

ABSTRACT
Colorectal cancer represents the second most common cause of cancer-related death. The human A33 transmembrane glycoprotein is a validated tumor-associated antigen, expressed in 95% of primary and metastatic colorectal cancers. Using phage display technology, we generated a human monoclonal antibody (termed A2) specific to human A33 and we compared its epitope and performance to those of previously described clinical-stage anti-human A33 antibodies. All antibodies recognized a similar immunodominant epitope, located in the V-domain of A33, as revealed by SPOT analysis. The A2 antibody homogenously stained samples of poorly, moderately, and well differentiated colon adenocarcinomas. All antibodies also exhibited an intense staining of healthy human colon sections. The A2 antibody, reformatted in murine IgG2a format, preferentially localized to A33-transfected CT26 murine colon adenocarcinomas in immunocompetent mice with a homogenous distribution within the tumor mass, while other antibodies exhibited a patchy uptake in neoplastic lesions. A2 efficiently induced killing of A33-expressing cells through antibody-dependent cell-mediated cytotoxicity in vitro and was able to inhibit the growth of A33-positive murine CT26 and C51 lung metastases in vivo. Anti-A33 antibodies may thus represent useful vehicles for the selective delivery of bioactive payloads to colorectal cancer, or may be used in IgG format in a setting of minimal residual disease.
KEYWORDS: colorectal cancer, A33 glycoprotein, tumor-associated antigens, antibody therapeutics, ADCC
Introduction
Colorectal cancer (CRC) is one of the most common cancer types worldwide and ranks second in terms of cancer-related deaths.1 Approximately 20% of newly diagnosed CRC patients have already developed metastases in the liver, lungs, lymph nodes, peritoneum or soft tissues.2 Despite the substantial progress made in treating the disease, including the development of antibody-based therapeutics inhibiting angiogenesis (e.g., anti-vascular endothelial growth factor) or tumor growth factors (e.g., anti-endothelial growth factor receptor), the prognosis of patients with metastatic CRC remains poor.3 Immune checkpoint inhibitors, which provide a clinical benefit for patients with many different types of malignancies, are typically not active for the treatment of colorectal cancer,4 with the exception of DNA mismatch repair-deficient or microsatellite instability-high metastatic CRC patients, which respond positively to the combination treatment of nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4).5 For these reasons, there is an urgent need to develop more efficacious strategies for the treatment of patients with metastatic CRC.
Intense research efforts are currently being devoted to the implementation of antibody-based therapeutic strategies, in which the antibody moiety serves as a delivery vehicle for bioactive agents. Radionuclides,6 immunostimulatory cytokines,7,8 and T cells (delivered via bispecific antibodies with one binding site that targets CD39) have been considered as payloads for the treatment of metastatic CRC. In this context, the carcinoembryonic antigen and transmembrane glycoprotein A33 represent the most commonly used target antigens, as these have received extensive nuclear medicine validation in patients, using radiolabeled preparations of monoclonal antibody reagents.6,10–13
A33 is expressed in epithelia of the lower gastrointestinal tract and in 95% of primary and metastatic colorectal cancers.14 Clinical-stage antibodies have been developed against A33 and have been used for CRC treatment either as “naked” immunoglobulins,15 antibodies engineered for the delivery of beta-emitting radionuclides,6 or T-cell-engaging antibody moieties.16 The first anti-A33 antibody to be used for a tumor-targeting application was developed by the group of Lloyd J. Old.17 Here, we refer to this antibody as “K”. A humanized version of K (here described as K.hu) was reported in 1995.18 Recently, MacroGenics described the isolation and characterization of a novel humanized monoclonal antibody (here described as “MG”), which has been used for the development of bispecific antibodies in DART® format.16
Here we describe the isolation and characterization of a novel human antibody (termed A2), specific to A33. A2, which was compared to the K, K.hu, and MG antibodies in terms of biochemical properties and tumor-targeting characteristics, was found to avidly bind to CRC specimens in vitro and in vivo. The novel anti-A33 antibody was able to kill A33-expressing murine adenocarcinoma cells by antibody-dependent cell-mediated cytotoxicity (ADCC) in vitro and prevent the formation of murine CRC lung metastases in vivo. The results described in this article suggest that A2 may be applied as intact antibody able to kill colorectal tumor cells through ADCC, or as a building block for the implementation of antibody-based therapeutic strategies for the treatment of metastatic CRC.
Results
Murine model of colorectal cancer expressing human A33
The human transmembrane glycoprotein A33 is expressed in 95% of human colorectal cancers but has only 67% amino acid identity with its murine counterpart. In order to establish an immunocompetent murine model of colorectal cancer expressing A33, we stably transfected the murine colorectal carcinoma cell lines CT26wt and C51wt with the gene coding for A33 [Figure 1]. The resulting clones CT26A33.C3 and C51A33.A5, selected by antibiotic resistance and single-cell sorting, showed a shift in fluorescence intensity upon fluorescence-activated cell sorting (FACS) analysis using A33-specific antibodies, compared to isotype controls [Figure 1]. The staining intensities were comparable with that observed using the LS174T cells human colorectal cancer cell line.
Figure 1.
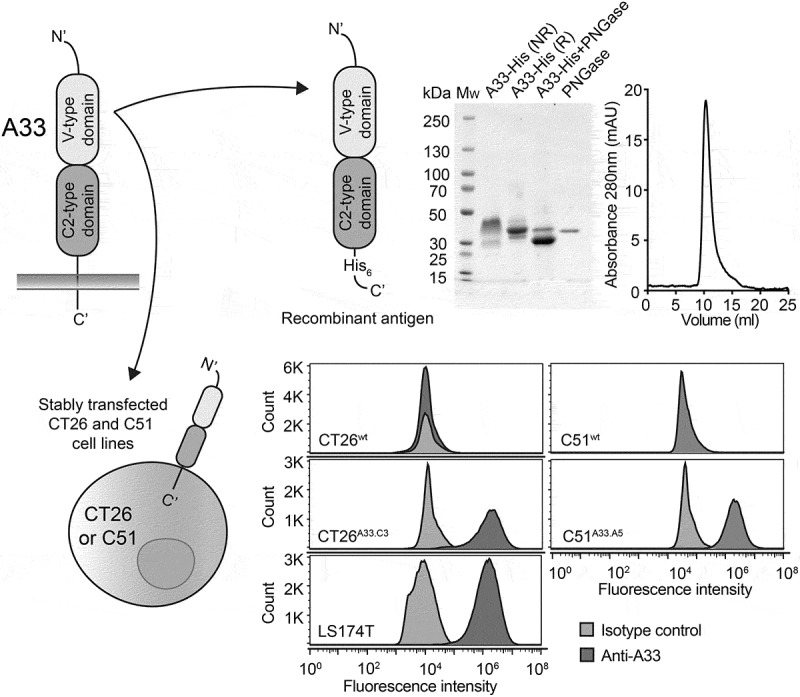
Recombinant A33-His antigen and murine A33-expressing adenocarcinoma cell lines. Top left: Schematic representation of the transmembrane A33 antigen. Top right: Schematic representation of the recombinant A33-His protein expressed in CHO cells. Only the extracellular V-type and C2-type Ig-like domains were expressed with a C-terminal Histidine Tag (His6). The recombinant antigen was characterized by SDS-PAGE analysis (MW: molecular weight, NR: non-reducing, R: reducing) and size exclusion chromatography. Bottom left: Representative image of murine colorectal cell lines CT26 and C51 expressing the A33 antigen as transmembrane protein. Bottom right: FACS analysis detecting expression of A33 on CT26wt, C51wt, human colorectal tumor cell line LS174T and transfected CT26A33.C3 and C51A33.A5 cells. Staining was performed with a murine anti-A33 specific antibody and the corresponding signal was amplified with an anti-mouse Alexa Fluor 488 secondary antibody. An isotype-matched antibody was used as negative control.
Expression of recombinant A33-his and antibody isolation
The extracellular Ig-like domains of A33 were cloned with a His-tag at the C-terminus into the mammalian expression vector pcDNA3.1(+) for protein expression in Chinese hamster ovary (CHO) cells [Figure 1]. The purified recombinant glycoprotein A33-His eluted as a single peak in size exclusion chromatography and migrated as a large band in non-reducing SDS-PAGE analysis, revealing a higher molecular weight (Mw) than the calculated Mw of 24.4 kDa. The sequence of A33 [Supplementary Figure 1] contains three possible sites for N-linked glycosylation. Deglycosylation of A33-His with PNGase led to the formation of a smaller and uniform band, with apparent Mw of 30 kDa in SDS-PAGE analysis [Figure 1].
Isolation of an A33-specific monoclonal antibody by phage display
The human antibody A2 in single-chain variable fragment (scFv) format was isolated by panning the human synthetic antibody phage library ETH-2-Gold19 against A33-His, immobilized on a solid support. The ETH-2-Gold library features scFv antibodies based on the germline VH segment DP47 and either the Vλ segment DPL16 or the Vk segment DPK22, which represent 12%, 16%, and 25%, respectively, of the antibody repertoire in humans. ScFv(A2) (VL germline DPL16) was reformatted into a chimeric murine IgG2a format for mammalian cell expression [Figure 2a]. Figure 2b indicates the amino acid sequence of scFv(A2), highlighting the portions of the CDR3 loops of VH and VL domains that had been combinatorially mutated in the ETH-2-Gold library.
Figure 2.
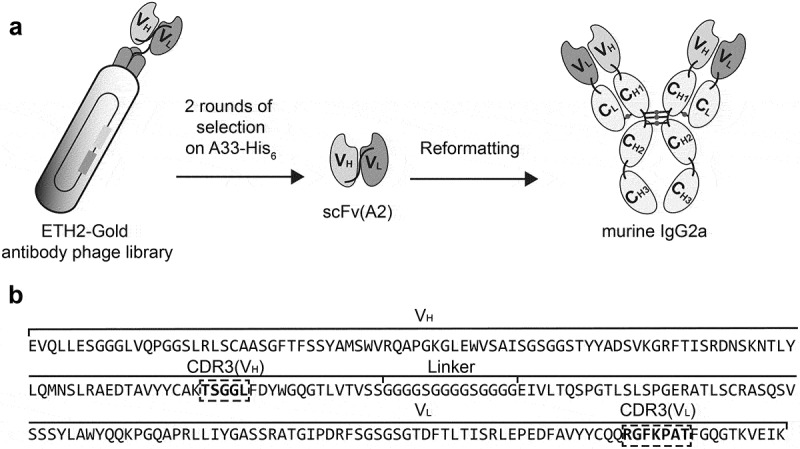
A monoclonal antibody specifically binding the human colorectal cancer antigen A33 was selected from the ETH-2-Gold phage display library. a) Schematic representation of the selection of the A33-specific antibody A2, as scFv fragment, from the ETH-2-Gold phage display library. The scFv(A2) was reformatted into murine IgG2a for further characterization. b) Amino acid sequence of scFv(A2) as selected from the ETH-2-Gold library, with the portions of the CDR3 loops of the VH and VL domains highlighted by dashed squares.
Expression and characterization of anti-A33 antibodies in the IgG2a format
In order to compare the antibody A2 to the three previously described clinical-stage anti-A33 antibodies [K 17, K.hu 18, and MG 9], all antibodies were reformatted in murine IgG2a format. The proteins were expressed in CHO cells and purified to homogeneity, using Protein A chromatography. The amino acid sequences of K, K.hu, and MG are reported in Supplementary Figure 2. Figure 3 illustrates the biochemical properties and the BIAcore profiles of the four antibodies. A slow dissociation from the antigen was observed for the A2 and MG antibodies, while a biphasic dissociation profile was seen for K and K.hu (the latter antibody showing a more rapid dissociation from A33 immobilized on the BIAcore sensor chip) [Figure 3]. Analyses of the functional affinities on CT26A33.C3 cells with serial dilutions of the anti-A33 IgG2a antibodies revealed apparent KD values ranging from 2 to 20 nM [Figure 4a]. The stronger functional affinity of K and K.hu to A33 on CT26A33.C3 cells compared to A2 and MG does not match with the BIAcore profiles on the immobilized A33. TA99, specific to a melanoma antigen,20 was used as negative control of irrelevant specificity in this context.
Figure 3.
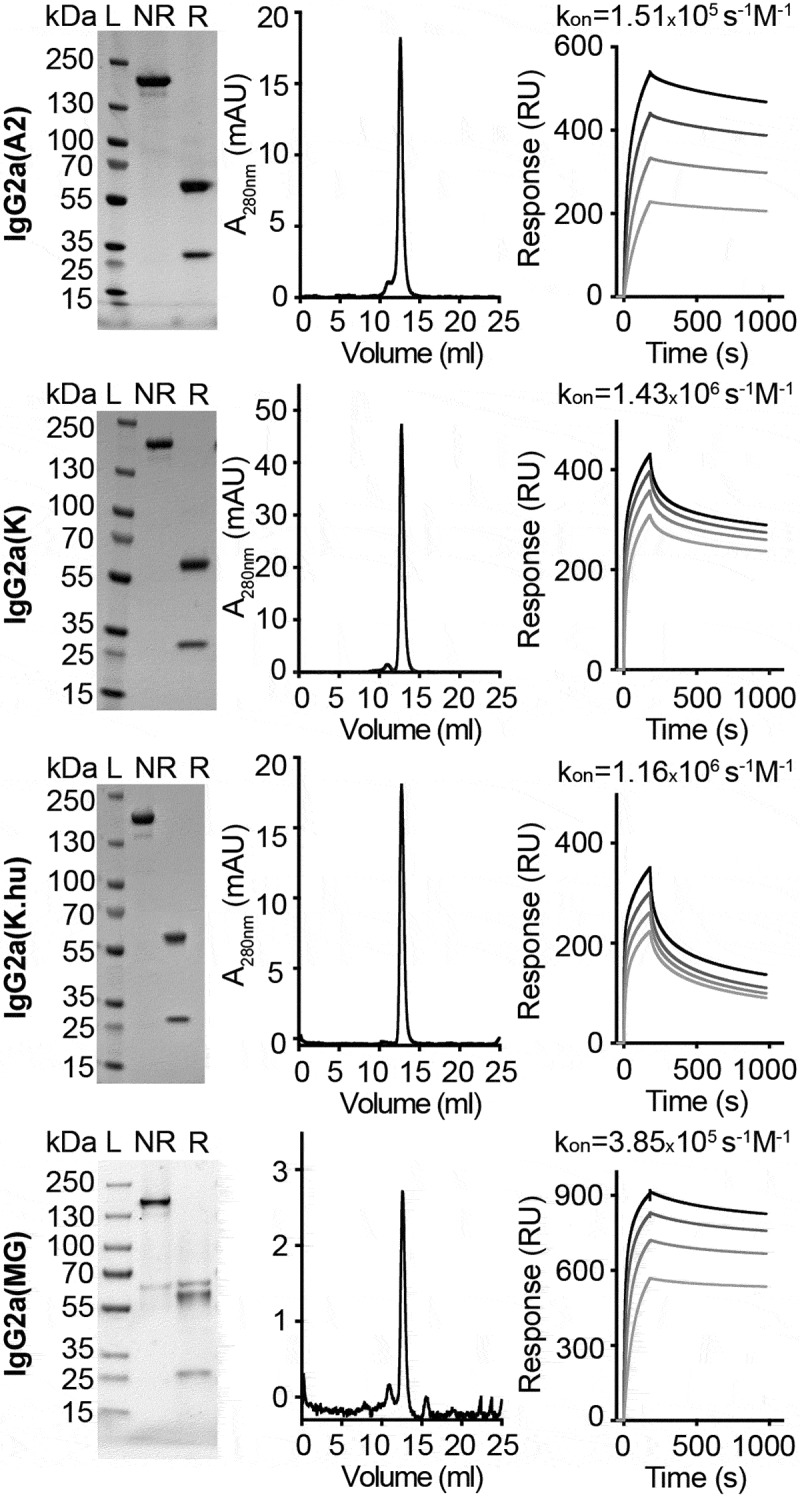
In vitro characterization of anti-A33 antibodies in the murine IgG2a format. SDS-PAGE analyses under non-reducing (NR) and reducing (r) conditions, size exclusion chromatography profiles, and SPR analyses of IgG2a(A2), IgG2a(k), IgG2a(K.hu), and IgG2a(MG). SPR analyses performed at antibody concentrations of 63 nM, 125 nM, 250 nM, and 500 nM.
Figure 4.
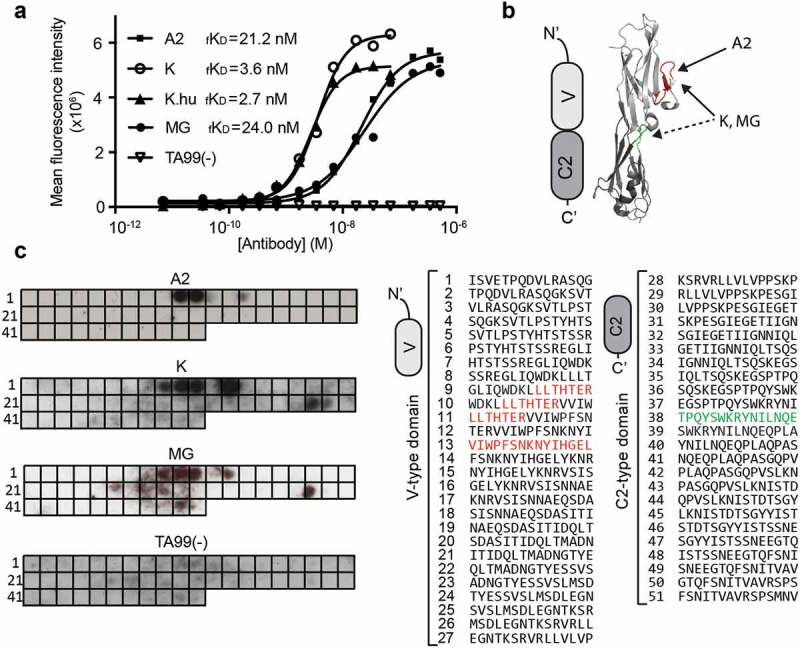
Binding affinity of the anti-A33 antibodies on CT26A33.C3 and epitope mapping. a)Functional binding affinity (fKD) of the antibodies A2, K, K.hu,MG,and negative control TA99(-), measured by flow cytometry on CT26A33.C3cells.b)Schematic representation and crystal structure of V-type and C2-type Ig-like domains, indicating the binding epitopes of A2 (red),K,and MG (discontinuous epitope red + green). c)Left:Binding epitopes of the anti-A33 antibodies A2,K,and MG,and the negative control TA99(-) on the PepSpot membrane (as black spots). Each spot (1–51) on the membrane is coated with 15 amino acid-long peptides, spanning the amino acid sequence of A33.Right: Amino acidsequence of every spot, with binding epitopes on the Ig-like V-type domain (for all three anti-A33 antibodies) in red and binding epitopes on the Ig-like C2-type domain (for the antibodies K and MG) in green.
Characterization of binding epitopes by SPOT technology
In order to identify the binding site of the anti-A33 antibodies, we used SPOT technology21 and immunodetection of antibody binding events to 15 amino acid-long peptides on a cellulose membrane, spanning the antigen sequence. All three antibodies (A2, K, and MG) recognized an immunodominant site of the A33 antigen [Figure 4b,c]. At a four times higher concentration, K and MG antibodies (2 µg/ml) generated multiple signals on the A33 peptide membrane, possibly indicating discontinuous binding epitopes on the Ig-like V-type (red) and Ig-like C2-type (green) domains [Figure 4b,c]. The TA99 antibody, serving as negative control of irrelevant specificity in this setting, did not exhibit binging on the SPOT assay [Figure 4c].
Comparison of A33 detection on human tissues
The A2, K, and MG antibodies were studied by immunofluorescence staining on a tissue array, which contained 37 colorectal cancer sections and 3 normal colon sections. The three antibodies were used at identical concentrations for the immunofluorescence analysis, while the TA99 antibody was used as negative control [Supplementary Figure 3]. Figure 5 shows sections stained in green with individual anti-A33 antibodies, while the vasculature was stained in red with an anti-von Willebrand Factor reagent. The A2 antibody exhibited a brighter and more homogenous staining of tissue sections, compared to the K and MG antibodies, regardless of the tumor differentiation status [Figure 5]. A complete analysis of the tissue arrays can be found in Supplementary Figure 3. No optimization of the concentrations for the immunofluorescence analyses was performed.
Figure 5.
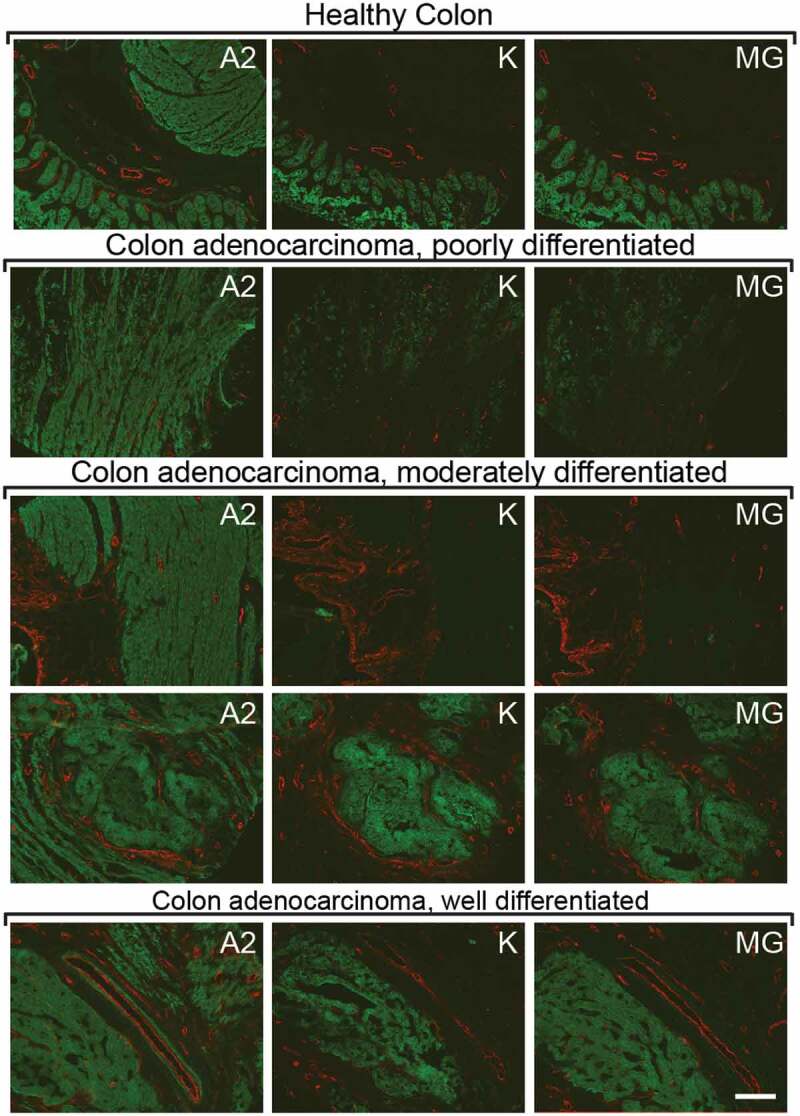
Immunofluorescence staining of A33 on human colon and human adenocarcinoma sections. Detection of A33 on human colon tissue sections and on human colon adenocarcinoma sections with the antibodies A2, K, and MG (green, Alexa Fluor 488). Blood vessels stained with an anti-Von Willebrand Factor antibody (red, Alexa Fluor 595). Each horizontal line represents sequential sections from the same tumor sample, stained with a different antibody. Tumor samples vary in degree of differentiation. Scale bar = 150 µm.
In vivo biodistribution of anti-A33 antibodies
The tumor-homing properties of the anti-A33 antibodies were assessed in immunocompetent BALB/c mice bearing subcutaneously grafted CT26A33.C3 carcinomas. An ex vivo immunofluorescence analysis, performed 24 h after intravenous (i.v.) administration of A2, K, and MG, respectively (150 µg/mouse, no dose optimization), revealed a selective homing of the antibodies to the tumor cells [Figure 6]. By contrast, the negative control antibody TA99 did not exhibit any detectable accumulation at the tumor site at the same time point. A2 exhibited a more homogeneous and less patchy distribution within the tumor mass, compared to the K and MG counterparts.
Figure 6.
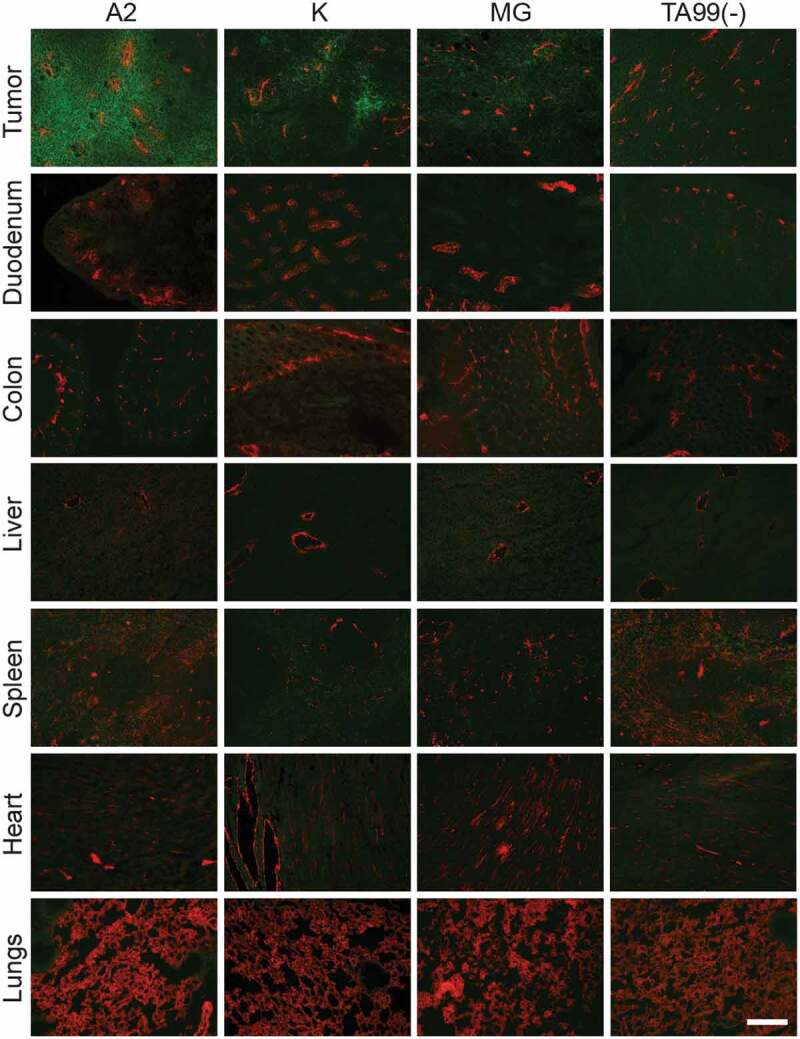
In vivo A2, K, and MG selectively accumulate in subcutaneous CT26A33.C3 tumors.Microscopic fluorescence analysis of organs from CT26A33.C3 tumor-bearing mice, 24 h after intravenous administration of FITC-labeled A2, K, MG, or TA99 antibodies (green, Alexa Fluor 488). Blood vessels stained with anti-CD31 (red, Alexa Fluor 594). Magnification 10x, Scale bar = 125 µm.
In vitro and in vivo A2-mediated cytotoxicity against C51 A33.C3 and CT26A33.C3 colorectal cell lines
The anti-A33 antibody A2 was reformatted into human IgG1, in order to test its ability to induce ADCC in vitro, using human peripheral blood mononuclear cells (PBMCs). The sequence of IgG1(A2) and the protein characterization are reported in Supplementary Figure 4. The target cell lines C51A33.C3 and CT26A33.C3 were stained with 5(6)-carboxyfluorescein diacetate N-succinimidyl ester (CFSE) dye and incubated with different concentrations of IgG1(A2) and human PMBCs, at 1:50 ratio. IgG1(KSF), directed against hen egg lysozyme,22 was used as negative control in this setting. Flow cytometric analyses after 24-h incubation revealed a specific and concentration-dependent depletion of CFSE-labeled target cells by the IgG1(A2) antibody [Figure 7a]. In vivo, the A2 antibody was used in the chimeric murine IgG2a format, in a setting aimed at preventing lung metastases. IgG2a(A2) was administered immediately after the i.v. injection of C51A33.C3 or CT26A33.C3 murine adenocarcinoma cells into BALB/c mice. The treatment significantly inhibited lung metastasis formation in both CT26 and C51 models, compared to saline or isotype-matched negative control antibodies (p < .05 for C51A33.C3 against isotype control and saline, p < .01 for CT26A33.C3 against isotype control and p < .001 against saline) [Figure 7b].
Figure 7.
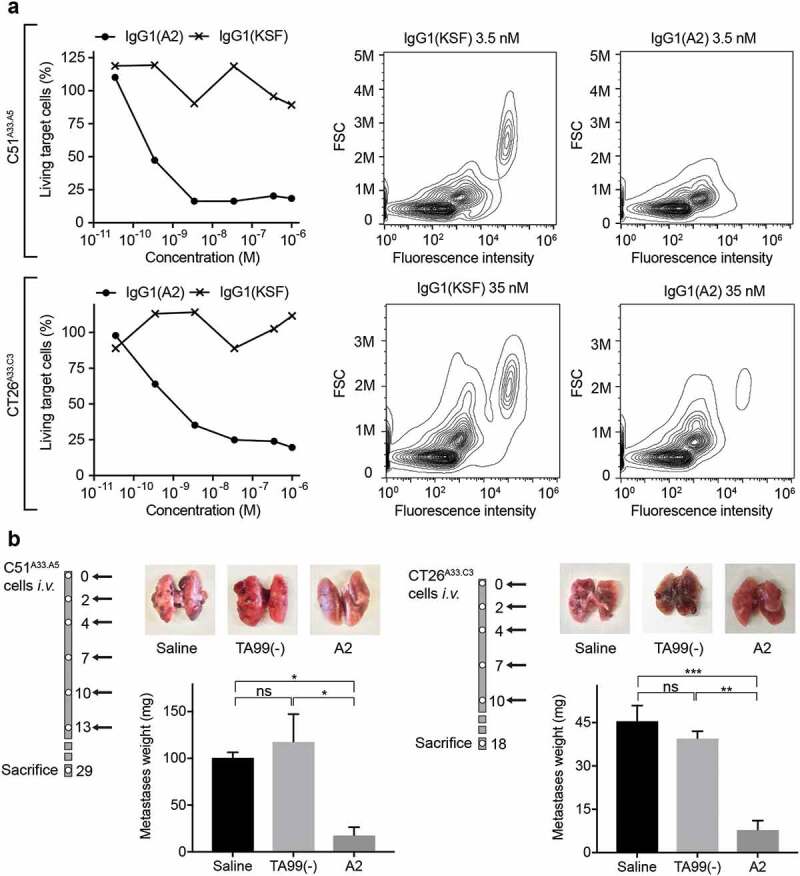
The antibody A2 induces ADCC in vitro and in vivo. a) In vitro ADCC killing of C51A33.A5 and CT26A33.C3 murine adenocarcinoma cells induced by the antibody A2 in IgG1 format, when incubated with human PBMCs for 24 h. Left: Percentage of living tumor (CFSE positive) cells plotted against increasing antibody concentrations. Right: Representative FACS panels showing the specific lysis of tumor cells (CFSE stained). b) Inhibition of lung metastases formation upon treatment with the antibody A2 in the IgG2a format in mice injected intravenously with C51A33.A5 (left) CT26A33.C3 (right). Schematic representation of the therapy schedule indicating i.v. tumor cell injection, followed by 200 µg A2, TA99, or saline i.v. injections. Representative images of lungs for each treatment group. Metastases were excised and weighted. Data represent mean metastases weight in mg ± SEM. ***, p < .001; **, p < .01; *, p < .05; ns, p > .05 (two-way ANOVA test, followed by Bonferroni posttest). C51A33.A5: Saline n = 3, TA99(-) and A2 n = 4. CT26A33.C3 n = 3.
Discussion
We have reported the generation of a novel human antibody specific to the human tumor-associated antigen A33. The antibody, named A2, was compared in vitro and in vivo with two previously described anti-A33 antibodies, here called K 17 and MG.9,16
The murine antibody K was tested in the early 1990s in Phase 1/2 clinical trials, in which the product was used as a “delivery vehicle” for 131I23 or 125I24 in patients with metastatic CRC. A humanized version of the K antibody (referred to as K.hu), with longer serum half-life, was also used for radioimmunotherapy in radioiodinated form. The product did not induce objective tumor responses in patients, but a high accumulation in CRC metastases could be detected using nuclear medicine techniques.25 Gastrointestinal toxicity was not reported, in spite of the fact that labeled K.hu antibody was found to also localize to the normal gastrointestinal tract.26 After repeated monthly administrations, the humanized K.hu antibody induced a human anti-human antibody response in the majority of CRC patients (73%),15 as evidenced by BIAcore analysis of sera. These data suggest that a less immunogenic anti-A33 antibody may be required for future clinical projects.
The MG antibody, generated by MacroGenics, has recently entered a Phase 1/2 clinical trial (NCT03531632) in a bispecific antibody format [A33 x CD3 DART™ (MGD007)], aimed at recruiting T cells to the tumor site. In this study, MGD007 is being evaluated in combination with an anti-PD-1 monoclonal antibody (MGA012) for the treatment of patients with metastatic CRC. The bispecific T-cell engager MGD007 induced tumor growth inhibition in mouse models of colorectal cancer (LS174T and Colo205) (LS174T and Colo205),16 but clinical results have not yet been disclosed.
The novel human anti-A33 antibody A2 selectively and homogenously accumulated into subcutaneously grafted CT26A33.C3 tumors in BALB/c mice upon i.v. injection [Figure 6]. Immunofluorescence staining of human colon adenocarcinoma and normal human colon tissue sections with A2 showed the presence of A33 in areas that were negative for A33 when stained with identical concentrations of the antibodies K and MG [Figure 5]. The three antibodies were shown to bind an immunodominant epitope on the Ig-like V-type domain of A33 [Figure 4], even though the antibodies K and MG displayed binding on a discontinuous epitope. Binding of A2 to A33-expressing murine adenocarcinoma cells induced immune-mediated cell killing through ADCC as demonstrated with human PBMCs in vitro [Figure 7a] and by inhibition of lung metastases formation in vivo [Figure 7b]. Based on our results, minimal contribution on in vivo efficacy of the A2 antibody by direct gpA33 blockade, by complement-dependent cytotoxicity or antibody-dependent cellular phagocytosis cannot be excluded.
ADCC represents an elegant avenue for re-directing the cytotoxic potential of immune effector cells to malignant cells expressing a target antigen. We and others have previously shown that the main limitation for an efficient implementation of ADCC in a solid tumor setting relates to the lack of active immune effector cells in the neoplastic mass.27 Clinical applications of monoclonal antibodies inducing ADCC for the treatment of disseminated solid tumors have been unsuccessful in most cases.28,29 However, the use of trastuzumab as adjuvant treatment in metastatic breast cancer was shown to provide a substantial benefit to patients,30 suggesting that ADCC strategies may be better suited for the treatment of minimal residual disease. The good tolerability of intact antibodies makes them suitable for adjuvant therapy, when long‐term treatment is required. Alternatively, ADCC activity in solid tumors may be boosted by combination therapies with tumor-targeted immunostimulatory cytokines27,31,32 or by Fc engineering strategies.33–36
A33 expression in human CRC has been described to be heterogeneous,37,38 as also observed by immunofluorescent staining of human CRC tissue sample [Supplementary Figure 3]. Therapeutic strategies capable of inducing a bystander effect on antigen-negative cells39-41 may be particularly suited when targeting the A33 antigen.
The novel anti-A33 A2 antibody was able to successfully deliver a fluorescein isothiocyanate (FITC) payload to the tumor site and to inhibit cancer dissemination in a minimal residual disease setting. A2 in IgG format may therefore be considered for adjuvant treatment applications in CRC patients or as a versatile “vehicle” for the delivery of bioactive payloads.
Materials and methods
Cell lines
CHO cells, the murine colorectal carcinoma cell line CT26wt, and the human colorectal cell lines HT-29 and LS174T were obtained from the American Type Culture Collection between 2015 and 2017, expanded, and stored as cryopreserved aliquots in liquid nitrogen. Cells were grown according to the manufacturer’s protocol. Transfected CT26 cells were kept in the same culture conditions as CT26.wt cells. Authentication of the cell lines, including check of post-freeze viability, growth properties, and morphology, test for mycoplasma contamination, isoenzyme assay, and sterility test, was performed by the cell bank before shipment.
Cloning, expression, biotinylation, and characterization of the antigen
The cDNA encoding for the A33 construct was obtained by total RNA extraction from the HT-29 colorectal carcinoma cell line (High Pure RNA Isolation Kit, Roche), reverse transcription (Transcriptor reverse transcriptase, Roche) and amplification of A33 cDNA by Taq DNA Polymerase (Sigma-Aldrich). The extracellular domains of the antigen were PCR amplified, extended with a nucleic acid sequence encoding for six histidine residues (His-tag) and cloned into the mammalian expression vector pcDNA3.1(+) (Invitrogen). The soluble antigen was produced by transient gene expression in CHO cells as described previously42 and purified from the cell culture medium by Ni-NTA resin (Roche). Quality control of A33-His was performed by SDS-PAGE and size exclusion chromatography (Superdex75 10/300GL, GE Healthcare). The protein was digested with PNGase F (New England BioLabs) under denaturing conditions. A33-His was biotinylated with Sulfo-NHS-LC-Biotin (Pierce). Biotinylation of the antigen was tested through mass spectrometry (MS) analysis and band shift assay [Supplementary Figure 5].
Transfection of human A33 in CT26 tumor cells and monoclonal selection
The gene for human A33 was cloned into the mammalian expression vector pcDNA3.1(+) containing an antibiotic resistance for G418 Geneticin. The plasmid was digested with PvuI (HF) for linearization prior to transfection. CT26wt and C51wt cells were transfected with the A33 gene in pcDNA3.1(+) using the Amaxa™ 4D-Nucleofector (Lonza) and the SG Cell Line 4D-Nucleofector® X Kit L (Lonza) and reseeded. Three days after the transfection, the growing medium was supplemented with 0.5 mg/ml G418 (Merck) for the selection of stably transfected polyclonal cells. To obtain a monoclonal cell line, single-cell sorting of stably transfected cells was performed using BD FACSAria III. Different clones were expanded and checked for antigen expression by FACS analysis and immunofluorescence. Clones CT26A33.C3 and C51A33.A5 were selected as the best clone based on A33 surface expression and cell viability.
Selection of A33.A2 from the ETH-2-Gold library by phage display
The recombinant biotinylated A33-His antigen was immobilized on streptavidin- (StreptaWells, Roche) or avidin-coated wells (Avidin, Sigma-Aldrich on MaxiSorp plates, Sigma). Monoclonal antibodies were isolated from the synthetic human scFv antibody library ETH-2-Gold19 by two rounds of biopanning as previously described.43,44 Supernatants of single bacterial colonies expressing the selected scFvs were screened by ELISA on immobilized A33-His. Clones yielding positive ELISA signals were sequenced and binding to the cognate antigen was confirmed by FACS analysis on the human colorectal carcinoma cell line LS174T and the transfected murine colorectal carcinoma cell line CT26A33.C3.
Cloning, expression, and characterization of the anti-A33 antibodies
Sequences of the variable region of the light and heavy chain (VH and VL) of A33.K and A33.K.hu were taken from King et al.18 Sequences of A33.MG VH and VL were found in the patent file from MacroGenics.9 The genes encoding for VH and VL of the antibodies A2, K, K.hu, and MG, and the constant regions of the IgG2a immunoglobulin were PCR amplified, PCR assembled, and cloned into the mammalian expression vector pMM137, as previously described.27 The IgG2a molecules were produced using transient gene expression in CHO cells following standard protocols22,42 and purified from the cell culture medium to homogeneity by Protein A chromatography (Thermo Scientific). Quality control of the proteins was performed by SDS-PAGE and size exclusion chromatography (Superdex200 10/300GL, GE Healthcare). The full amino acid sequences are presented in Supplementary Figure 2. The A2 antibody was additionally expressed in the human IgG1 format, for testing ADCC activity with human PBMCs. The sequence and characterization of IgG1(A2) are reported in Supplementary Figure 4.
Surface plasmon resonance analysis on A33-his coated chip
Biotinylated A33-His was immobilized on SA-sensor chip with a density of 1000 RU using a BIAcore 200 System. Real-time interaction analysis was performed with serial dilutions of the anti-A33 antibodies. Phosphate-buffered saline (PBS; pH 7.4) was used as mobile phase and 10 mM HCl as regeneration solution. The binding curves were analyzed with the BIAevaluation 3.2 software to obtain Kon values.
Flow cytometry
CT26wt, CT26A33.C3, C51wt, C51A33.A5, and LS174T cells were detached with 2 mM ethylenediaminetetraacetic acid (EDTA) from the culture flask and stained with different concentrations (75–0.001µg/ml) of the respective anti-A33 antibody or the negative control TA99. The antibodies were detected using donkey anti-mouse Alexa Fluor 488 (Invitrogen). All staining and washing steps were performed in 2 mM EDTA 0.5% bovine serum albumin (BSA) in PBS. Cells were sorted by FACS (CytoFLEX, Beckman Coulter) and analyzed using FlowJo software.
Epitope mapping using peptide array
The binding epitope of the anti-A33 antibodies on the A33 extracellular domain was determined using a peptide array (PepSpot, JPT). Sequences of 15 amino acid-long peptides covering the whole sequence of the A33 extracellular domain with overlapping linear sequences were covalently bound to a cellulose membrane on 51 different spots. The assay was performed according to the manufacturer’s instructions and the binding spots were detected by a chemiluminescence imager (Agfa Curix 60, Agfa Healthcare). The respective IgG2a antibodies were incubated with the PepSpot membrane at concentrations ranging from 0.5 µg/ml to 2 µg/ml and detected by protein A-HRP (GE Healthcare).
Ex-vivo immunofluorescences on CT26A33.C3 tumor bearing mice
Seven weeks old female BALB/c mice were obtained from Janvier. About 4 × 106 CT26A33.C3 were suspended in 100 µl Hanks’ Balanced Salt Solution (HBSS) (Gibco) and injected subcutaneously in the left flank of each mouse using 0.5 ml 29G insulin syringes (MicroFine™+, BD medical). The anti-A33 antibodies A2, K, and MG were labeled with FITC as described in the manufacturer’s protocol (Sigma). For ex vivo immunofluorescence analysis, BALB/c mice bearing CT26A33.C3 subcutaneous tumors (100 mm3) were injected with sterile-filtered 150 µg FITC-labeled IgG2a antibody in 100 µl PBS (Gibco). Mice were sacrificed 24 h after injection. The organs were excised, embedded in cryoembedding medium (Thermo Scientific), and cryostat sections (8 mm) were stained using rabbit anti-FITC (BioRad; 4510–780) and rat anti-CD31 (BD Biosciences; 553370) as primary antibody and donkey anti-rabbit Alexa Fluor 488 (Invitrogen; A21206) and anti-rat Alexa Fluor 594 (Invitrogen; A21209) as secondary antibodies. Slides were mounted with fluorescent mounting medium (Dako) and analyzed with Axioscop2 mot plus microscope (Zeiss). For the biodistribution of the fluorescently labeled antibodies, one mouse was used per tumor model, per antibody. Immunoreactivity of the anti-gpA33 antibodies K, A2, and MG to the recombinant gpA33 antigen was preserved after FITC-labeling, as assessed by ELISA (data not shown).
Immunofluorescence analysis on human colorectal tumor sections
Arrays of freshly frozen human colorectal tumor (37) and healthy colon tissues (3) were obtained from Amsbio. A list of the 40 different tissues, including pathology grade, is shown in Supplementary Figure 3. For immunofluorescence analysis, the tissue arrays were fixed in chilled acetone before staining. As primary staining antibodies, A2, K, MG, and TA99 were used at a concentration of 1 µg/ml together with a rabbit anti-human Von Willebrand Factor (1:800, Dako) in 3% BSA in PBS. Detection was performed using a donkey anti-mouse IgG Alexa Fluor 488 (Invitrogen, A21202) and goat anti-rabbit IgG (Invitrogen, A11037).
In vitro ADCC assay on CT26A33.C3 and C51A33.A5 cells
CT26A33.C3 and C51A33.A5 were stained with CFSE dye (BioLegend) as described by the manufacturer’s protocol and seeded in a 48-well plate (2⋅104/well). After informed consent was given, peripheral blood samples were obtained from healthy donors from the Blutspendedienst SRK, Zurich. PBMCs were isolated by density gradient centrifugation on Ficoll Paque Plus (GE Healthcare) following the manufacturer’s protocol. The peripheral blood was diluted 1:3 in 2 mM EDTA/PBS. Thirty milliliters of diluted peripheral blood was layered on 12.9 ml Ficoll and centrifuged at 400 g for 40 min at RT. PBMCs were collected, washed twice with PBS/EDTA, and incubated with the tumor target cells (106/well, 1:50 target:effector ratio) in CT26A33.C3 and C51A33.A5 culture media. The antibody IgG1(A2) was added at different concentrations to the wells (total volume of 200µl/well) and the plate was incubated at 37°C, 5% CO2 for 24 h. On the next day, the samples with IgG1(A2) + PBMCs + target cells were transferred to a 96-well plate, washed twice with PBS and stained Fixable Viability Dye (Invitrogen) for 30 min. The samples were washed twice with 2 mM EDTA 0.5% BSA in PBS, before being analyzed by FACS (CytoFLEX, Beckman Coulter). The percentage of living target cells was derived from the mean fluorescence intensity of the CFSE-positive cell population normalized based on the negative control samples containing PBMCs and target cells only.
In vivo prevention of CT26A33.C3 and C51A33.A5 lung metastases formation
About 5⋅105 CT26A33.C3 or 5⋅104 C51A33.A5 cells were suspended in 100 µl HBSS (Gibco) and injected (i.v.) through the tail vein of 7 weeks old female BALB/c mice (Janvier) using 0.5 ml 29G insulin syringes (MicroFine™+, BD medical). Mice were injected (i.v.) with 100 µl PBS (Gibco), 200 µg of A2 or TA99 antibody (in 100 µl PBS, pH 7.4) in the IgG2a format as indicated in Figure 7b (black arrows). After euthanasia, mice were perfused with 1% paraformaldehyde (PFA) through the pulmonary artery and the aorta after. Lungs were excised and fixed for 1 h in 4% PFA. Lung metastases were excised and weighted. Unresectable, visible metastases were counted and added to the final weight (1 mg each). For in vivo ADCC experiments with C51A33.A5 cells, three mice were treated with saline and four mice with IgG2a(A2) and IgG2a(TA99), respectively. Three mice per treatment group were injected with CT26A33.C3.
Animal experiments
Experiments were performed under a project license (license number 04/2018), granted by the Veterinäramt des Kanton Zürich, Switzerland, in compliance with the Swiss Animal Protection Act (TSchG) and the Swiss Animal Protection Ordinance (TSchV). BALB/c mice (Janvier, 7–8 weeks old female) were kept in the OHB Animal Facility of the Rodent Center ETH Hönggerberg in cages of 2–5 mice, left for at least one-week acclimatization upon arrival and, after that, handled under sterile BL2 workbenches. Mice were injected with tumor cells suspended in 100 µl HBSS (Gibco) and injected subcutaneously in the right flank or i.v. using 0.5 ml 29G insulin syringes (MicroFine™+, BD medical). All antibodies were diluted in PBS (Gibco) and sterile-filtered before i.v. injection in the lateral tail vein of mice.
Funding Statement
The work was financially supported by ETH Zürich, the ERC Advanced Grant “Zauberkugel” (Grant Agreement 670603), the Swiss National Science Foundation (project number: 310030_182003/1), the Swiss Federal Commission for Technology and Innovation (Grant number: 17072.1) and the “Stiftung zur Krebsbekämpfung”.
Abbreviations
| ADCC | Antibody-dependent cell-mediated cytotoxicity |
| CDR | Complementarity-determining region |
| CFSE | 5(6)-carboxyfluorescein diacetate N-succinimidyl ester |
| CRC | Colorectal cancer |
| Mw | Molecular weight |
| PBMC | Peripheral blood mononuclear cell |
| scFv | Single-chain variable fragment |
| VL | Variable domain of the light chain |
| VH | Variable domain of the heavy chain |
Acknowledgments
We thank Dr. Renier Myburgh for providing the PBMCs for in vitro ADCC evaluation. The authors gratefully acknowledge financial support from ETH Zürich, the ERC Advanced Grant “Zauberkugel” (Grant Agreement 670603), the Swiss National Science Foundation (project number: 310030_182003/1), the Swiss Federal Commission for Technology and Innovation (Grant number: 17072.1), and the “Stiftung zur Krebsbekämpfung”.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Conflict of interest
Prof. Dr. Dario Neri is a board member and shareholder of Philogen AG. The remaining authors declare no conflict of interest.
Ethics approval and consent to participate
Experiments were performed under a project license (license number 04/2018), granted by the Veterinäramt des Kanton Zürich, Switzerland, in compliance with the Swiss Animal Protection Act (TSchG) and the Swiss Animal Protection Ordinance (TSchV).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A.. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–11. doi: 10.3322/caac.v68.6. [DOI] [PubMed] [Google Scholar]
- 2.Riihimaki M, Hemminki A, Sundquist J, Hemminki K. Patterns of metastasis in colon and rectal cancer. Sci Rep. 2016;6:29765. doi: 10.1038/srep29765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marley AR, Nan H. Epidemiology of colorectal cancer. Int J Mol Epidemiol Genet. 2016;7:105–14. [PMC free article] [PubMed] [Google Scholar]
- 4.Kalyan A, Kircher S, Shah H, Mulcahy M, Benson A. Updates on immunotherapy for colorectal cancer. J Gastrointest Oncol. 2018;9:160–69. doi: 10.21037/jgo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, Morse MA, Van Cutsem E, McDermott R, Hill A, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–79. doi: 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 6.Zanzonico P, Carrasquillo JA, Pandit-Taskar N, O’Donoghue JA, Humm JL, Smith-Jones P, Ruan S, Divgi C, Scott AM, Kemeny NE, et al. PET-based compartmental modeling of (124)I-A33 antibody: quantitative characterization of patient-specific tumor targeting in colorectal cancer. Eur J Nucl Med Mol Imaging. 2015;42:1700–06. doi: 10.1007/s00259-015-3061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwegler C, Dorn-Beineke A, Nittka S, Stocking C, Neumaier M. Monoclonal anti-idiotype antibody 6G6.C4 fused to GM-CSF is capable of breaking tolerance to carcinoembryonic antigen (CEA) in CEA-transgenic mice. Cancer Res. 2005;65:1925–33. doi: 10.1158/0008-5472.CAN-04-3591. [DOI] [PubMed] [Google Scholar]
- 8.Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology. 2017;6:e1277306. doi: 10.1080/2162402X.2016.1277306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore PA, Li J, Zhifen Chen F, Johnson LS, Shah K, Bonvini E. Bi-specific diabodies that are capable of binding gpA33 and CD3 and uses thereof, patent WO2015026894A3 MacroGenics Inc. 2015.
- 10.Behr T, Becker W, Hannappel E, Goldenberg DM, Wolf F. Targeting of liver metastases of colorectal cancer with IgG, F(ab’)2, and Fab’ anti-carcinoembryonic antigen antibodies labeled with 99mTc: the role of metabolism and kinetics. Cancer Res. 1995;55:5777s–85s. [PubMed] [Google Scholar]
- 11.Goldenberg DM, Wlodkowski TJ, Sharkey RM, Silberstein EB, Serafini AN, Garty II, Van Heertum RL, Higginbotham-Ford EA, Kotler JA, Balasubramanian N, et al. Colorectal cancer imaging with iodine-123-labeled CEA monoclonal antibody fragments. J Nucl Med. 1993;34:61–70. [PubMed] [Google Scholar]
- 12.Murray JL, Rosenblum MG, Zhang HZ, Podoloff DA, Kasi LP, Curley SA, Chan JC, Roh M, Hohn DC, Brewer H, et al. Comparative tumor localization of whole immunoglobulin G anticarcinoembryonic antigen monoclonal antibodies IMMU-4 and IMMU-4 F(ab’)2 in colorectal cancer patients. Cancer. 1994;73:850–57. [DOI] [PubMed] [Google Scholar]
- 13.Carrasquillo JA, Pandit-Taskar N, O’Donoghue JA, Humm JL, Zanzonico P, Smith-Jones PM, Divgi CR, Pryma DA, Ruan S, Kemeny NE, et al. (124)I-huA33 antibody PET of colorectal cancer. J Nucl Med. 2011;52:1173–80. doi: 10.2967/jnumed.110.086165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garinchesa P, Sakamoto J, Welt S, Real F, Rettig W, Old L. Organ-specific expression of the colon cancer antigen A33, a cell surface target for antibody-based therapy. Int J Oncol. 1996;9:465–71. doi: 10.3892/ijo.9.3.465. [DOI] [PubMed] [Google Scholar]
- 15.Welt S, Ritter G, Williams C Jr., Cohen LS, John M, Jungbluth A, Richards EA, Old LJ, Kemeny NE. Phase I study of anticolon cancer humanized antibody A33. Clin Cancer Res. 2003;9:1338–46. [PubMed] [Google Scholar]
- 16.Moore PA, Shah K, Yang Y, Alderson R, Roberts P, Long V, Liu D, Li JC, Burke S, Ciccarone V, et al. Development of MGD007, a gpA33 x CD3-bispecific DART protein for T-cell immunotherapy of metastatic colorectal cancer. Mol Cancer Ther. 2018;17:1761–72. doi: 10.1158/1535-7163.MCT-17-1086. [DOI] [PubMed] [Google Scholar]
- 17.Welt S, Divgi CR, Real FX, Yeh SD, Garin-Chesa P, Finstad CL, Sakamoto J, Cohen A, Sigurdson ER, Kemeny N, et al. Quantitative analysis of antibody localization in human metastatic colon cancer: a phase I study of monoclonal antibody A33. J Clin Oncol. 1990;8:1894–906. doi: 10.1200/JCO.1990.8.11.1894. [DOI] [PubMed] [Google Scholar]
- 18.King DJ, Antoniw P, Owens RJ, Adair JR, Haines AM, Farnsworth AP, Finney H, Lawson A, Lyons A, Baker TS, et al. Preparation and preclinical evaluation of humanised A33 immunoconjugates for radioimmunotherapy. Br J Cancer. 1995;72:1364–72. doi: 10.1038/bjc.1995.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silacci M, Brack S, Schirru G, Marlind J, Ettorre A, Merlo A, Viti F, Neri D. Design, construction, and characterization of a large synthetic human antibody phage display library. Proteomics. 2005;5:2340–50. doi: 10.1002/(ISSN)1615-9861. [DOI] [PubMed] [Google Scholar]
- 20.Welt S, Mattes MJ, Grando R, Thomson TM, Leonard RW, Zanzonico PB, Bigler RE, Yeh S, Oettgen HF, Old LJ, et al. Monoclonal antibody to an intracellular antigen images human melanoma transplants in nu/nu mice. Proc Natl Acad Sci U S A. 1987;84:4200–04. doi: 10.1073/pnas.84.12.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frank R. The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports–principles and applications. J Immunol Methods. 2002;267:13–26. doi: 10.1016/S0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- 22.Pasche N, Woytschak J, Wulhfard S, Villa A, Frey K, Neri D. Cloning and characterization of novel tumor-targeting immunocytokines based on murine IL7. J Biotechnol. 2011;154:84–92. doi: 10.1016/j.jbiotec.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 23.Welt S, Divgi CR, Kemeny N, Finn RD, Scott AM, Graham M, Germain JS, Richards EC, Larson SM, Oettgen HF, et al. Phase I/II study of iodine 131-labeled monoclonal antibody A33 in patients with advanced colon cancer. J Clin Oncol. 1994;12:1561–71. doi: 10.1200/JCO.1994.12.8.1561. [DOI] [PubMed] [Google Scholar]
- 24.Welt S, Scott AM, Divgi CR, Kemeny NE, Finn RD, Daghighian F, Germain JS, Richards EC, Larson SM, Old LJ, et al. Phase I/II study of iodine 125-labeled monoclonal antibody A33 in patients with advanced colon cancer. J Clin Oncol. 1996;14:1787–97. doi: 10.1200/JCO.1996.14.6.1787. [DOI] [PubMed] [Google Scholar]
- 25.Chong G, FT Lee, Hopkins W, Tebbutt N, JS Cebon, AJ Mountain, Chappell B, Papenfuss A, Schleyer P, Paul U, et al. Phase I trial of 131I-huA33 in patients with advanced colorectal carcinoma. Clin Cancer Res. 2005;11:4818–26. doi: 10.1158/1078-0432.CCR-04-2330. [DOI] [PubMed] [Google Scholar]
- 26.Scott AM, Lee FT, Jones R, Hopkins W, MacGregor D, Cebon JS, Hannah A., Chong G, Paul U, Papenfuss A, et al. A phase I trial of humanized monoclonal antibody A33 in patients with colorectal carcinoma: biodistribution, pharmacokinetics, and quantitative tumor uptake. Clin Cancer Res. 2005;11:4810–17. doi: 10.1158/1078-0432.CCR-04-2329. [DOI] [PubMed] [Google Scholar]
- 27.Murer P, Kiefer JD, Pluss L, Matasci M, Blumich SL, Stringhini M, Neri D. Targeted delivery of TNF potentiates the antibody-dependent cell-mediated cytotoxicity of an anti-melanoma immunoglobulin. J Invest Dermatol. 2019;139(6):1339–1348. doi:10.1016/j.jid.2018.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 29.Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pintér T, Lim R, Bodoky G, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–17. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 30.Baselga J, Perez EA, Pienkowski T, Bell R. Adjuvant trastuzumab: a milestone in the treatment of HER-2-positive early breast cancer. Oncologist. 2006;11(Suppl 1):4–12. doi: 10.1634/theoncologist.11-90001-4. [DOI] [PubMed] [Google Scholar]
- 31.Schliemann C, Palumbo A, Zuberbuhler K, Villa A, Kaspar M, Trachsel E, Klapper W, Menssen HD, Neri D. Complete eradication of human B-cell lymphoma xenografts using rituximab in combination with the immunocytokine L19-IL2. Blood. 2009;113:2275–83. doi: 10.1182/blood-2008-05-160747. [DOI] [PubMed] [Google Scholar]
- 32.Gutbrodt KL, Schliemann C, Giovannoni L, Frey K, Pabst T, Klapper W, Berdel WE, Neri D. Antibody-based delivery of interleukin-2 to neovasculature has potent activity against acute myeloid leukemia. Sci Transl Med. 2013;5:201ra118. doi: 10.1126/scitranslmed.3006221. [DOI] [PubMed] [Google Scholar]
- 33.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. 2006;103:4005–10. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peipp M, Lammerts van Bueren JJ, Schneider-Merck T, Bleeker WW, Dechant M, Beyer T, Repp R, van Berkel PHC, Vink T, van de Winkel JGJ, et al. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood. 2008;112:2390–99. doi: 10.1182/blood-2008-03-144600. [DOI] [PubMed] [Google Scholar]
- 35.Pereira NA, Chan KF, Lin PC, Song Z. The “less-is-more” in therapeutic antibodies: afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs. 2018;10:693–711. doi: 10.1080/19420862.2018.1466767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Umana P, Jean-Mairet J, Bailey J. Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity. AG RG ed. USA: Roche Glycart AG; 2003. Patent Nr. US8258198P. [Google Scholar]
- 37.Baptistella AR, Salles Dias MV, Aguiar S Jr., Begnami MD, Martins VR. Heterogeneous expression of A33 in colorectal cancer: possible explanation for A33 antibody treatment failure. Anticancer Drugs. 2016;27:734–37. doi: 10.1097/CAD.0000000000000379. [DOI] [PubMed] [Google Scholar]
- 38.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 39.Bremer E, Samplonius D, Kroesen BJ, van Genne L, de Leij L, Helfrich W. Exceptionally potent anti-tumor bystander activity of an scFv:sTRAIL fusion protein with specificity for EGP2 toward target antigen-negative tumor cells. Neoplasia. 2004;6:636–45. doi: 10.1593/neo.04229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kovtun YV, Audette CA, Ye Y, Xie H, Ruberti MF, Phinney SJ, Leece BA, Chittenden T, Blättler WA, Goldmacher VS, et al. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006;66:3214–21. doi: 10.1158/0008-5472.CAN-05-3973. [DOI] [PubMed] [Google Scholar]
- 41.Ross SL, Sherman M, McElroy PL, Lofgren JA, Moody G, Baeuerle PA, Coxon A, Arvedson T. Bispecific T cell engager (BiTE(R)) antibody constructs can mediate bystander tumor cell killing. PLoS One. 2017;12:e0183390. doi: 10.1371/journal.pone.0183390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajendra Y, Kiseljak D, Baldi L, Hacker DL, Wurm FM. A simple high-yielding process for transient gene expression in CHO cells. J Biotechnol. 2011;153:22–26. doi: 10.1016/j.jbiotec.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 43.Ravenni N, Weber M, Neri D. A human monoclonal antibody specific to placental alkaline phosphatase, a marker of ovarian cancer. MAbs. 2014;6:86–94. doi: 10.4161/mabs.27230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viti F, Nilsson F, Demartis S, Huber A, Neri D. Design and use of phage display libraries for the selection of antibodies and enzymes. Methods Enzymol. 2000;326:480–505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.