

Abstract
Background
The pathophysiology of osteonecrosis of the femoral head (ONFH) is poorly understood, and the diagnosis is idiopathic in as many as 40% of patients. Genetic and epigenetic etiologies have been postulated, yet no single nucleotide polymorphisms (SNPs) with intuitive biologic implications have been elucidated.
Questions/purposes
(1) Do individuals with ONFH share common biologically relevant genetic variants associated with disease development? (2) What is the mechanism by which these SNPs may impact the expression or function of the affected gene or protein?
Methods
This retrospective genome-wide association study (GWAS) evaluated participants from the Mayo Clinic Biobank and Mayo Clinic Genome Consortium between August 2009 and March 2017. We included every patient with atraumatic ONFH in each of these respective registries and every control patient in a previous GWAS with an acceptable platform to perform statistical imputation. The study was performed in two phases, with an initial discovery cohort and a subsequent validation cohort. The initial discovery cohort consisted of 102 patients with ONFH and 4125 controls. A logistic regression analysis was used to evaluate associations between SNPs and the risk of ONFH, adjusted for age and sex. Seven SNPs were identified in a gene of biological interest, peroxisome proliferator-activated receptor gamma (PPARG), which were then evaluated in a subsequent validation cohort of 38 patients with ONFH and 464 controls. Age, sex, race, and previous steroid exposure were similar between patients with ONFH and controls in both the discovery and validation cohorts. Separate from the two-phase genetic investigation, we performed targeted pharmacosurveillance to evaluate the risk association between the use of antidiabetic thiazolidinediones, a class of PPARG agonists, and development of ONFH by referencing 9,638,296 patient records for individuals treated at Mayo Clinic.
Results
A combined analysis of the discovery and validation cohorts revealed that seven SNPs were tightly clustered adjacent to the 3’ end of PPARG, suggesting an association with the risk of ONFH (p = 1.58 x 10-2-5.50 x10-6). PPARG gene-level significance was achieved (p = 3.33 x 10-6) when all seven SNPs were considered. SNP rs980990 had the strongest association with the risk of ONFH (odds ratio [OR], 1.95; 95% CI, 1.46-2.59; p = 5.50 x 10-6).
The seven identified SNPs were mapped to a region near the PPARG gene and fell in a highly conserved region consisting of several critical transcription factor binding sites. Nucleotide polymorphisms at these sites may compromise three-dimensional chromatin organization and alter PPARG 3’ end interactions with its 5’ promoter and transcription start site. Pharmacosurveillance identified that patients who were exposed to thiazolidinediones had an increased relative risk of developing ONFH of 5.6 (95% CI, 4.5-7.1).
Conclusions
We found that disruption of PPARG regulatory domains is linked to an increased risk of ONFH. Mechanistically, aberrant regulation of PPARG compromises musculoskeletal differentiation because this master regulator creates a proadipogenic and antiosteogenic state. Furthermore, PPARG alters steroid metabolism and vasculogenesis, processes that are inextricably linked with ONFH. Pharmacologically, predisposition to ONFH was further exposed with thiazolidinedione use, which upregulates the expression of PPARG and is known to alter bone metabolism. Collectively, these findings provide a foundation to perform confirmatory studies of our proposed mechanism in preclinical models to develop screening diagnostics and potential therapies in patients with limited options.
Level of Evidence
Level III, prognostic study.
Introduction
In the United States, osteonecrosis of the femoral head (ONFH) occurs in an estimated 20,000 new patients every year, predominantly in those younger than 40 years [36,44,45]. ONFH occurs when trabecular bone osteocytes undergo necrosis and fail to regenerate appropriately [13,46]. Severe ONFH is marked by loss of bony architecture, leading to subchondral collapse and progressive degenerative joint changes [22]. The pathophysiology of ONFH remains poorly understood; however, once the femoral head has collapsed, patients often undergo THA for pain relief and improvement in daily function. Importantly, patients with ONFH typically undergo THA at a younger age than is characteristic among patients with primary osteoarthritis, generating interest in improved diagnostics and therapeutics [4,21,27,46].
Risk factors associated with ONFH include alcohol use, coagulopathies, sickle cell disease, HIV, radiation exposure, smoking, pregnancy, and autoimmune conditions [7,8,24]. Although corticosteroid use has been identified as perhaps the strongest primary risk factor, ONFH develops in only a minority of patients using high-dose corticosteroid regimens [44,46]. Despite the multitude of etiologic associations, the disease of up to 40% of patients is eventually classified as idiopathic [46]. This large proportion of patients with idiopathic disease and the fact that ONFH develops in only 6% of patients exposed to steroids indicates that some patients may be genetically predisposed to ONFH.
Previous investigations have shown derangements in pathways potentially related to the development of ONFH. Studies examining the effect of coagulation have noted that up to 82% of patients with ONFH had at least one coagulation abnormality [28,29]. Likewise, polymorphisms in genes regulating blood vessel tone, specifically endothelial nitric oxide synthase [15-18,34], have been associated with ONFH. Genetic polymorphisms have also been described in collagen production and the metabolism of steroids and alcohol, two major risk factors of ONFH development [5,32,39,60]. Although insightful, these early studies were performed with older techniques, were limited by small population sizes, and remain unvalidated.
Rapid advances have occurred in technologies capable of probing the contribution of genetic variation to disease. One such technique is genome-wide association study (GWAS), which identifies single nucleotide polymorphisms (SNPs) in the genome and establishes their relative association to a particular phenotype [40]. Leveraging the power of this technology for complex diseases can help elucidate pathophysiology and provide a basis for more-targeted and selective study [41,49]. In particular, GWAS has recently been proposed as a critical early-stage investigation to understand the contribution of genetics to the potentiation of musculoskeletal pathology [48]. In multifactorial and largely idiopathic diseases such as ONFH, a GWAS generates an invaluable roadmap to lead investigators in the right direction during the initial study. SNPs often impact the expression levels of the genes they are located adjacent to or within. By identifying SNPs associated with a specific phenotype, researchers are provided with signposts throughout the genome that mark areas and genes of interest for further investigation. SNPs detected via GWAS are germline variations at the DNA level; therefore, they remain consistent throughout every cell within an individual and do not change over time. Thus, GWAS provides several advantages over other global “omics” approaches such as mRNA- or ChIP sequencing because these assays assess metrics that are highly variable between cell types and become less informative when extracted from whole tissues (such as bone). Additionally, GWAS can be conducted on easily accessible, peripheral sources of DNA (such as blood), which lends itself well as a diagnostic tool. Other approaches require that specimens be collected directly from the tissue of interest and may not always be feasible. We believe GWAS can help us narrow our search for an underlying molecular mechanism of ONFH and therefore chose to begin our studies with a GWAS. Using this roadmap, we might subsequently turn our attention to specific locations and genes within the genome to inform the use of other diagnostic tools such as RNA sequencing and ChIP sequencing. Identification of genetic variants associated with the risk of ONFH could provide valuable insight for disease pathophysiology, risk-stratification screening tests, and targeted interventions for at-risk patients.
We performed a large discovery cohort screening GWAS that identified several SNPs related to the gene peroxisome proliferator-activated receptor-γ (PPARG). This gene was a potentially attractive target, given its known roles in the differentiation of musculoskeletal tissue, metabolism of steroids and lipids, and vasculogenesis. It is also a pharmacologic target for a class of diabetes drugs known as the thiazolidinediones (TZD), which have been linked to an increased proclivity for fractures. Given the potential biological implications of this gene in relation to the pathophysiology of ONFH, we performed a second-phase targeted validation analysis of SNPs related to PPARG. Herein, we report this process and subsequent analyses used to determine the mechanistic relationship of PPARG to ONFH.
In this study, we used GWAS to ask: (1) Do individuals with ONFH share common biologically relevant genetic variants associated with disease development? (2) What is the mechanism by which these SNPs may impact the expression or function of the affected gene or protein?
Methods
This institutional review board-approved study was a two-stage retrospective GWAS using participants enrolled in the Mayo Clinic Biobank and Mayo Clinic Genome Consortium from August 2009 to March 2017. The Mayo Clinic Biobank is a tissue database of more than 50,000 volunteers or patients prescheduled for medical examination in the divisions of community internal medicine, family medicine, or general internal medicine [47]. The Mayo Clinic Genome Consortium is a database of approximately 10,000 patients who were part of a historical GWAS at Mayo Clinic [3]. A power analysis was not performed for this study. We included every patient with atraumatic ONFH in each of these respective registries and every control patient in a previous GWAS performed on an acceptable platform, to perform imputation as detailed below.
The study was performed in two phases with an initial discovery cohort and a subsequent validation cohort (Fig. 1). The discovery cohort was established in a stepwise fashion. First, the Mayo Clinic Biobank was queried using ICD-9 code 733.4 for patients with a history of ONFH. The medical records of identified patients were reviewed to determine the anatomical location of ONFH as well as previous corticosteroid exposure and trauma history. In this study, we did not assess each patient’s ONFH stage as a variable against genetic association. Eighty-eight patients with atraumatic ONFH were identified. Fifty of these 88 patients had a history of high-dose corticosteroid use (≥ 20 mg/day x ≥ 1 month) before ONFH was diagnosed. Controls were also selected from the Mayo Clinic Biobank and were 2:1 frequency matched (176 patients) to patients based on age (± 5 years), sex, BMI (±3 kg/m2), the patient’s self-reported race (exact match), and previous steroid exposure without subsequent development of documented ONFH. Cryopreserved white blood cells from these patients were then genotyped with the Illumina Omni 5.0 platform (Illumina, San Diego, CA).
Fig. 1.
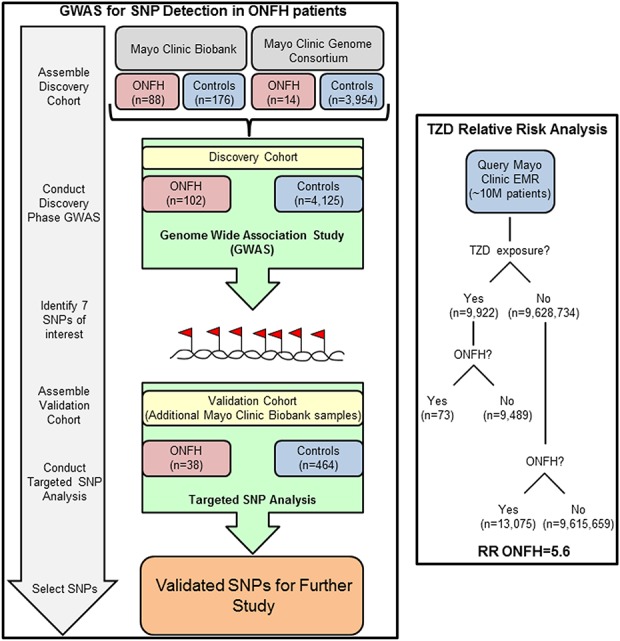
This schematic diagram outlines the phases of investigation including establishment of the discovery and validation cohorts for genetic evaluation as well as the complementary pharmacologic impact data from patient record review.
The discovery cohort’s data were then enriched with institutional historical GWAS data from patients in the Mayo Clinic Genome Consortium to increase the sample size. Patients in this registry were identified in a similar fashion to those from the Mayo Clinic Biobank. Fourteen patients with atraumatic ONFH were identified. Seven of those 14 patients had a history of high-dose corticosteroid use before ONFH was diagnosed. Instead of matching controls as we did with the Mayo Clinic Biobank patients, we included all available control patients from the Mayo Clinic Genome Consortium in a GWAS performed on platforms suitable to perform imputation. This constituted 3954 patients, including 909 with a history of oral or intravenous corticosteroid use and no subsequent ONFH. All patients with ONFH and controls identified in the Mayo Clinic Genome Consortium were then combined with the Mayo Clinic Biobank patients to arrive at the final cohort for the discovery analysis, which included the following: 102 patients with ONFH (53 with a history of high-dose corticosteroid use) and 4125 controls (1001 with a history of corticosteroid use). The median age was 57 years, 56% were women, and > 99% were white (Table 1).
Table 1.
Demographics of the discovery and validation cohorts

Imputation was used to combine and compare data from patient samples in the discovery cohort. Each GWAS platform was assessed for standard quality-control metrics; only platforms meeting thresholds for high-fidelity imputation were included. We imputed each GWAS platform separately using IMPUTE2 and the 1000 Genomes Project version 3 (March 2012 release) reference panel.
The discovery cohort GWAS revealed that seven SNPs were related to PPARG. Given the biological interest in this gene, a cluster of seven SNPs became the focus of a targeted validation cohort and subsequent investigations of potential mechanistic involvement with ONFH (Fig. 1). For the validation cohort, we queried the Mayo Clinic Biobank again for new patients with ONFH and controls because the database had grown between the discovery phase and the validation analysis. We identified 38 new patients with atraumatic ONFH who had a history of steroid use and 464 controls with a history of steroid use but who did not have symptomatic ONFH. DNA from validation Mayo Clinic Biobank samples were isolated from cryopreserved white blood cells and genotyped using a Sequenom custom-designed panel to assess the seven SNPs of interest that had been identified during the discovery phase. In the validation cohort, the median age was 64 years, 52% were women, and > 99% were white (Table 1).
The evolutionary conservation of the newly identified SNPs was assessed by viewing the 100 Vertebrate Conservation Track using the Track Data Hubs Feature [53] on the Human GRCh37/hg19 Assembly in the UCSC Genome Browser (University of California Santa Cruz, Santa Cruz, CA, USA) [33].
The location of protein binding sites was determined using the Human GRCh37/hg19 Assembly in the UCSC Genome Browser [33] with the Transcription Factor Chip Track [14,61,62] overlaid using the Track Data Hubs Feature [53].
The chromatin structure was assessed using the 15-state chromatin model within the Roadmap Epigenomics Project [35] on the Human hg19 assembly. The three-dimensional chromatin structure was assessed on the same gene track using experimental HiC data collected in HEK293 cells [66].
Datasets curated by The Genotype-Tissue Expression (GTEx) Project [19] were used to assess the functional impact of the newly identified SNPs on gene expression in varying tissues. The expression data and boxplots described here were obtained from the GTEx Portal (GTEx Analysis Release V7 [dbGaP Accession phs000424.v7.p2]).
When PPARG emerged as a potential candidate for involvement in the pathogenesis of ONFH, we used the Mayo Clinic electronic medical record and data abstraction interfaces to evaluate the clinical records of 9,638,296 individuals treated at Mayo Clinic (Fig. 1).
Risk association was tested between the use of antidiabetic thiazolidinediones, a class of PPARG agonists, and the development of ONFH. Specifically, the drugs rosiglitazone and pioglitazone were captured by validated medicine reconciliation platforms and evaluated against subsequent development of atraumatic ONFH as determined by ICD9 and ICD10 codes (Fig. 2D).
Fig. 2.
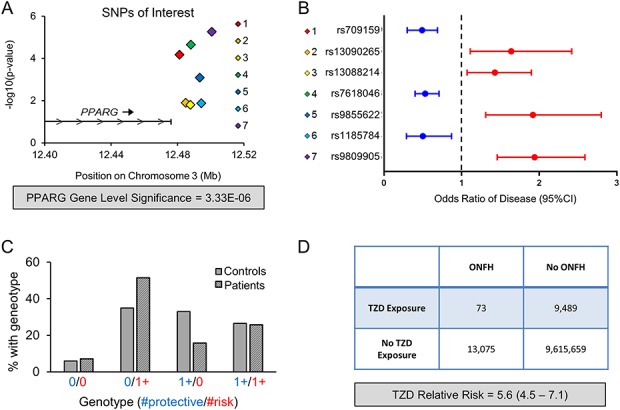
The GWAS and clinical cohort assessment associate modulation of PPARG with increased ONFH risk is shown. (A) The schematic shows the location on chromosome 3 and –log10(p value) for the seven SNPs of interest with respect to the 3’ end of the PPARG locus with cumulative PPARG gene-level significance indicated below the graph. (B) Plot outlining the respective OR for ONfh development in patients harboring the indicated SNP. (C) The percentage of patients in the control or ONFH group found to have the indicated combinations of “Protective” (OR < 1) or “Risk” (OR > 1) alleles. (D) Table outlining the increased risk of ONFH with the use of TZD drugs that are PPARG agonists for diabetes management.
Population stratification was assessed using STRUCTURE (Stanford University, Palo Alto, CA) software. We used a logistic regression within Plink to test for an association with ONFH, assuming a log-additive genetic model adjusting for age, sex, subject source, and the top three principal components. We performed a gene-based level analyses using SNPs within PPARG. All genotyped and imputed SNPs were tested within PPARG (defined by its gene boundaries per Genome Browser +/- 75 kb) using sequence kernel association tests [25] with an unweighted linear kernel implemented in the sequence kernel association test package v1.0.9 [55] in R v3.1.1 [52]. Then, the top seven significant PPARG SNPs were used to form haplotypes. Associations between the haplotypes and ONFH were tested with logistic regression implemented in the haplo.stats package v1.6.11 [57] in R v3.1.1. The package locusZoom v1.3 [51] was used to visualize the single SNP GWAS results in the PPARG gene region. Logistic regression was also used in the validation cohort, but age and sex were the only covariates included in the models because the subjects were drawn from a single source (Mayo Clinic Biobank). Continuous covariates were compared using the Wilcoxon rank sum test and categorical covariates were compared using the Pearson chi-squared test. Data from the discovery cohorts and validation cohorts were analyzed separately as well as in aggregate through a meta-analysis of the two cohorts. For all three analyses, gene-level significance was set at p < 3.33 x 10-5 and individual SNP significance was set at p < 5 x 10-8, consistent with standard GWAS definitions [49].
Results
Genetic Variants Associated with ONFH
In the discovery cohort, we identified a cluster of seven SNPs related to PPARG that were associated with the differential risk of ONFH (Fig. 2A). These seven SNPs were in the top 1000 most genetically different SNPs on chromosome 3. Individual p values for these seven SNPs ranged from 1.03 x 10-5 to 1.56 x 10-2; thus, no SNP achieved significance (cutoff p < 5 x 10-8). Gene-level analysis of PPARG achieved a p value of 3.33 x 10-6, which was significant after whole-genome Bonferroni correction (cutoff = p < 3.33 x 10-5) (Fig. 2A). Thus, although no single SNP met the genome-wide significance cutoff of 5 x 10-8, PPARG showed considerable variance associated with disease (Table 2).
Table 2.
GWAS analysis of significant SNPs in PPARγ

All seven SNPs identified in the discovery cohort demonstrated similar p values and odds ratios (ORs) for disease when assessed in the validation cohort (Table 2). Four of the seven SNPs were associated with an increased risk of ONFH (OR, 1.43-1.94; p = 1.58 x 10-2-5.50 x 10-6) and three were associated with a decreased risk of ONFH (OR, 0.49-0.53, p = 1.32 x 10-2-2.27 x 10-5) (Table 2, Fig. 2B). Next, we analyzed patient genotypes at the individual level to investigate the prevalence of SNPs associated with increased versus decreased risk in the control and ONFH populations. We noted that a higher proportion of patients with ONFH (51%) possessed at least one SNP that was associated with increased risk and no SNPs that were associated with decreased risk compared with controls (35%, p < 0.001). Conversely, we found that a greater number of patients in the control group (33%) possessed at least one SNP associated with decreased risk and had no SNPs associated with increased risk than did patients with ONFH (16%, p < 0.001) (Fig. 2C).
Possible Mechanism by Which PPARG May Impact the Risk of ONFH
Given the strong association between PPARG genetic variance and ONFH development, a large institutional database was evaluated to determine differences in the risk of ONFH based on exposure to TZDs. These drugs are used widely for managing diabetes with a primary mechanism of action as PPARG agonists. In a database of nearly 10 million patients, TZD use increased the risk of ONFH development by a factor of 5.6 (95% CI, 4.5-7.1; p < 0.001) (Fig. 2D).
Although unexplored in relation to musculoskeletal tissue, four of the seven SNPs demonstrated an association with genes including PPARG in other tissues, suggesting their presence or absence had functional consequences (see Figure, Supplemental Digital Content 1, http://links.lww.com/CORR/A158) [19]. To better understand how newly identified SNPs may be associated with the risk of ONFH, we observed their location within the genome in depth. First, we mapped the location of the variants in relation to evolutionarily conserved regions among vertebrates (Fig. 3A). We noted that SNP 1 (rs709159) and SNP 7 (rs9809905) fall in highly conserved regions. These regions share similar patterns of conservation among ambulatory vertebrates but diverge in birds and lower organisms (see Table, Supplemental Digital Content 2, http://links.lww.com/CORR/A164, and Figure, Supplemental Digital Content 3, http://links.lww.com/CORR/A165). Noting several evolutionary conserved regions proximal to the 3’ end of PPARG, next, we searched for the presence of transcription factor binding domains in relation to our SNPs of interest (see Figure, Supplemental Digital Content 4, http://links.lww.com/CORR/A166). This demonstrated a substantial number of binding sites for the protein CTCF, an important regulator that establishes domain boundaries between accessible and non-accessible DNA in 3-D chromatin structures (Fig. 3B). We found that SNP 7 (rs9809905) is in the center of a highly enriched CTCF and CEBPB binding region (Fig. 3C). Because of the critical role of CTCF in controlling 3-D chromatin organization, we used the Epigenomics Roadmap [35] to assess the chromatin structure around the PPARG locus (Fig. 3D). In osteoblasts and bone marrow-derived mesenchymal stromal or stem cells (BMSCs), we observed regions of chromatin compaction and quiescence. However, in adipose tissue, adipose tissue-derived mesenchymal stromal or stem cells (AMSCs), and AMSCs directed into the adipogenic lineage, we observe regions of open chromatin and active transcription. Furthermore, HiC data show 3-D chromatin looping that links the 3’ end of PPARG, which contain the newly identified SNPs with the 5’ promoter region and transcriptional start site (Fig. 3D). Together, these observations suggest that the newly identified polymorphisms may have a functional association with the expression levels of PPARG. SNP 7 (rs9809905) is of particular interest because it has the highest OR of disease (OR = 1.95) and lowest p value (p = 5.05 x 10-6), and is located in an evolutionarily conserved protein binding region shown to be involved in 3-D chromatin interaction.
Fig. 3.
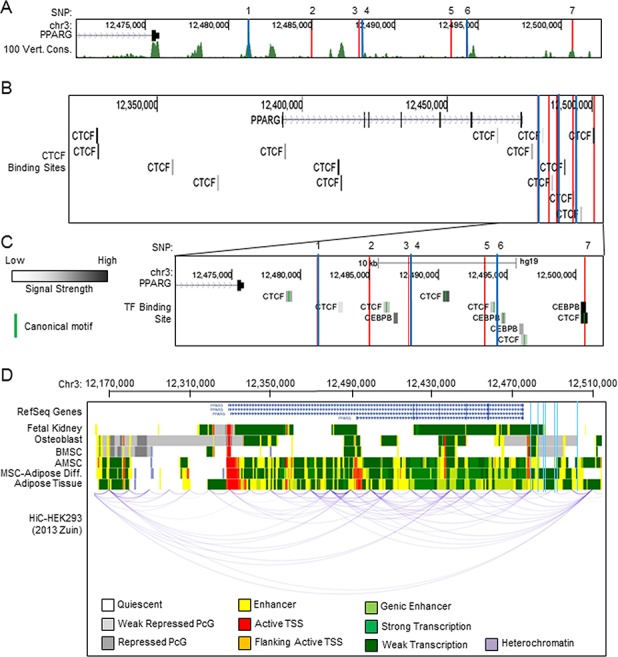
Evolutionary conservation of SNP loci and chromatin structure with three-dimensional organization of PPARG is shown. (A) UCSC Genome Browser display of the 3’ PPARG region with SNP locations indicated (blue = protective, red = risk). The 100 vertebrate conservation track (green) and individual species conservation (black) is indicated below. (B) SNP locations are indicated (blue vertical lines) and track colors correspond to the chromatin state indicated at the bottom of the panel. UCSC Genome Browser display of CTCF binding sites with respect to the PPARG locus and SNP locations are indicated (blue = protective, red = risk). (C) An enhanced view of the 3’ end of PPARG depicting CTCF and CEBPβ binding sites is shown. (D) A chromatin state model for the indicated cell type (left) and HiC long-range interaction in HEK293 cells with respect to the PPARG locus is shown. Signal strength determined by ChIP assay and canonical binding motif regions are indicated as specified (left).
Discussion
ONFH is a complex hip disorder with many described risk factors, yet up to 40% of patients are ultimately considered to have idiopathic disease [22]. The large number of patients with idiopathic disease is consistent with the concept we validated in this study, that genetic susceptibility may play a role in the pathogenesis of disease. GWAS was used as a screening tool to identify loci within the genome that may be linked with the risk of ONFH. GWAS is an ideal technology for preliminary work of this nature and provides a foundation for subsequent validation with complementary techniques. The identification of PPARG in this study as a critical modulator of the risk of ONFH is based on strong corroborating lines of evidence, including (1) genetic variance in loci governing the expression of PPARG and (2) pharmacologic modulation with common antidiabetic agents functioning through PPARG agonism. PPARG provides both a novel genetic marker and a potential pathway-targeting strategy for modulating disease progression.
This study must be interpreted in light of important limitations. Most importantly, the sample size of ONFH patients (n = 140) was prohibitive for a GWAS capable of meeting individual SNP p value cutoffs < 5 x 10-8 thought to represent unequivocal whole-genome significance [49]. Nevertheless, several SNPs achieved p values that are remarkable in the context of this initial cohort’s sample size. On the contrary, PPARG achieved a high level of significance even after Bonferroni correction, despite the aforementioned modest sample size of patients, lending further credence to the strength of these findings. Furthermore, the results are supported by biologically intuitive and relevant implications for the identified gene and strong complementary clinical data confirming pharmacologic risk is mediated through PPARG. Second, the GWAS platforms from the discovery cohort were not uniform and thus required imputation and rigid exclusion criteria. We demonstrated excellent data quality through this process, but increased caution must be exercised when variant SNPs are identified through imputation at specific loci versus through uniform genotyping. Third, the genetic component of this study is restricted to GWAS evaluation. This method has strengths as an initial screening tool, but it lacks the capability to detect disease-related or SNP-induced changes in gene expression or chromatin modifications. Now that we have used GWAS to focus our search, we aim to use the advantages of RNA or ChIP sequencing technologies in future studies to complement the presented data. Furthermore, this study did not prove our proposed mechanism. Additional corroborative evidence will be required to substantiate these relationships. Investigations of the expression of PPARG in the femoral head tissue of patients with ONFH as well as cell line manipulation and experimentation with animal models are possible lines of exploration.
GWAS is a powerful screening tool for understanding the genetic contribution to disease. Although this technology may yield SNPs that are highly associated with a particular condition, findings are often difficult to interpret if the SNP is not related to a gene or pathway with known biological activity. Although our study lacks statistical power, a strength of our preliminary work lies in the molecular potential of the identified polymorphisms. PPARG has multiple intuitive biologic implications for the pathophysiology of ONFH (Fig. 4). First, PPARG functions in conjunction with the WNT pathway and governs whether mesenchymal stem cells differentiate toward adipose tissue or bone [37,59,64]. When active, PPARG controls the lineage allocation of stem cells by promoting adipogenic differentiation and suppressing osteogenic differentiation. This molecular ability is directly relevant to ONFH, which results from a combination of sentinel necrosis followed by an inability to regenerate trabecular bone [22,23,63]. Our findings also provide insight into likely mechanisms by which TZDs, as potent agonists of PPARG, increase the risk of ONFH. These agents are PPARG-activating and thus might shift the balance of the local regenerative cell population toward formation of adipose tissue instead of restorative bone [56]. Clinically, this principle was partly demonstrated before our study in large-scale evaluations of TZD use showing an elevated risk of atypical fracture [6,20,30,31,42,65]. Animal and in vitro studies have further confirmed that TZD use yields increased bone adiposity and decreased bone mineralization [2,43,54,58]. In retrospect, these collective findings might suggest that the earlier findings could be attributed to a previously unrecognized ONFH-like syndrome secondary to TZDs.
Fig. 4.
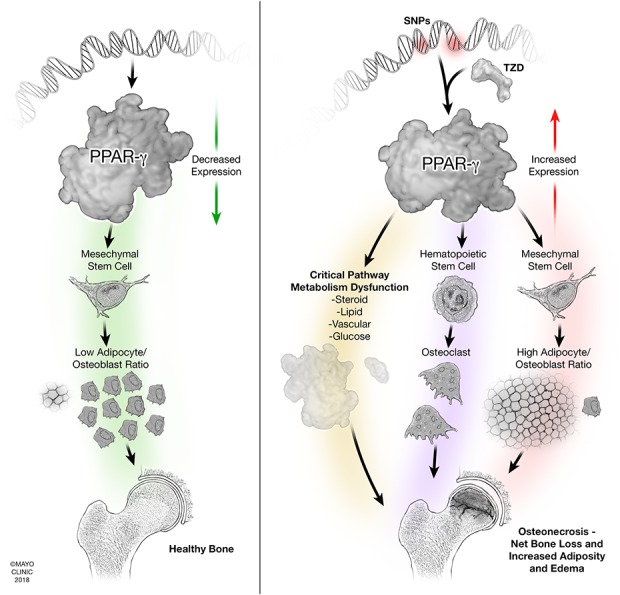
Proposed mechanisms for PPARG involvement in the pathophysiology of ONFH are shown. (A) PPARG function in a healthy state with low levels of expression yielding normal bone architecture. (B) Pathologic upregulation of PPARG, either through SNPs or pharmacologic modulation with TZD drugs, altering pathways leading to osteonecrosis in bone.
Steroids are a primary risk factor of ONFH development. PPARG has a high affinity for steroid receptors [26]. Therefore, it is possible that genetic- or pharmacologically-induced alterations in PPARG cause patient-specific sensitivity to steroids. Epigenetic modifications that depend on PPARG may explain why an even greater proportion of patients have ONFH and could impact the emerging landscape of individualized medicine for these patients. Furthermore, PPARG is a key regulator of lipid metabolism and vasculogenesis [1]. Previous reports have shown that statins are protective for patients with ONFH, and avascular lesions are pathognomonic for the disease [44,46,50]. These critical relationships warrant further investigation to further clarify the extent of PPARG’s role in the pathophysiology of ONFH.
Although a great deal of work is required to clarify the role of PPARG in ONFH, we summarize our current proposed mechanism as follows (Fig. 4). In a healthy state, PPARG maintains low levels of expression, yielding normal bone architecture. In ONFH, we suggest that pathologic upregulation of PPARG, either through SNPs or pharmacologic modulation with TZDs, may alter pathways leading to osteonecrosis in bone. In normal bone physiology, PPARG is expressed at low levels, which encourages a decreased adipocyte/osteoblast ratio for healthy bone formation. However, upregulation of PPARG leads to a high adipocyte/osteoblast ratio in the MSC lineage and simultaneously acts on the hematopoietic lineage to increase osteoclastogenesis; together, these mechanisms yield net bone loss and increased adiposity and edema. Substantial adiposity in bone may also lead to increased pressure in the femoral head with a subsequent “compartment-syndrome-like condition” and disruption of the vascular supply. Furthermore, PPARG acts on a variety of critical metabolic pathways that, if disrupted, have intuitive biologic implications for the pathophysiology of osteonecrosis. PPARG interacts with steroid receptors, holding particular relevance because steroids are the primary exogenous risk factor of the development of osteonecrosis. PPARG also interacts with lipid metabolism, which has long been proposed as a critical pathway for osteonecrosis susceptibility as evidenced by studies demonstrating attenuated osteonecrosis progression with the administration of statin drugs. PPARG also plays a critical role in local vascular formation; indeed, “avascular necrosis” is the historical term for this disease because a disrupted vascular supply is uniformly responsible for traumatic osteonecrosis and has been shown histologically in a subset of patients with atraumatic disease.
Multilevel molecular analysis of the chromosomal regions in which the SNPs reside shows that our newly identified SNPs are in a region that has many molecular properties consistent with a crucial regulatory region for controlling the expression of PPARG. Specifically, we mapped SNP 7 (rs9809905) to an evolutionarily conserved DNA region containing binding sites for the key regulators CTCF and CEBPB. Previous studies have elucidated the dynamic binding patterns of CTCF during adipogenesis and identified it as a key regulator of PPARG transcriptional activation [11,12]. Additionally, analysis of HiC 3-D chromatin interaction data demonstrates that this region is looped back to the 5’ promoter region of PPARG. CTCF has been well-established as a mediator of chromatin looping between regulatory and promoter regions [9], and augmented CTCF binding has been shown to reactivate previously silenced genes [10]. These data suggest that the increased OR associated with SNP 7 (rs9809905) may stem from its functional impact on the expression of PPARG. It is also important to note the tissue and cell-specific variability of the chromatin state near PPARG when considering the functional impact of these SNPs. In bone or bone-derived cells (osteoblasts and BMSCs), chromatin is compacted at both the 3’ and 5’ region, resulting in quiescent or weak transcription of PPARG. Conversely, in adipose or adipose-derived cells, chromatin is open and active at the 3’ and 5’ regions of PPARG, resulting in active gene transcription. This dichotomy in chromatin states is reflected in the PPARG expression levels within respective tissues. PPARG is well-described as a master regulator of adipogenesis [38] that drives adipocyte differentiation and development of fat tissue. Therefore, the expression of PPARG must be tightly regulated depending on the cell phenotype. This is likely why we observed chromatin compaction in nonadipogenic bone cells and open chromatin in adipose-derived cells. Our results suggest that SNPs that alter the appropriate regulation and expression of PPARG could lead to improper differentiation of mesenchymal stem cells, giving rise to ONFH. This could explain the combination of human and animal data showing atypical fractures following TZD administration, marked by increased bone marrow adiposity and edema leading to fracture. We reason that the location of SNP 7 (rs9809905) in a highly conserved CTCF binding site may augment the binding of CTCF in this region. In osteoblasts, this may lead to relaxation of an otherwise compacted region of chromatin at the 3’ end of PPARG and promote looping to the 5’ promoter. In osteoblasts and preosteoblasts, which express low basal PPARG mRNA levels, SNP-induced expression of PPARG may be detrimental to proper cell differentiation or function, resulting in compromised bone integrity. Therefore, it is intuitive that SNP 7 (rs9809905) has the highest OR for ONFH and has the highest incidence among the diseased population. Our followup pharmacosurveillance using the Mayo Clinic’s electronic medical record yielded complementary results in that patients receiving TZD (a PPARG agonist) had an increased risk of ONFH development by a factor of 5.6 (95% CI, 4.5-7.1; p < 0.001). This finding confirms that elevated PPARG expression is associated with ONFH. In light of this finding, we reason that these newly identified SNPs, particularly rs980990, may have some association with the risk of ONFH. Because PPARG is already capable of being manipulated pharmacologically with TZDs, theoretically, an inhibitor of PPARG either locally or systemically, could benefit patients with early-stage ONFH.
This study identified PPARG as a potential modulator of the risk of ONFH through genetic variance and pharmacologic upregulation with TZDs. The primary mechanism by which PPARG is known to function is through a shift in mesenchymal stem cell differentiation towards the formation of adipose tissue rather than bone formation. Furthermore, known interactions of PPARG with the well-established risk factors of ONFH—steroid use and impaired vasculogenesis—provide further interest for a possible role in the pathogenesis of ONFH. Nevertheless, considerable additional investigation is needed to confirm or refute these proposed relationships. RNA sequencing of femoral head tissues with and without ONFH as well as cellular manipulation and animal model recapitulation of disease are potential avenues for exploration. Given previous success with the pharmacologic modulation of PPARG, therapeutic intervention and prevention of ONFH may be possible for patients with this condition, who are typically younger and often have limited options.
Acknowledgments
We thank the Mayo Clinic Biobank, Mayo Clinic Genome Consortium, and Mayo Clinic Center for Individualized Medicine for contributing patient samples, data, and infrastructure for performance of the presented work. We thank Melissa Larson and Brian Kabat for their assistance in bioinformatics analysis. This study was supported by an Accelerated Regenerative Medicine Grant from the Mayo Clinic Center for Regenerative Medicine (Rochester, MN, USA). This publication was made possible by CTSA Grant Number UL1 TR000135 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
Footnotes
The institution of one or more of the authors (RJS, CCW, MTH, AB) has received, during the study period, funding from the Mayo Clinic Center for Regenerative Medicine in the form of an Accelerated Regenerative Medicine Grant to support this work.
All ICMJE Conflict of Interest Forms for authors and Clinical Orthopaedics and Related Research® editors and board members are on file with the publication and can be viewed on request.
Each author certifies that his or her institution approved or waived approval for the human protocol for this investigation and that all investigations were conducted in conformity with ethical principles of research.
Clinical Orthopaedics and Related Research® neither advocates nor endorses the use of any treatment, drug, or device. Readers are encouraged to always seek additional information, including FDA approval status, of any drug or device before clinical use.
This study was supported by an Accelerated Regenerative Medicine Grant from the Mayo Clinic Center for Regenerative Medicine (Rochester, MN, USA). This publication was made possible by CTSA Grant Number UL1 TR000135 from the National Center for Advancing Translational Sciences (NCATS), a component of the NIH. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
This investigation was performed at the Mayo Clinic, Rochester, MN, USA.
References
- 1.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146:1226-1235. [DOI] [PubMed] [Google Scholar]
- 3.Bielinski SJ, Chai HS, Pathak J, Talwalkar JA, Limburg PJ, Gullerud RE, Sicotte H, Klee EW, Ross JL, Kocher JP, Kullo IJ, Heit JA, Petersen GM, de Andrade M, Chute CG. Mayo Genome Consortia: a genotype-phenotype resource for genome-wide association studies with an application to the analysis of circulating bilirubin levels. Mayo Clin Proc. 2011;86:606-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bozic KJ, Zurakowski D, Thornhill TS. Survivorship analysis of hips treated with core decompression for nontraumatic osteonecrosis of the femoral head. J Bone Joint Surg Am. 1999;81:200-209. [DOI] [PubMed] [Google Scholar]
- 5.Chao YC, Wang SJ, Chu HC, Chang WK, Hsieh TY. Investigation of alcohol metabolizing enzyme genes in Chinese alcoholics with avascular necrosis of hip joint, pancreatitis and cirrhosis of the liver. Alcohol Alcohol. 2003;38:431-436. [DOI] [PubMed] [Google Scholar]
- 6.Chen HH, Horng MH, Yeh SY, Lin IC, Yeh CJ, Muo CH, Sung FC, Kao CH. Glycemic control with thiazolidinedione is associated with fracture of T2DM patients. PLoS One. 2015;10:e0135530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruess RL. Experience with steroid-induced avascular necrosis of the shoulder and etiologic considerations regarding osteonecrosis of the hip. Clin Orthop Relat Res. 1978:86-93. [PubMed] [Google Scholar]
- 8.Cruess RL. Steroid-induced osteonecrosis: a review. Can J Surg. 1981;24:567-571. [PubMed] [Google Scholar]
- 9.de Wit E, Vos ES, Holwerda SJ, Valdes-Quezada C, Verstegen MJ, Teunissen H, Splinter E, Wijchers PJ, Krijger PH, de Laat W. CTCF Binding polarity determines chromatin looping. Mol Cell. 2015;60:676-684. [DOI] [PubMed] [Google Scholar]
- 10.Deng W, Rupon JW, Krivega I, Breda L, Motta I, Jahn KS, Reik A, Gregory PD, Rivella S, Dean A, Blobel GA. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014;158:849-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubois-Chevalier J, Oger F, Dehondt H, Firmin FF, Gheeraert C, Staels B, Lefebvre P, Eeckhoute J. A dynamic CTCF chromatin binding landscape promotes DNA hydroxymethylation and transcriptional induction of adipocyte differentiation. Nucleic Acids Res. 2014;42:10943-10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubois-Chevalier J, Staels B, Lefebvre P, Eeckhoute J. The ubiquitous transcription factor CTCF promotes lineage-specific epigenomic remodeling and establishment of transcriptional networks driving cell differentiation. Nucleus. 2015;6:15-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ficat RP. Idiopathic bone necrosis of the femoral head. Early diagnosis and treatment. J Bone Joint Surg Br. 1985;67:3-9. [DOI] [PubMed] [Google Scholar]
- 14.Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, Min R, Alves P, Abyzov A, Addleman N, Bhardwaj N, Boyle AP, Cayting P, Charos A, Chen DZ, Cheng Y, Clarke D, Eastman C, Euskirchen G, Frietze S, Fu Y, Gertz J, Grubert F, Harmanci A, Jain P, Kasowski M, Lacroute P, Leng JJ, Lian J, Monahan H, O'Geen H, Ouyang Z, Partridge EC, Patacsil D, Pauli F, Raha D, Ramirez L, Reddy TE, Reed B, Shi M, Slifer T, Wang J, Wu L, Yang X, Yip KY, Zilberman-Schapira G, Batzoglou S, Sidow A, Farnham PJ, Myers RM, Weissman SM, Snyder M. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glueck CJ, Freiberg R, Glueck HI, Tracy T, Stroop D, Wang Y. Idiopathic osteonecrosis, hypofibrinolysis, high plasminogen activator inhibitor, high lipoprotein(a), and therapy with Stanozolol. Am J Hematol. 1995;48:213-220. [DOI] [PubMed] [Google Scholar]
- 16.Glueck CJ, Freiberg RA, Boppana S, Wang P. Thrombophilia, hypofibrinolysis, the eNOS T-786C polymorphism, and multifocal osteonecrosis. J Bone Joint Surg Am. 2008;90:2220-2229. [DOI] [PubMed] [Google Scholar]
- 17.Glueck CJ, Freiberg RA, Sieve L, Wang P. Enoxaparin prevents progression of stages I and II osteonecrosis of the hip. Clin Orthop Relat Res. 2005:164-170. [DOI] [PubMed] [Google Scholar]
- 18.Glueck CJ, Glueck HI, Welch M, Freiberg R, Tracy T, Hamer T, Stroop D. Familial idiopathic osteonecrosis mediated by familial hypofibrinolysis with high levels of plasminogen activator inhibitor. Thromb Haemost. 1994;71:195-198. [PubMed] [Google Scholar]
- 19.GTExConsortium The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Habib ZA, Havstad SL, Wells K, Divine G, Pladevall M, Williams LK. Thiazolidinedione use and the longitudinal risk of fractures in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2010;95:592-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernigou P, Bachir D, Galacteros F. The natural history of symptomatic osteonecrosis in adults with sickle-cell disease. J Bone Joint Surg Am. 2003;85-A:500-504. [DOI] [PubMed] [Google Scholar]
- 22.Houdek MT, Wyles CC, Martin JR, Sierra RJ. Stem cell treatment for avascular necrosis of the femoral head: current perspectives. Stem Cells Cloning. 2014;7:65-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houdek MT, Wyles CC, Packard BD, Terzic A, Behfar A, Sierra RJ. Decreased osteogenic activity of mesenchymal stem cells in patients with corticosteroid-induced osteonecrosis of the femoral head. J Arthroplasty. 2016;31:893-898. [DOI] [PubMed] [Google Scholar]
- 24.Hungerford DS, Zizic TM. Alcoholism associated ischemic necrosis of the femoral head. Early diagnosis and treatment. Clin Orthop Relat Res. 1978:144-153. [PubMed] [Google Scholar]
- 25.Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet. 2013;92:841-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645-650. [DOI] [PubMed] [Google Scholar]
- 27.Ito H, Matsuno T, Omizu N, Aoki Y, Minami A. Mid-term prognosis of non-traumatic osteonecrosis of the femoral head. J Bone Joint Surg Br. 2003;85:796-801. [PubMed] [Google Scholar]
- 28.Jones LC, Hungerford DS. Osteonecrosis: etiology, diagnosis, and treatment. Curr Opin Rheumatol. 2004;16:443-449. [DOI] [PubMed] [Google Scholar]
- 29.Jones LC, Mont MA, Le TB, Petri M, Hungerford DS, Wang P, Glueck CJ. Procoagulants and osteonecrosis. J Rheumatol. 2003;30:783-791. [PubMed] [Google Scholar]
- 30.Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O'Neill MC, Zinman B, Viberti G, Group AS. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427-2443. [DOI] [PubMed] [Google Scholar]
- 31.Kahn SE, Zinman B, Lachin JM, Haffner SM, Herman WH, Holman RR, Kravitz BG, Yu D, Heise MA, Aftring RP, Viberti G, Diabetes Outcome Progression Trial Study G. Rosiglitazone-associated fractures in type 2 diabetes: an Analysis from A Diabetes Outcome Progression Trial (ADOPT). Diabetes Care. 2008;31:845-851. [DOI] [PubMed] [Google Scholar]
- 32.Kaneshiro Y, Oda Y, Iwakiri K, Masada T, Iwaki H, Hirota Y, Kondo K, Takaoka K. Low hepatic cytochrome P450 3A activity is a risk for corticosteroid-induced osteonecrosis. Clin Pharmacol Ther. 2006;80:396-402. [DOI] [PubMed] [Google Scholar]
- 33.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koo KH, Lee JS, Lee YJ, Kim KJ, Yoo JJ, Kim HJ. Endothelial nitric oxide synthase gene polymorphisms in patients with nontraumatic femoral head osteonecrosis. J Orthop Res. 2006;24:1722-1728. [DOI] [PubMed] [Google Scholar]
- 35.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lavernia CJ, Sierra RJ, Grieco FR. Osteonecrosis of the femoral head. J Am Acad Orthop Surg. 1999;7:250-261. [DOI] [PubMed] [Google Scholar]
- 37.Lee MJ, Chen HT, Ho ML, Chen CH, Chuang SC, Huang SC, Fu YC, Wang GJ, Kang L, Chang JK. PPARgamma silencing enhances osteogenic differentiation of human adipose-derived mesenchymal stem cells. J Cell Mol Med. 2013;17:1188-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lefterova MI, Haakonsson AK, Lazar MA, Mandrup S. PPARgamma and the global map of adipogenesis and beyond. Trends Endocrinol Metab. 2014;25:293-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu YF, Chen WM, Lin YF, Yang RC, Lin MW, Li LH, Chang YH, Jou YS, Lin PY, Su JS, Huang SF, Hsiao KJ, Fann CS, Hwang HW, Chen YT, Tsai SF. Type II collagen gene variants and inherited osteonecrosis of the femoral head. N Engl J Med. 2005;352:2294-2301. [DOI] [PubMed] [Google Scholar]
- 40.Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166-176. [DOI] [PubMed] [Google Scholar]
- 41.Manolio TA. Bringing genome-wide association findings into clinical use. Nat Rev Genet. 2013;14:549-558. [DOI] [PubMed] [Google Scholar]
- 42.McDonough AK, Rosenthal RS, Cao X, Saag KG. The effect of thiazolidinediones on BMD and osteoporosis. Nat Clin Pract Endocrinol Metab. 2008;4:507-513. [DOI] [PubMed] [Google Scholar]
- 43.Mieczkowska A, Basle MF, Chappard D, Mabilleau G. Thiazolidinediones induce osteocyte apoptosis by a G protein-coupled receptor 40-dependent mechanism. J Biol Chem. 2012;287:23517-23526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mont MA, Hungerford DS. Non-traumatic avascular necrosis of the femoral head. J Bone Joint Surg Am. 1995;77:459-474. [DOI] [PubMed] [Google Scholar]
- 45.Mont MA, Jones LC, Einhorn TA, Hungerford DS, Reddi AH. Osteonecrosis of the femoral head. Potential treatment with growth and differentiation factors. Clin Orthop Relat Res. 1998:S314-335. [PubMed] [Google Scholar]
- 46.Mont MA, Zywiel MG, Marker DR, McGrath MS, Delanois RE. The natural history of untreated asymptomatic osteonecrosis of the femoral head: a systematic literature review. J Bone Joint Surg Am. 2010;92:2165-2170. [DOI] [PubMed] [Google Scholar]
- 47.Olson JE, Ryu E, Johnson KJ, Koenig BA, Maschke KJ, Morrisette JA, Liebow M, Takahashi PY, Fredericksen ZS, Sharma RG, Anderson KS, Hathcock MA, Carnahan JA, Pathak J, Lindor NM, Beebe TJ, Thibodeau SN, Cerhan JR. The Mayo Clinic Biobank: a building block for individualized medicine. Mayo Clin Proc. 2013;88:952-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paria N, Copley LA, Herring JA, Kim HK, Richards BS, Sucato DJ, Rios JJ, Wise CA. The impact of large-scale genomic methods in orthopaedic disorders: insights from genome-wide association studies. J Bone Joint Surg Am. 2014;96:e38. [DOI] [PubMed] [Google Scholar]
- 49.Pearson TA, Manolio TA. How to interpret a genome-wide association study. Jama. 2008;299:1335-1344. [DOI] [PubMed] [Google Scholar]
- 50.Pritchett JW. Statin therapy decreases the risk of osteonecrosis in patients receiving steroids. Clin Orthop Relat Res. 2001:173-178. [DOI] [PubMed] [Google Scholar]
- 51.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.R Core Team (2014). R: A language and environment for statistical computing. R Foundation for Statistical Computing V, Austria: URL http://www.R-project.org/. Date Accessed: January 15, 2018. [Google Scholar]
- 53.Raney BJ, Dreszer TR, Barber GP, Clawson H, Fujita PA, Wang T, Nguyen N, Paten B, Zweig AS, Karolchik D, Kent WJ. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 2014;30:1003-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samadfam R, Awori M, Benardeau A, Bauss F, Sebokova E, Wright M, Smith SY. Combination treatment with pioglitazone and fenofibrate attenuates pioglitazone-mediated acceleration of bone loss in ovariectomized rats. J Endocrinol. 2012;212:179-186. [DOI] [PubMed] [Google Scholar]
- 55.wcfLMaMWSS-SSKATRpvhC Seunggeun Lee.
- 56.Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B. PPARgamma2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J Cell Biochem. 2009;106:232-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sinnwell JP, Schaid DJ. haplo.stats: Statistical analysis of haplotypes with traits and covariates when linkage phase is ambiguous. Available at: https://cran.r-project.org/web/packages/haplo.stats/index.html. 2014. Date Accessed: January 15, 2018.
- 58.Smith SY, Samadfam R, Chouinard L, Awori M, Benardeau A, Bauss F, Guldberg RE, Sebokova E, Wright MB. Effects of pioglitazone and fenofibrate co-administration on bone biomechanics and histomorphometry in ovariectomized rats. J Bone Miner Metab. 2015;33:625-641. [DOI] [PubMed] [Google Scholar]
- 59.Takada I, Kouzmenko AP, Kato S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol. 2009;5:442-447. [DOI] [PubMed] [Google Scholar]
- 60.Tokuhara Y, Wakitani S, Oda Y, Kaneshiro Y, Masada T, Kim M, Kadoya Y, Azuma T, Takaoka K. Low levels of steroid-metabolizing hepatic enzyme (cytochrome P450 3A) activity may elevate responsiveness to steroids and may increase risk of steroid-induced osteonecrosis even with low glucocorticoid dose. J Orthop Sci. 2009;14:794-800. [DOI] [PubMed] [Google Scholar]
- 61.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, Rando OJ, Birney E, Myers RM, Noble WS, Snyder M, Weng Z. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798-1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Zhuang J, Iyer S, Lin XY, Greven MC, Kim BH, Moore J, Pierce BG, Dong X, Virgil D, Birney E, Hung JH, Weng Z. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013;41:D171-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wyles CC, Houdek MT, Crespo-Diaz RJ, Norambuena GA, Stalboerger PG, Terzic A, Behfar A, Sierra RJ. Adipose-derived mesenchymal stem cells are phenotypically superior for regeneration in the setting of osteonecrosis of the femoral head. Clin Orthop Relat Res. 2015;473:3080-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuan Z, Li Q, Luo S, Liu Z, Luo D, Zhang B, Zhang D, Rao P, Xiao J. PPARgamma and Wnt signaling in adipogenic and osteogenic differentiation of mesenchymal stem cells. Curr Stem Cell Res Ther. 2016;11:216-225. [DOI] [PubMed] [Google Scholar]
- 65.Zhu ZN, Jiang YF, Ding T. Risk of fracture with thiazolidinediones: an updated meta-analysis of randomized clinical trials. Bone. 2014;68:115-123. [DOI] [PubMed] [Google Scholar]
- 66.Zuin J, Dixon JR, van der Reijden MI, Ye Z, Kolovos P, Brouwer RW, van de Corput MP, van de Werken HJ, Knoch TA, van IWF, Grosveld FG, Ren B, Wendt KS. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A. 2014;111:996-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]