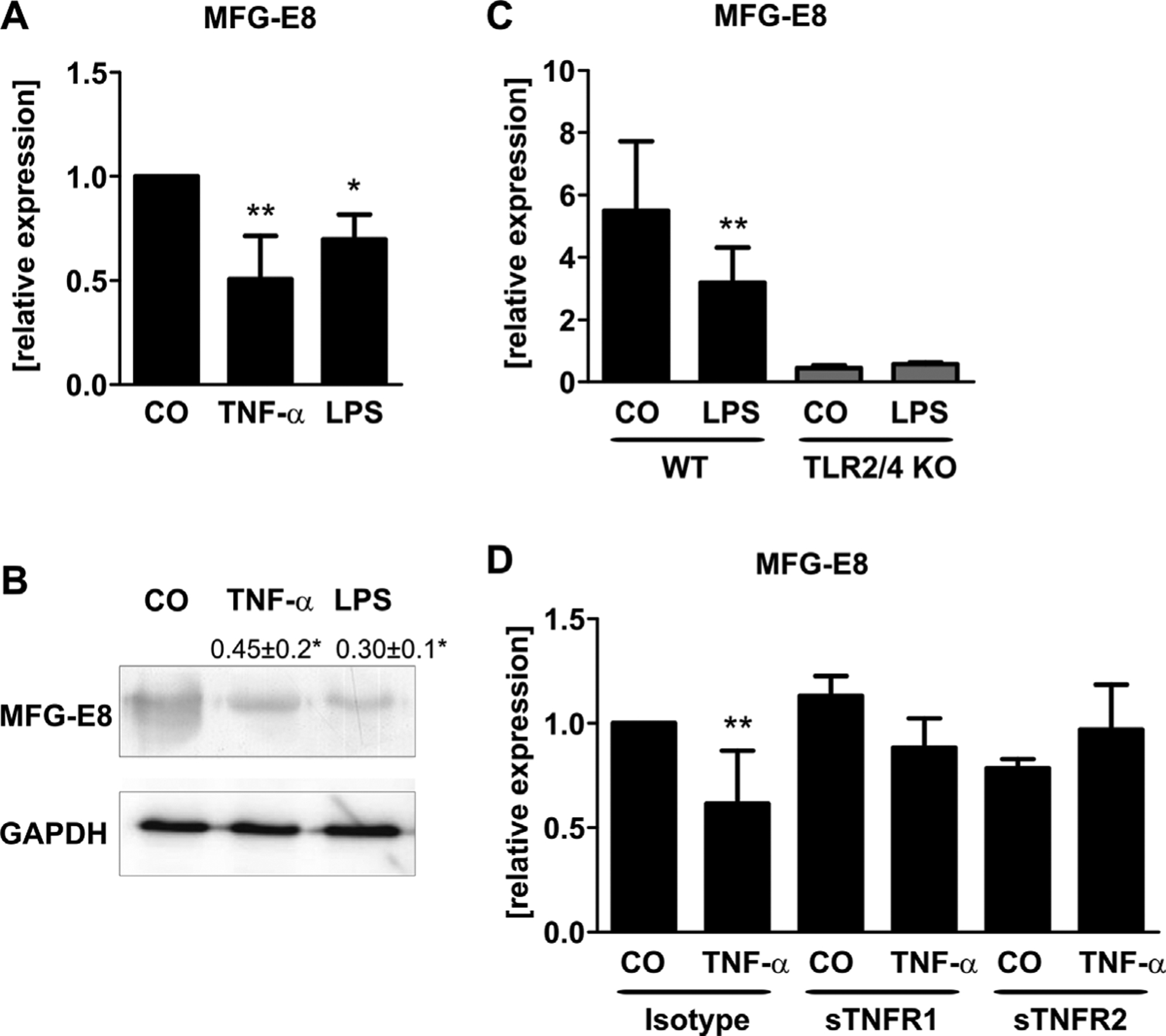
Lipopolysaccharide (LPS) and tumor necrosis factor-a (TNF-α) suppress MFG-E8 expression in vitro. Bone marrow stromal cells were differentiated for 7 days into osteoblasts and treated with LPS (1 mg/mL) or TNF-α (50 ng/mL) for 48 hours. Vehicle-treated cells were used as controls (CO). (A) Gene expression of MFG-E8 was determined using qPCR and normalized to β-actin. (B) Protein levels were determined using Western blot analysis. One representative blot is shown. Numbers indicate mean ± standard deviation of 4 independent experiments. (C) Cells were isolated from wild-type (WT) or Toll-like receptor 2/4 knock-out (TLR2/4 KO) mice and stimulated with LPS (1 μg/mL, 48 hours). Gene expression of MFG-E8 was determined using qPCR and was normalized to β-actin. (D) Cells were pretreated with sTNFR1 (1 μg/mL) or sTNFR2 (1 μg/mL) for 1 hour before cells were treated with 50 ng/mL TNF-α for 48 hours. MFG-E8 expression was determined using qPCR. n = 4–6 mice per experiment. *p < 0.05; **p < 0.01.
MFG-E8 regulates pro-inflammatory cytokine expression
To further investigate whether MFG-E8 is not only regulated by pro-inflammatory stimuli but also regulates the inflammatory response itself, we treated osteoblasts from WT, MFG-E8 KO, and TLR2/4 KO mice with LPS for 24 hours and measured the increase in IL-6 and TNF-α expression. The response of osteoblasts lacking MFG-E8 was increased 2.5-fold (IL-6) and 2-fold (TNF-α) compared with WT (Fig. 2A, B). Thus, the absence of MFG-E8 leads to a stronger response to inflammatory mediators in osteogenic cells.
Fig. 2.
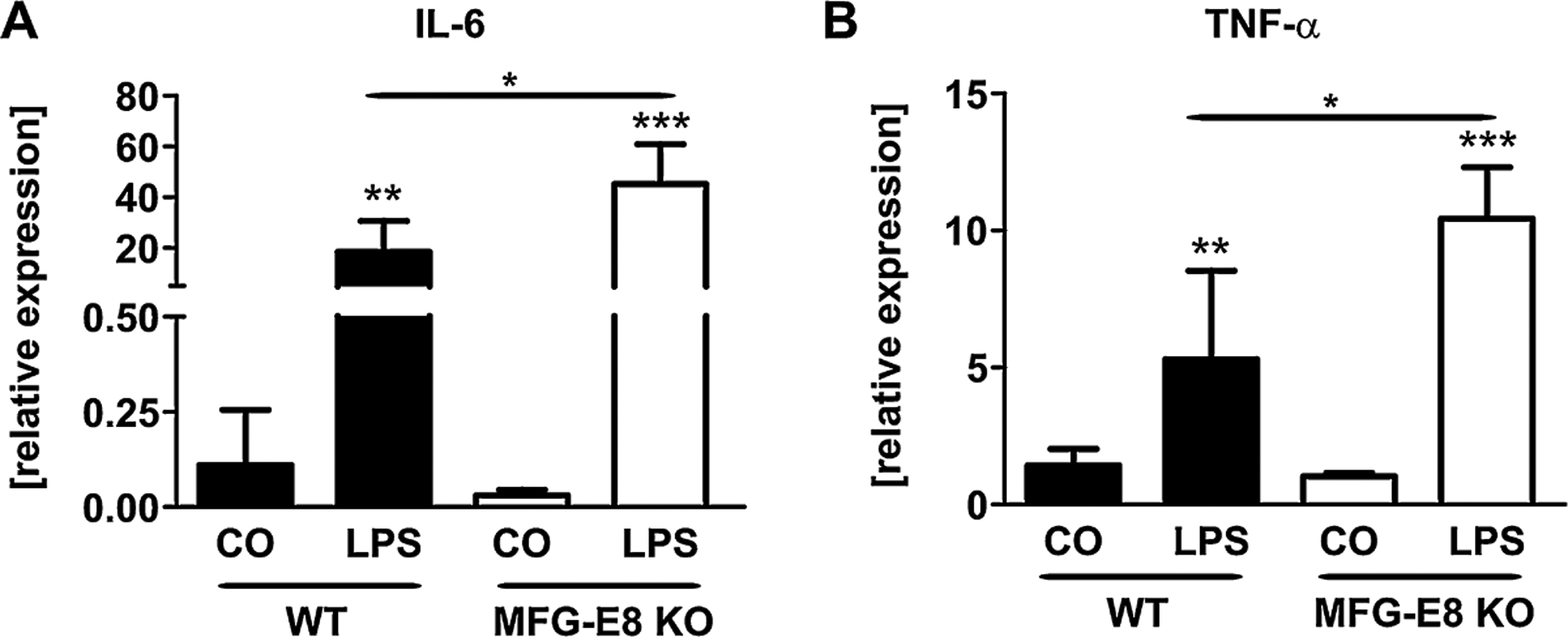
MFG-E8 deficiency enhances the inflammatory response in vitro. Bone marrow stromal cells, isolated from wild-type (WT) and MFG-E8 knock-out (MFG-E8 KO) mice were differentiated for 7 days into osteoblasts. Cells were treated with LPS (1 μg/mL) or vehicle (CO) for 48 hours. (A) Gene expression of interleukin-6 (IL-6) and (B) tumor necrosis factor-a (TNF-α) was determined using qPCR and normalized to β-actin. n = 4–5. *p < 0.05;**p < 0.01; ***p < 0.001.
MFG-E8 is downregulated in arthritic mice
Because in vitro experiments indicated a negative regulation of MFG-E8 under inflammatory conditions, we investigated the level of MFG-E8 in the serum and paws from arthritic mice. At the time of analysis, mice with CIA had displayed signs of arthritis for 10 days. A subset of mice was additionally treated with DEX starting at arthritis onset. Arthritic mice had a higher disease burden than healthy or DEX-treated animals as indicated by the arthritis score (CO: 0.0 ± 0.0; CIA: 5.6 ± 2.3; CIA+ DEX: 1.1 ± 0.6, p < 0.01).(15) Serum concentrations of MFG-E8 were 42% lower in arthritic mice compared with healthy controls, whereas DEX treatment tended to increase MFG-E8 levels again (+18%, p = 0.06) (Fig. 3A). In addition to serum levels, we also assessed the expression of MFG-E8 in the paws of arthritic mice and found that although several pro-inflammatory cytokines were upregulated (TNF-α: 1.8-fold; IL-6: 35.9-fold; CCL2: 4.43-fold), MFG-E8 expression was downregulated by 52% (Fig. 3B). To validate our findings in another arthritis mouse model, we used the K/BxN serum transfer model. Similar to the CIA model, mice with K/BxN STA had a higher disease activity score than healthy control mice (CO: 0.0 ± 0.0; STA: 6.6 ± 2.7, p < 0.05), reduced serum concentrations of MFG-E8 (–24%, p < 0.05) (Fig. 3C), and a reduced expression of MFG-E8 in the paw tissue (–46%, p < 0.05) (Fig. 3D). Thus, in line with our in vitro experiments, the systemic inflammatory milieu of arthritis suppresses MFG-E8 in vivo.
MFG-E8 serum levels are altered in patients with rheumatoid arthritis
To investigate whether MFG-E8 production is also decreased in patients with RA, we measured serum concentrations of MFG-E8 in patients with RA (n = 93) and healthy controls (n = 140). Patient characteristics are given in Table 1. MFG-E8 serum levels were decreased by 17% in patients with RA compared with healthy controls (Fig. 3D). In addition, serum levels of the bone remodeling markers P1NP (–14%, p = 0.01), osteocalcin (–39%, p < 0.01), and CTX (–37%, p < 0.01) were decreased in patients with RA (Table 1). MFG-E8 did not correlate with any of the bone remodeling markers (P1NP: r = 0.02, p = 0.76; osteocalcin: r = 0.045, p = 0.5; CTX: r = 0.092, p = 0.16). In a subgroup of 21 patients with moderate to high disease activity (CDAI >10), MFG-E8 serum concentrations increased by 67% (p < 0.001) after disease activity was lowered (CDAI <4) (Fig. 3E, G). In line with the decreased disease activity, also serum concentrations of CRP decreased after successful treatment (Fig. 3F). Together, these data support the findings in mice showing that MFG-E8 is negatively regulated by inflammation.
Loss of MFG-E8 exacerbates arthritis and neutrophil infiltration in inflamed joints
To further analyze whether MFG-E8 plays a role in the development of arthritis, we induced arthritis in WT and MFG-E8 KO mice using the K/BxN serum transfer model. MFG-E8 KO animals showed a more severe course of arthritis and reached significantly higher arthritic scores compared with WT (two-way ANOVA: p < 0.001) (Fig. 4A). Moreover, the arthritis lasted longer and the resolution phase was delayed. Although WT mice barely had swollen paws 2 weeks after arthritis induction, MFG-E8 KO mice still had an average arthritis score of 3.5 (Fig. 4A). The more severe course of arthritis was further confirmed using thermal imaging, which was applied to assess the paw temperature as another feature of inflammation. Arthritic mice presented with higher temperatures compared with healthy mice (arthritic: 27.2 ± 2.4°C versus healthy 25.4 ± 1.2°C, p < 0.01) and, in line with the arthritis score, arthritic MFG-E8 KO mice had a higher paw temperature compared with arthritic WT mice (+11%, 30.1 ± 2.8°C, p < 0.05) (Fig. 4B). Finally, we confirmed a higher inflammatory reaction in the paws of MFG-E8 KO mice by determining the expression of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6, as well as the chemokine CCL2. For all investigated genes, MFG-E8 KO mice showed a three- to fivefold higher expression (Fig. 4C).
Fig. 4.
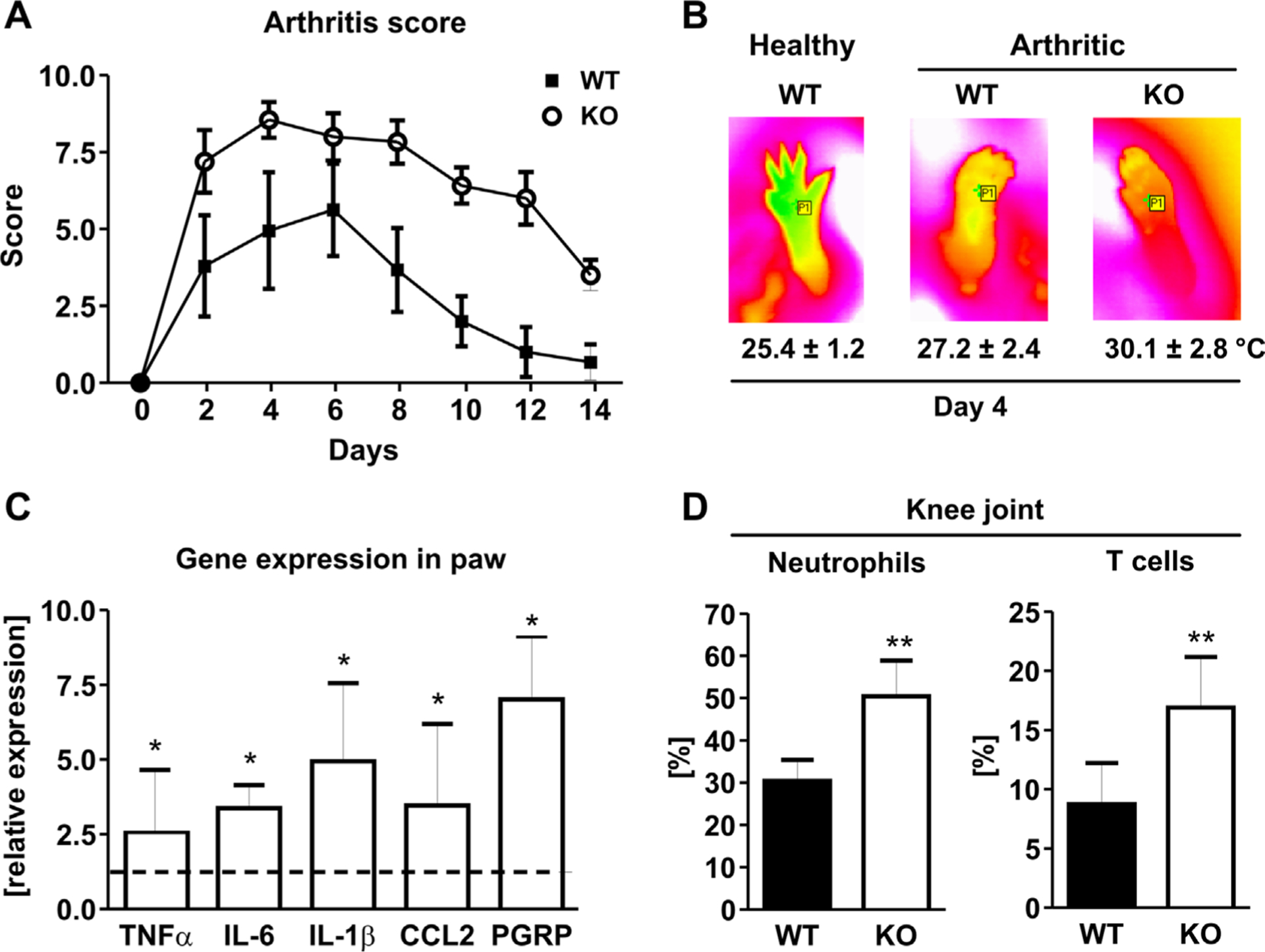
MFG-E8 deficiency increases arthritis severity in mice. (A) Arthritis was induced in wild-type (WT) and MFG-E8 knock-out (KO) mice using the K/BxN serum transfer model. Disease severity was assessed by the extent of paw swelling (0 [no swelling] to 3 [strong swelling] per paw) every other day. (B) Paw temperature was assessed at the hind paw at day 4 after disease onset using an infrared camera. Representative pictures are shown per group. Numbers indicate mean ± standard deviation of 8 to 10 mice per group. (C) RNA was isolated from the paws and subjected to qPCR analyses for inflammatory genes (tumor necrosis factor-α [TNF-α], interleukin-6 [IL-6], interleukin-1β [IL-1β], chemokine [C-C motif] ligand 2 [CCL2]), peptidoglycan recognition protein [PGRP]). Values are shown relative to the WT control. (D) Percentage of neutrophils (CD11b/Gr1-positive) and T cells (CD3-positive) that invaded the inflamed joints in WT and MFG-E8 KO mice. n = 8–10. *p < 0.05; **p < 0.01.
One of the early events in the development of arthritis is the infiltration of neutrophils into the affected joints. Thus, we also analyzed the expression of peptidoglycan recognition protein (PGRP), a neutrophil marker, in the paw extract. The expression of PGRP was increased sevenfold in MFG-E8 KO mice compared with arthritic WT mice, suggesting an increased presence of neutrophils in the inflamed joints (Fig. 4C). To further investigate this, we performed flow cytometric analyses of the knee joints of arthritic mice at day 4 after arthritis induction. After discriminating duplets and gating for CD45-positive hematopoietic cells, we found an increased percentage of CD11b/Gr1-positive neutrophils in the joints of MFG-E8 KO mice (KO: 50.4 ± 8.4% versus WT: 30.4 ± 5.0%, p < 0.01; Fig. 4D). Interestingly, also CD3-positive T cells invaded the joints and showed a twofold higher prevalence in MFG-E8 KO mice (KO: 16.9 ± 4.3% versus WT: 8.8 ± 3.5%, p < 0.05; Fig. 4D). No B220-positive B cells were detected in the inflamed joints, indicating that the synovial cells were not contaminated with bone marrow cells. Thus, these data suggest that an increased recruitment of neutrophils to the inflamed joints may lead to a stronger inflammatory reaction in this mouse arthritis model.
Loss of MFG-E8 leads to more severe bone loss in K/BxN serum transfer arthritic mice
Because local bone erosions and systemic bone loss are both major complications of RA and MFG-E8 has been implicated in bone remodeling, we further assessed the extent of bone loss in these mice. MFG-E8-deficient mice had a more severe inflammation-induced systemic bone loss at the spine compared with their WT controls (total bone mineral density: −14%, p < 0.05 two-way ANOVA; trabecular bone mineral density: −15%, p < 0.05, two-way ANOVA) (Fig. 5A, B). In addition, arthritic MFG-E8 KO mice showed more severe bone erosions than arthritic WT mice as determined by histology and μCT analyses (Fig. 5C–H). Histology also revealed a decreased number and bone-covered surface of osteoblasts in MFG-E8 KO mice, whereas osteoclast parameters were increased (Fig. 5C–F). Moreover, MFG-E8 KO mice displayed a larger extent of bone erosions and cartilage destruction in the paws compared with WT animals (Fig. 5I). In line with these data, the expression of osteoclastic markers, tartrate-resistant acid phosphatase (TRAP), osteoclast-associated receptor (OSCAR), cathepsin K (CTSK), and receptor activator of NF-κB ligand (RANKL) was increased in mRNA extracts from paws of MFG-E8 KO mice (Fig. 5H). Osteoprotegerin (OPG) expression was not altered (Fig. 5H). Moreover, serum levels of CTX were increased in arthritic MFG-E8 KO mice (WT: 38.8 ± 5.6 ng/mL; KO: 52.0 ± 10.0 ng/mL, p < 0.01). These data demonstrate that MFG-E8 is not only regulated by inflammation but also that it plays an active role in the pathogenesis of arthritis and arthritis-induced bone loss.
Fig. 5.
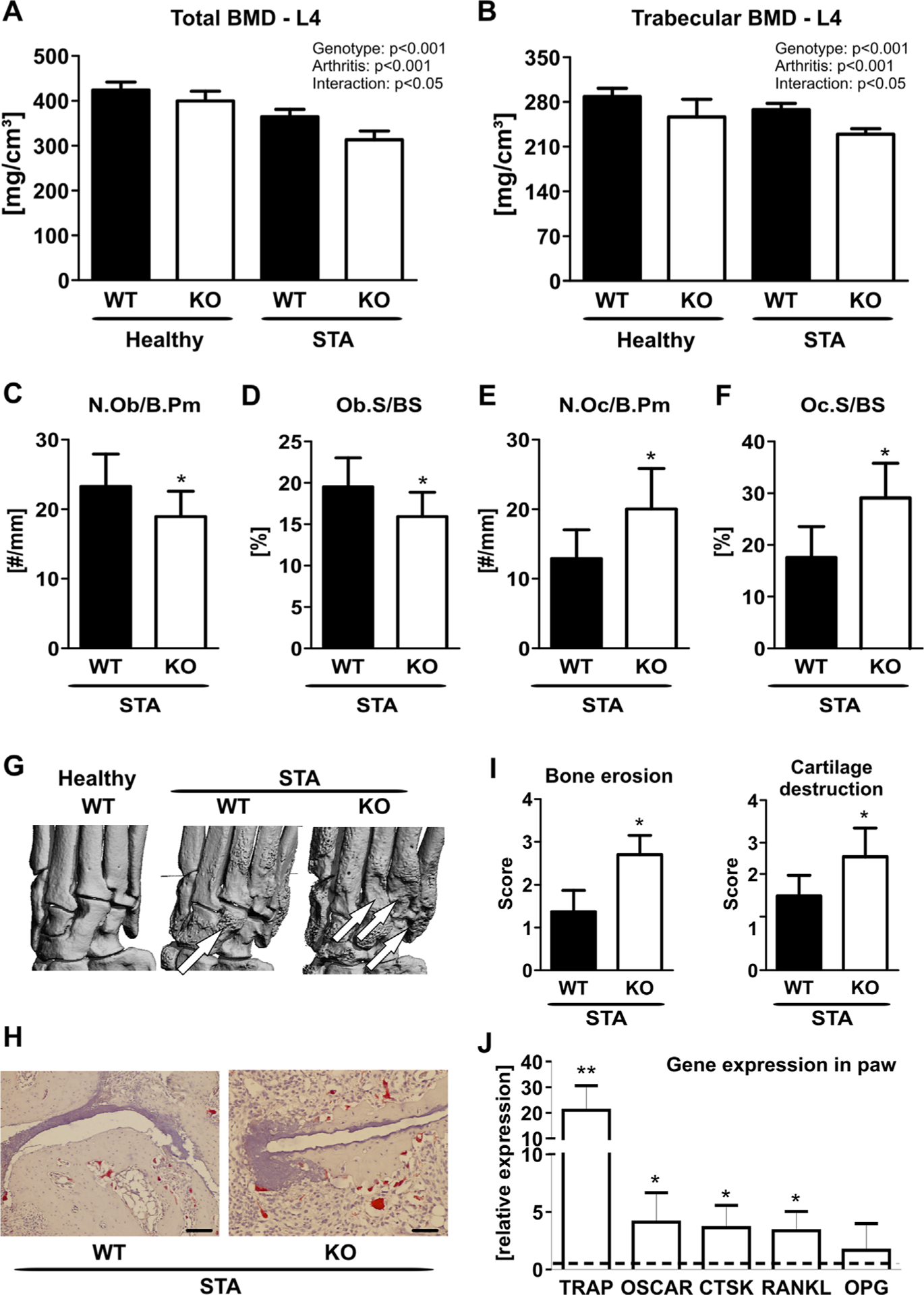
Loss of MFG-E8 is associated with more severe arthritis-induced bone loss. Peripheral quantitative computed tomography (pQCT) was performed to analyze (A) the total and (B) the trabecular bone mineral density (BMD) at the fourth lumbar vertebra. (C–F) Histology from the paws of K/BxN arthritic WT and MFG-E8 knock-out (KO) mice showing (C) osteoblast number per bone perimeter (N.Ob/B.Pm), (D) osteoblast surface per bone surface (Ob.S/BS), (E) osteoclast number per bone perimeter (N.Oc/B.Pm), and (F) osteoclast surface per bone surface (Oc.S/BS). (G) Local bone erosions were visualized using μCT. Arrows indicate areas of extensive bone erosions. (H) Representative TRAP staining of the paws of arthritic WT and MFG-E8 KO mice. Scale bar = 20 μm. (I) Quantification of bone erosions and cartilage integrity on TRAP-stained sections of the paw. (J) RNA was isolated from the paws. qPCR analyses was performed for osteoclast-related genes (TRAP = tartrate-resistant acid phosphatase; OSCAR = osteoclast-associated receptor; CTSK = cathepsin K; RANKL = receptor activator of NF-κB ligand; OPG = osteoprotegerin) in paws from MFG-E8 KO (white bars) compared with gene expression in paws from WT mice (dashed line). n = 7–9 per group. *p < 0.05; **p < 0.01; ***p < 0.001. ##p < 0.01 versus healthy; ###p < 0.001 versus healthy.
Discussion
Here we show that MFG-E8 is involved in the pathogenesis of RA and subsequent bone destruction and acts as an anti-inflammatory mediator, thereby extending previous reports showing that MFG-E8 has a protective role in inflammation-induced tissue injury in the colon, lungs, liver, and periodontium.(7,8,10,24–27) In addition, our data from isolated cells, mouse studies, and human patient materials show that an inflammatory milieu reduces MFG-E8 levels, which renders cells more responsive to inflammatory stimuli as shown by the increased production of IL-6 and TNF-α in MFG-E8-deficient cells. Although numerous studies have shown that MFG-E8 suppresses LPS-induced cytokine production by blocking NF-κB signaling,(28,29) only few studies have addressed the regulation of MFG-E8 by inflammatory stimuli in vitro.(30,31) Our study shows that both LPS and TNF-α reduce MFG-E8 expression in osteogenic cells, which are well known for their immunomodulatory function at immature stages (such as used in this study). This is consistent with reports showing that MFG-E8 expression and promoter activity is decreased by LPS.(29,32) Interestingly, MFG-E8 expression was barely detectable in cells lacking TLR2/4, suggesting that intact TLR signaling may be required for maintaining basal MFG-E8 expression. In contrast to our results, a recently published study found that TNF-α increases MFG-E8 expression in epithelial cells.(33) The reason for this discrepancy is currently unclear but may lie in the nature of the investigated cells, mesenchymal cells in our case, and thus harboring an immunomodulatory function, as opposed to epithelial origin.(33)
A low expression of MFG-E8 in vivo has been consistently detected in several inflamed tissues.(11,34) Here, we also found a low expression of MFG-E8 in the inflamed paws. Importantly, low levels of MFG-E8 were not only found locally but also systemically in the serum of arthritic mice and patients with RA. Furthermore, our data suggest that serum levels of MFG-E8 can be reversed again after relieving inflammation, as shown by the restored MFG-E8 serum concentrations in arthritic mice treated with DEX or in patients who achieved a low disease activity after changing their therapy. Of note, our study involving RA patients is limited by the retrospective design of the study. We only had a small number of patients with a moderate to high disease activity (CDAI >10; 21/93, 23%). Nevertheless, in those patients a marked increase in MFG-E8 serum levels was detected after inflammation was reduced. The increase in MFG-E8serum levels was independent of the therapeutic approach (conventional versus biological DMARDs). It should be kept in mind, however, that our study is limited by a small sample size and we cannot rule out that MFG-E8 levels may be regulated differently by different treatment strategies. Nevertheless, the robustness of the effect suggests that the serum levels of MFG-E8 in our patient cohort might be overestimated because most of the patients had a low disease activity or were in remission. It remains to be determined if a more homogenous group of patients with a high disease activity might present with even lower MFG-E8 serum levels. Larger studies are needed to determine the value of MFG-E8 as a biomarker of inflammation or as an indicator of therapy efficacy in RA and across other inflammatory diseases.
Using MFG-E8-deficient mice, we further show that MFG-E8 contributes to the pathogenesis of RA. As our in vitro data propose, one mechanism might be the increased production of pro-inflammatory cytokines in the absence of MFG-E8. Indeed, higher levels of TNF-α, IL-6, IL-1β, and CCL2, factors that are crucial for the induction and maintenance of RA, were found in the inflamed paw tissue.(35,36) These factors may also facilitate the infiltration of the joints with immune cells such as neutrophils and T cells. Increased numbers of neutrophils have also been detected in other inflammatory models in MFG-E8-deficient mice, underlining the critical role of MFG-E8 in mediating cellular activities.(10) In addition, MFG-E8 has been shown to support M2 polarization of macrophages, promoting an anti-inflammatory macrophage phenotype.(37,38) Considering the many functions of MFG-E8, it is likely that not only one but several functions, including the clearance of apoptotic cells, the regulation of pro- and anti-inflammatory signals, and thus the control of immune cell functions, may add to the protective effect of MFG-E8 in inflammatory diseases.
Finally, we addressed the role of MFG-E8 in inflammation-induced bone destruction because previous reports have identified MFG-E8 as a critical regulator of bone homeostasis in mice.(8,12) In these studies, MFG-E8 was shown to regulate osteoclastogenesis in mice(8,12) and in human cells (KS, LCH, and MR, unpublished data), as well as osteoblastogenesis. Arthritis-induced bone loss was more prominent in MFG-E8-deficient mice. Interestingly, not only osteoclast numbers and activity were increased, but also the number of osteoblasts was decreased, further enhancing bone loss. Of note, despite the lower number of osteoblasts, RANKL expression was increased in the paw tissue of MFG-E8 KO mice, suggesting that MFG-E8 may not only directly influence osteoclast differentiation but also indirectly through the production of RANKL in osteoblasts (and osteocytes). Because of these results, we investigated the serum concentrations of bone turnover markers in our patient cohort. However, MFG-E8 serum concentrations did not correlate with any of the markers, suggesting that MFG-E8 may not be sensitive enough to reflect bone turnover in vivo.
In conclusion, our results demonstrate that MFG-E8 is a critical player in the pathogenesis of RA and subsequent bone loss. Whether MFG-E8 qualifies as an indicator of therapy efficacy or as a therapeutic target remains to be determined in future studies.
Acknowledgments
This work was supported by a grant from the Else Kröner Fresenius-Stiftung, the start-up grant of the German Rheumatology Society, the Boehringer Ingelheim Fonds, and the German Research Foundation (RA1923/4-2 and HO1875/8-2, Immunobone) and funding from the Excellence Initiative by the German Federal and State Governments (Institutional Strategy, measure “Support the best”) to MR and LCH. MCU is supported by funds from the Center for Cancer Research, National Cancer Institute in the Intramural Research Program of the National Institutes of Health.
SHIP (Study of Health in Pomerania) is part of the Community Medicine Research net of the University of Greifswald, Germany, which is funded by the Federal Ministry of Education and Research (grants no. 01ZZ9603, 01ZZ0103, and 01ZZ0403), the Ministry of Cultural Affairs, as well as the Social Ministry of the Federal State of Mecklenburg-West Pomerania. This work is also part of the research project Greifswald Approach to Individualized Medicine (GANI_MED), which is funded by the Federal Ministry of Education and Research and the Ministry of Cultural Affairs of the Federal State of Mecklenburg-West Pomerania(03IS2061A). Furthermore, this work is supported by an independent research grant from Immunodiagnostic Systems for determination of bone turnover concentrations in SHIP-1.
Footnotes
Disclosures
All authors state that they have no conflicts of interest.
References
- 1.Charles P, Elliott MJ, Davis D, et al. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J Immunol. 1999;163(3): 1521–8. [PubMed] [Google Scholar]
- 2.Stubbs JD, Lekutis C, Singer KL, et al. cDNA cloning of a mouse mammary epithelial cell surface protein reveals the existence of epidermal growth factor-like domains linked to factor VIII-like sequences. Proc Natl Acad Sci USA. 1990;87(21):8417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aziz M, Jacob A, Matsuda A, Wang P. Review: milk fat globule-EGF factor 8 expression, function and plausible signal transduction in resolving inflammation. Apoptosis. 2011;16(11):1077–86. [DOI] [PubMed] [Google Scholar]
- 4.Borisenko GG, Iverson SL, Ahlberg S, Kagan VE, Fadeel B. Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: implications for macrophage clearance of apoptotic cells. Cell Death Differ. 2004;11(8):943–5. [DOI] [PubMed] [Google Scholar]
- 5.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417(6885):182–7. [DOI] [PubMed] [Google Scholar]
- 6.Hanayama R, Tanaka M, Miyasaka K, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304(5674):1147–50. [DOI] [PubMed] [Google Scholar]
- 7.Aziz MM, Ishihara S, Mishima Y, et al. MFG-E8 attenuates intestinal inflammation in murine experimental colitis by modulating osteopontin-dependent avb3 integrin signaling. J Immunol. 2009; 182(11):7222–32. [DOI] [PubMed] [Google Scholar]
- 8.Abe T, Shin J, Hosur K, Udey MC, Chavakis T, Hajishengallis G. Regulation of osteoclast homeostasis and inflammatory bone loss by MFG-E8. J Immunol. 2014;193(3):1383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang F, Shah KG, Qi L, et al. Milk fat globule epidermal growth factor-factor 8 mitigates inflammation and tissue injury after hemorrhagic shock in experimental animals. J Trauma Acute Care Surg. 2012;72(4):861–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aziz M, Matsuda A, Yang W-L, Jacob A, Wang P. Milk fat globule-epidermal growth factor-factor 8 attenuates neutrophil infiltration in acute lung injury via modulation of CXCR2. J Immunol. 2012;189(1):393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Q, Yu Y, Zuo X, Dong Y, Li Y. Milk fat globule-epidermal growth factor 8 is decreased in intestinal epithelium of ulcerative colitis patients and thereby causes increased apoptosis and impaired wound healing. Mol Med. 2012;18:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinningen K, Albus E, Thiele S, et al. Loss of milk fat globule-epidermal growth factor 8 (MFG-E8) in mice leads to low bone mass and accelerates ovariectomy-associated bone loss by increasing osteoclastogenesis. Bone. 2015;76:107–14. [DOI] [PubMed] [Google Scholar]
- 13.Han X, Bolcato AL, Amar S. Identification of genes differentially expressed in cultured human osteoblasts versus human fibroblasts by DNA microarray analysis. Connect Tissue Res. 2002;43(1):63–75. [PubMed] [Google Scholar]
- 14.Harre U, Keppeler H, Ipseiz N, et al. Moonlighting osteoclasts as undertakers of apoptotic cells. Autoimmunity. 2012;45(8):612–9. [DOI] [PubMed] [Google Scholar]
- 15.Rauner M, Thiele S, Sinningen K, et al. Effects of the selective glucocorticoid receptor modulator compound A on bone metabolism and inflammation in male mice with collagen-induced arthritis. Endocrinology. 2013;154(10):3719–28. [DOI] [PubMed] [Google Scholar]
- 16.Monach PA, Mathis D, Benoist C. The K/BxN arthritis model. Curr Protoc Immunol. 2008. May;Chapter 15:Unit 15.22. [DOI] [PubMed]
- 17.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87(5):811–22. [DOI] [PubMed] [Google Scholar]
- 18.Motegi S, Garfield S, Feng X, Sárdy M, Udey MC. Potentiation of platelet-derived growth factor receptor-β signaling mediated by integrin-associated MFG-E8. Arterioscler Thromb Vasc Biol. 2011;31(11):2653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neutzner M, Lopez T, Feng X, Bergmann-Leitner ES, Leitner WW, Udey MC. MFG-E8/lactadherin promotes tumor growth in an angiogenesis-dependent transgenic mouse model of multistage carcinogenesis. Cancer Res. 2007;67(14):6777–85. [DOI] [PubMed] [Google Scholar]
- 20.Thiele S, Ziegler N, Tsourdi E, et al. Selective glucocorticoid receptor modulation maintains bone mineral density in mice. J Bone Miner Res. 2012;27(11):2238–41. [DOI] [PubMed] [Google Scholar]
- 21.Volzke H, Alte D, Schmidt CO, et al. Cohort profile: the study of health in Pomerania. Int J Epidemiol. 2010;40(2):294–307. [DOI] [PubMed] [Google Scholar]
- 22.Bao L, Lindgren JU, Zhu YU, Ljunggren H-G, Zhu J. Exogenous soluble tumor necrosis factor receptor type I ameliorates murine experimental autoimmune neuritis. Neurobiol Dis. 2003;12(1): 73–81. [DOI] [PubMed] [Google Scholar]
- 23.Kohno T, Brewer MT, Baker SL, et al. A second tumor necrosis factor receptor gene product can shed a naturally occurring tumor necrosis factor inhibitor. Proc Natl Acad Sci USA. 1990;87(21): 8331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuda A, Jacob A, Wu R, Zhou M, Aziz M, Wang P. Milk fat globule-EGF factor VIII ameliorates liver injury after hepatic ischemia-reperfusion. J Surg Res. 2013;180(1):e37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai W, Li Y, Lv Y, Wei C, Zheng H. The roles of a novel anti-inflammatory factor, milk fat globule-epidermal growth factor 8, in patients with coronary atherosclerotic heart disease. Atherosclerosis. 2014;233(2):661–5. [DOI] [PubMed] [Google Scholar]
- 26.Uchiyama A, Yamada K, Ogino S, et al. MFG-E8 regulates angiogenesis in cutaneous wound healing. Am J Pathol. 2014; 184(7):1981–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuda A, Jacob A, Wu R, et al. Milk fat globule-EGF factor VIII in sepsis and ischemia-reperfusion injury. Mol Med. 2011;17(1–2):126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aziz M, Jacob A, Matsuda A, et al. Pre-treatment of recombinant mouse MFG-E8 downregulates LPS-induced TNF-α production in macrophages via STAT3-mediated SOCS3 activation. PLoS One. 2011;6(11):e27685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miksa M, Amin D, Wu R, et al. Maturation-induced downregulation of MFG-E8 impairs apoptotic cell clearance and enhances endotoxin response. Int J Mol Med. 2008;22(6):743–8. [PMC free article] [PubMed] [Google Scholar]
- 30.Miksa M, Wu R, Dong W, Das P, Yang D, Wang P. Dendritic cell-derived exosomes containing milk fat globule epidermal growth factor-factor VIII attenuate proinflammatory responses in sepsis. Shock. 2006;25(6):586–93. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Bu H-F, Liu SX, De Plaen IG, Tan X-D. Molecular mechanisms underlying the regulation of the MFG-E8 gene promoter activity in physiological and inflammatory conditions. J Cell Biochem. 2015;116(9):1867–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miksa M, Amin D, Wu R, Dong W, Ravikumar TS, Wang P. Fractalkine-induced MFG-E8 leads to enhanced apoptotic cell clearance by macrophages. Mol Med. 2007;13:553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu L, Anderson S, Oehninger S, Bocca S. Tumor necrosis factor α up-regulates endometrial milk fat globule-epidermal growth factor 8 protein production via nuclear factor κB activation, resulting in cell migration of epithelial cells. Fertil Steril. 2014;101(2):552–9. [DOI] [PubMed] [Google Scholar]
- 34.Cui T, Miksa M, Wu R, et al. Milk fat globule epidermal growth factor 8 attenuates acute lung injury in mice after intestinal ischemia and reperfusion. Am J Respir Crit Care Med. 2010;181(3):238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srirangan S, Choy EH. The role of interleukin 6 in the pathophysiology of rheumatoid arthritis. Ther Adv Musculoskelet Dis. 2010;2(5): 247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Németh T, Mócsai A. The role of neutrophils in autoimmune diseases. Immunol Lett. 2012;143(1):9–19. [DOI] [PubMed] [Google Scholar]
- 37.Brissette M-J, Lepage S, Lamonde A-S, et al. MFG-E8 released by apoptotic endothelial cells triggers anti-inflammatory macrophage reprogramming. PLoS One. 2012;7(4):e36368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soki FN, Koh AJ, Jones JD, et al. Polarization of prostate cancer-associated macrophages is induced by milk fat globule-EGF factor 8 (MFG-E8)-mediated efferocytosis. J Biol Chem. 2014;289(35): 24560–72. [DOI] [PMC free article] [PubMed] [Google Scholar]