

The mode of action of novel antimalarial, JPC-3210 has been revealed using biochemical assays combined with several multi-omics techniques, including proteomics, peptidomics and metabolomics. Metabolomics and peptidomics, in combination with hemoglobin fractionation assays and β-hematin polymerization assays, revealed JPC-3210 to inhibit the parasite's hemoglobin digestion pathway. Proteomics analysis of JPC-3210-treated parasites identified significant enrichment for proteins involved in the regulation of translation.
Keywords: Metabolomics, malaria, peptidomics, drug targets, metabolites, 2-aminomethylphenol, antimalarial drug discovery, mode of action, proteomics
Graphical Abstract

Highlights
Multi-omics analysis on mode of action of novel antimalarial, JPC-3210
JPC-3210 has rapid parasite killing kinetics.
Metabolomics and peptidomics demonstrated JPC-3210 inhibits hemoglobin digestion.
Proteomics demonstrated JPC-3210 enriches for translation regulation proteins.
Abstract
The increasing incidence of antimalarial drug resistance to the first-line artemisinin combination therapies underpins an urgent need for new antimalarial drugs, ideally with a novel mode of action. The recently developed 2-aminomethylphenol, JPC-3210, (MMV 892646) is an erythrocytic schizonticide with potent in vitro antimalarial activity against multidrug-resistant Plasmodium falciparum lines, low cytotoxicity, potent in vivo efficacy against murine malaria, and favorable preclinical pharmacokinetics including a lengthy plasma elimination half-life. To investigate the impact of JPC-3210 on biochemical pathways within P. falciparum-infected red blood cells, we have applied a “multi-omics” workflow based on high resolution orbitrap mass spectrometry combined with biochemical approaches. Metabolomics, peptidomics and hemoglobin fractionation analyses revealed a perturbation in hemoglobin metabolism following JPC-3210 exposure. The metabolomics data demonstrated a specific depletion of short hemoglobin-derived peptides, peptidomics analysis revealed a depletion of longer hemoglobin-derived peptides, and the hemoglobin fractionation assay demonstrated decreases in hemoglobin, heme and hemozoin levels. To further elucidate the mechanism responsible for inhibition of hemoglobin metabolism, we used in vitro β-hematin polymerization assays and showed JPC-3210 to be an intermediate inhibitor of β-hematin polymerization, about 10-fold less potent then the quinoline antimalarials, such as chloroquine and mefloquine. Further, quantitative proteomics analysis showed that JPC-3210 treatment results in a distinct proteomic signature compared with other known antimalarials. While JPC-3210 clustered closely with mefloquine in the metabolomics and proteomics analyses, a key differentiating signature for JPC-3210 was the significant enrichment of parasite proteins involved in regulation of translation. These studies revealed that the mode of action for JPC-3210 involves inhibition of the hemoglobin digestion pathway and elevation of regulators of protein translation. Importantly, JPC-3210 demonstrated rapid parasite killing kinetics compared with other quinolones, suggesting that JPC-3210 warrants further investigation as a potentially long acting partner drug for malaria treatment.
Malaria is a potentially fatal tropical infectious disease caused by parasites of the Plasmodium spp, and is a major global health concern. While the global malaria burden has reduced substantially over the last decade, there were still 435,000 deaths and 219 million new malaria cases in 2017 (1). Chemotherapy with antiparasitic drugs is the basis for malaria treatment, and, along with vector reduction strategies (e.g. insecticides and bednets), is critical for the overall management of disease burden. Reports of emerging drug resistance to the current gold standard antimalarial drug class, the artemisinins (2, 3) and many currently used artemisinin partner drugs (3, 4) is of great concern. Therefore, new antimalarial drugs are urgently needed for the treatment, management and eventual eradication of malaria. Further, new drugs with novel modes of action (MoA)1 are desired, to minimize the potential for cross-resistance to existing antimalarials.
JPC-3210 (MMV 892646) is a promising new antimalarial (5) that is in its final stages of preclinical development prior to testing in humans. JPC-3210 has recently been reported to have potent in vitro antimalarial activity against the chloroquine-sensitive Plasmodium falciparum D6 line (Sierra Leone) and seven multidrug-resistant lines, i.e. 7G8 (Brazil), TM90-C2B, TM91-C235 and TM93–1088 (Thailand), and MRA1239, MRA1240, and MRA1241 (Cambodia), with mean 50% inhibition concentrations (IC50) less than 19 nm (6). These multidrug-resistant lines have been well characterized and have different levels of susceptibility to chloroquine (CQ), dihydroartemisinin (DHA) and mefloquine (MQ) (11–14). TM90-C2B and TM93–1088 are also highly resistant to atovaquone (ATQ) (12). JPC-3210 possesses high in vivo potency in the modified Thompson test with a curative 3-day dose of 4 mg/kg/day against P. berghei (7). A single oral dose of 5 mg/kg of JPC-3210 cured Aotus monkeys infected with the chloroquine-resistant P. falciparum FVO strain (McCallum et al. unpublished data). JPC-3210 also has high oral bioavailability (86%) in mice, is highly protein bound (97%), has little in vitro metabolism in mouse and human hepatocytes and has a lengthy plasma elimination half-life of ∼4.5 days in mice and 11.8 days in monkeys (5). The MoA of JPC-3210 is currently undefined, but its potency against multiple drug resistant strains supports the contention that JPC-3210 has a MoA distinct from currently used antimalarials.
Knowledge of MoA can help aid the selection of suitable partner drugs as well as give an insight into the potential for drug resistance. Drug resistance mechanisms for the major classes of currently used antimalarials have been extensively researched and reviewed elsewhere (8). There are many different antimalarial drug targets in P. falciparum that have been identified and are being exploited including the apicoplast (9), purine biosynthesis (10), kinases (11), proteases (12), mitochondria (13) and ribosomes (14). In particular, the parasite's heme detoxification pathway is a key target for some of the most commonly used antimalarial drugs. These include the natural product, quinine and its synthetic quinoline derivatives; N-desethylamodiaquine (AQm), CQ, piperaquine (PPQ) and pyronaridine (PYN), and likely includes the structurally related drugs MQ, halofantrine (HF) and lumefantrine (LF). Despite drug resistance reducing their efficacy and mandating their use as partner drugs with the artemisinin derivatives, these drugs have helped save many thousands of lives through treatment and chemoprophylaxis. The artemisinin derivatives also act through the hemoglobin degradation pathway, albeit via a different mechanism to the quinolines, as artemisinins are activated by free heme that is liberated from hemoglobin degradation, subsequently causing radical-mediated damage to a large number of proteins, leading to parasite death (15, 16). Elucidation of the MoA of a novel compound such as JPC-3210 is particularly challenging, as the unique structure does not provide an obvious mechanistic hypothesis to pursue. Metabolomics approaches have previously been demonstrated as an untargeted approached to reveal unique metabolic signatures for antimalarial compounds that are clustered according to their MoA, and in some cases to reveal specific metabolic pathways targeted by metabolic inhibitors (17–19).
In the present study, we have used several omics-based approaches to identify biochemical changes in in vitro cultures of P. falciparum malaria to gain an insight into the MoA of JPC-3210. The results from the untargeted metabolomics, peptidomics and hemoglobin fractionation analysis pointed to a dysfunction in hemoglobin digestion, which was further investigated using in vitro β-hematin polymerization assays that indicated intermediate inhibition of the heme detoxification pathway. Proteomics analysis significantly perturbed parasite proteins involved in the regulation of protein translation. Interestingly, JPC-3210 demonstrated rapid parasite killing kinetics compared with currently used quinoline antimalarials.
EXPERIMENTAL PROCEDURES
Drugs and P. falciparum In Vitro Culture
JPC-3210 was obtained from Jacobus Pharmaceutical Company, NJ. DHA was obtained from DK Pharma, Vietnam. AQm was obtained from AlsaChim, France. ATQ, CQ, MQ, PPQ, PYN, LF, quinine (QN) and tafenoquine (TQ) were obtained from Sigma, MO. All drugs were prepared as 10 mm stocks in dimethyl sulfoxide (DMSO, Sigma, MO) except CQ which was prepared in water, and stored at −80 °C prior to assay.
The P. falciparum (3D7) parasite line was maintained in continuous culture, using standard methods (20), with minor modifications as detailed in (17). Parasites were tightly synchronized by double sorbitol lysis 14 h apart and all experiments were conducted on trophozoite-stage parasites around 58 h following synchronization.
Parasite Viability Drug Pulse Assay
Parasite sensitivity to short exposures of JPC-3210, MQ and CQ was assessed as described previously (21). In short, 28–30 h post invasion trophozoite-stage parasites were exposed to drug for either 1 or 5 h and parasite viability was determined following 48 h after the drug was washed off. Drug dilutions were prepared in 100 μl of media using a flat bottom 96-well microplate via serial dilution from a 1 μm starting concentration (final hematocrit of 2%). The parasite culture was adjusted to 8% parasitaemia and 4% hematocrit before 100 μl was added to the prepared drug dilutions and incubated. At the end of the desired incubation period (either 1 or 5 h), drug was removed by transferring the cultures to V-bottom microplates and washed four times with 200 μl of complete RPMI culture medium containing 5% Albumax II. At this point smears were made and giemsa stained to check for parasite viability by light microscopy. Treatment with 1 μm DHA was also performed only for giemsa stained smears as a positive control for parasite death. Following washes, the cultures were adjusted to 1% parasitaemia and 2% hematocrit by resuspending a proportion of infected red blood cells (iRBCs) from each well in a flat-bottom 96-well microplate containing uninfected RBCs and complete RPMI medium so that the desired conditions were obtained. These cultures were incubated at 37 °C for 48 h. Parasites exposed to JPC-3210 for 48 h under matching conditions was used as a control for 100% parasite killing while untreated iRBCs were used as a control for parasite growth. The assays were performed in duplicate in three independent experiments.
After 48 h, parasite drug susceptibility was assessed by measurement of SYBR green I fluorescence as previously described (22). Briefly, lysis buffer (20 mm Tris, 5 mm EDTA, 0.008% w/v Saponin, 0.08% w/v Triton-X, in Milli-Q water pH 7.5) containing SYBR green dye (0.1 μl/ml) was added to each well and thoroughly mixed. After a 1 h incubation at room temperature in the dark the fluorescence was measured using a Novostar V fluorescence plate reader (BMG Labtech) with excitation and emission wavelengths of 485 nm and 530 nm respectively. The data was analyzed using Prism (v7.01; GraphPad Software, San Diego, CA) by the nonlinear regression [log(inhibitor) versus normalized response] to yield the drug concentration that produced 50% growth inhibition relative to the drug-free controls (IC50).
Experimental Design and Statistical Rationale
The metabolomics analysis included seven independent biological replicates for the DMSO control, three independent biological replicates for JPC-3210 and four independent biological replicates for all other compounds, with a total of 42 samples. For proteomics and peptidomics analysis three independent biological replicates were analyzed, totaling 15 samples for each analysis. DMSO treated infected red blood cells were used as a mock treated control to determine relevant drug induced changes. Sample injections within the experiment were randomized to avoid any impact of systematic instrument drift on metabolite signals. For the metabolomics analysis, a pooled biological quality control was used to monitor downstream sample stability and analytical reproducibility. A Welch's t test (α = 0.05) was used to determine statistical significance because of the unequal sample size and variance between the compound of interest and the DMSO control. For the metabolomics clustering analysis, sparse partial least squares-discriminant analysis was performed with metaboanalyst (23) with 10 metabolites per component to identify only the metabolites responsible for separation. For the proteomics and peptidomics analysis, a Student's t test (p value < 0.05) was used to determine statistical significance between the compound of interest and the DMSO control. For proteomics analysis connectivity was determined using the bioinformatics interaction network analysis tool STRINGdb (24). Connectivity was based on experimental, database and co-expression evidence and a strict minimum interaction score (> 0.7). Cytoscape 3.6 (25) and the ClusterONE algorithm was used to integrate and visualize relationships. Metaboanalyst was also used for principal-component analysis, sparse partial least squares - discriminant analysis and hierarchical clustering analysis to identify similarities between the different compounds.
Metabolomics Sample Preparation
The metabolism of mid-trophozoite-stage parasites (24–28 h post invasion) in response to treatment with a panel of antimalarial compounds was investigated. Cultures (200 μl) were adjusted to 8% parasitaemia and 2% hematocrit before being exposed to test compounds; JPC-3210, AQm, PYN, ATQ, DHA, CQ, MQ, LF, and TQ (1 μm) for 1 h in a flat bottom 96-well plate. At least three replicates of each compound and seven replicates of an untreated control which contained an equivalent volume of DMSO vehicle (0.01% final concentration) were analyzed.
Following drug incubation, metabolites were extracted as detailed in (17). After incubation with drug, culture medium was aspirated from each well, and the metabolism of iRBCs was quenched by the addition of ice-cold phosphate-buffered saline (PBS). Cells were pelleted by centrifugation for 5 min at 1000 × g, and the PBS supernatant was removed prior to the addition of 135 μl ice cold methanol (containing the internal standard compounds; CHAPS (3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate), CAPS [N-cyclohexyl-3-aminopropane-sulfonic acid], and PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)]). Samples were rapidly mixed by pipetting three times to extract iRBC metabolites. Samples were left on ice with gentle agitation for 60 min and then centrifuged at 3000 × g to remove insoluble material. Supernatants were transferred to glass high-performance liquid chromatography (HPLC) vials and stored at −80 °C until analysis. An aliquot (10 μl) of each sample was combined to generate a pooled biological quality control (PBQC) sample, which was used to monitor downstream sample stability and analytical reproducibility.
Metabolomics LC-MS Analysis and Data Processing
Metabolite analysis was performed by liquid chromatography-mass spectrometry (LC-MS), using hydrophilic interaction liquid chromatography (HILIC) and high-resolution (Q-Exactive Orbitrap, Thermo Fisher) MS as previously described (17). Briefly, samples (10 μl) were injected onto a Dionex RSLC U3000 LC system (Thermo) fitted with a ZIC-pHILIC column (5 μm particle size, 4.6 by 150 mm; Merck) and 20 mm ammonium carbonate (A) and acetonitrile (B) were used as the mobile phases. A 30 min gradient starting from 80% B to 40% B over 20 min, followed by washing at 5% B for 3 min and re-equilibration at 80% B, was used. MS used a Q-Exactive Orbitrap MS (Thermo) with a heated electrospray source operating in positive and negative modes (rapid switching) and a mass resolution of 35,000 from m/z 85 to 1050. Sample injections within the experiment were randomized to avoid any impact of systematic instrument drift on metabolite signals. Retention times for ∼350 authentic standards were checked manually to aid metabolite identification.
Metabolomics data sets were analyzed using IDEOM (26). Raw files were converted to mzXML with msconvert (27), extraction of LC-MS peak signals was conducted with the Centwave algorithm in XCMS, alignment of samples and filtering of artifacts with mzMatch, and additional data filtering and metabolite identification in IDEOM (supplementary Data file S1). A total number of 536 putative metabolites were putatively identified. Metabolite abundance was determined by LC-MS peak height and was normalized to the average for untreated samples. Statistical analyses used Welch's t test (α = 0.05) and Pearson's correlation (Microsoft Excel), as well as a principal-component analysis (PCA) using the Metaboanalyst web interface (23). Sparse partial least squares - discriminant analysis (sPLS-DA) algorithms were run in Metaboanalyst with increasing numbers of metabolites in each component (up to 10 metabolites). The final sPLS-DA plots were developed using 10 metabolites in each component.
Proteomics Sample Preparation
Proteomics samples were prepared as previously described with minor modifications (28, 29). Synchronized cultures of 3D7 parasites at 30 h post invasion were enriched to 82% parasitemia using MACS magnetic column purification. Parasites were adjusted to 0.5% hematocrit in fresh media and treated with JPC-3210, CQ, MQ (1 μm), DHA (10 nm), and DMSO control for 5 h. Mature trophozoite-stage parasites were isolated by resuspending the pellet in 0.05% (w/v) saponin in 1 x PBS containing protease and phosphatase inhibitors (Complete mini protease inhibitor mixture (Roche), 20 mm sodium fluoride, 0.1 mm sodium orthovanadate and 10 mm β-glycerolphosphate). The isolated trophozoite-stage parasites were pelleted (2000 × g for 8 min), washed (15,850 × g for 1 min) thrice in 1 ml of the above solution excluding saponin. The parasites were solubilized with lysis buffer (100 mm HEPES (N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid), 4% sodium deoxycholate, pH 8.1) supplemented with phosphatase and protease inhibitors for 10 min at 95 °C. Proteins were reduced and alkylated using tris(2-carboxyethyl)phosphine hydrochloride (Sigma) (TCEP) (10 mm final concentration) and iodoacetamide (Sigma) (40 mm final concentration) respectively. Proteins were then precipitated using 1:4 volume of trichloroacetic acid (TCA) and the supernatants were kept for peptidomics sample preparation. The protein pellets were resuspended in 1 ml of lysis buffer, and 500 μg of total protein, accurately determined using Pierce bicinchoninic acid (BCA) protein assay kit (Thermofisher Scientific) were incubated overnight with trypsin (1:50; Promega) at 37 °C. The following day, trypsin activity was quenched using 5% formic acid (FA), and detergent was removed as previously described (28). The samples were then subjected to desalting using in-house-generated StageTips as described previously (30). The samples were then dried and resuspended in 20 μl of 2% (v/v) acetonitrile (ACN) and 0.1% (v/v) FA containing indexed retention time (iRT) peptides (Biognosys) for LC-MS/MS analysis.
Proteomics LC-MS/MS Analysis and Data Processing
LC-MS/MS Analysis
LC-MS/MS was carried out as described previously (28), with minor modifications. Briefly, samples were loaded at a flow rate of 15 μl/min onto a reversed-phase trap column (100 μm × 2 cm), Acclaim PepMap media (Dionex), which was maintained at a temperature of 40 °C. Peptides were then eluted from the trap column at a flow rate of 0.25 μl/min through a reversed-phase capillary column (75 μm × 50 cm) (LC Packings, Dionex).
For data dependent analysis (DDA) acquisition, the HPLC gradient was set to 158 min using a gradient that reached 30% of ACN after 123 min, then 34% of ACN after 126 min, 79.2% of ACN after 131 min and 2% after 138 min for a further 20 min. The mass Spectrometer was operated in a data-dependent mode with 2 microscans FTMS scan event at 70,000 resolution over the m/z range of 375 to 1575 Da in positive ion mode. The 20 most intense precursors with charge states 2–6 were selected for fragmentation with normalized collision energy 27.0, activation time of 15 ms and enabled dynamic exclusion.
For data independent analysis (DIA) acquisition, the HPLC gradient was the same as above. The mass spectrometer was operated in a data-independent mode with a 25-fixed-window setup of 24 m/z effective precursor isolation over the m/z range of 375–975 Da.
Proteomics Data Analysis
DDA data analysis was performed using the Maxquant version 1.6.0.1 analysis software with previously described settings (31). The DDA files were searched against P. falciparum (UP000001450, release version 2016_04) and Homo sapiens (UP000005640, release version 2017_05) UniProt fasta database and the Biognosys iRT peptides database. The number of entries in the database searched were 3,970,852 with trypsin as enzyme specificity and 2 missed cleavages were permitted. Carbamidomethylation of cysteines was set as a fixed modification. Oxidation of methionine and protein N-terminal acetylation were set as variable modifications. Parent mass error tolerance and fragment mass tolerance were set to 20.0 ppm and 20 ppm respectively. For both peptide and protein identification, a false discovery rate of 1% was set. MaxQuant search results were imported as spectral libraries into Spectronaut with default settings.
Raw files were processed using SpectronautTM 13.0 against the in-house generated P. falciparum (3D7 line) iRBCs library. The consensus library contained 42,245 peptides corresponding to 4421 protein groups. For processing, raw files were loaded and spectronaut calculated the ideal mass tolerances for data extraction and scoring based on its extensive mass calibration with a correction factor of 1. Both at precursor and fragment level, the highest data-point within the selected m/z tolerance was chosen. Identification of peptides against the library was based on default Spectronaut settings (Manual for Spectronaut 13.0, available on Biognosis website), briefly precursor Qvalue Cut-off and Protein Qvalue Cut-off were as per default at 1%, therefore only those that passed the cut-off were considered as identified and used for other subsequent processes. RT prediction type was set to dynamic indexed RT. Interference correction was on MS2 level. For quantification, the interference correction was activated, and a cross run normalization was performed using the total peak area as normalization base. A significance filter of 0.01 was used. Fold-changes for the drug treated samples compared with the DMSO control sample were calculated in Microsoft Excel. The reliability of the quantification measurements between biological replicates were based on p values generated using standard student t test. The bioinformatics interaction network analysis tool STRINGdb (24) was used to build a protein-protein interaction network using the significantly perturbed proteins. Connectivity was based on experimental, database and co-expression evidence and a strict minimum interaction score (> 0.7) was applied to limit false positive associations in the predicted network. The STRINGdb protein connectivity output was exported to Cytoscape 3.6 (25) and the ClusterONE algorithm was used to integrate and visualize relationships between proteins that were significantly perturbed by drug treatment. sPLS-DA and hierarchical clustering algorithms were run in Metaboanalyst (23). The final sPLS-DA plots were developed using 40 proteins in each component. Hierarchical clustering analysis was developed using 60 differentially regulated proteins.
Peptidomics Sample Preparation
TCA soluble supernatants from the proteomics experiments described above, were subjected to centrifugal filtration (10 kDa cutoff, Amicon Ultra) and used for peptidomics analyses. The flow-through containing endogenous peptides was collected, and an equal amount of ethyl acetate was added to remove residual detergent. Equal quantities of total peptides (30 μg measured using Pierce BCA protein assay kit) were then used for peptidomics analyses. Peptide samples were subjected to desalting (30) and were then dried and resuspended in 20 μl of 2% (v/v) ACN and 0.1% (v/v) FA for LC-MS/MS analysis.
Peptidomics LC-MS/MS Analysis and Data Processing
LC-MS/MS was performed using the same method as the proteomics LC-MS/MS analysis (described above), with minor modifications. The HPLC gradient was set to 120 min using a gradient that reached 37.5% of ACN after 88 min, then 42.5% of ACN after 91 min, 99% of ACN after 93 min and 2% after 100 min for a further 20 min. The mass spectrometer was operated in a data-dependent mode with 2 microscan FTMS scan events at 70,000 resolution over the m/z range of 375–1575 Da in positive ion mode. The 20 most intense precursors with charge states 1–8 were selected for fragmentation with normalized collision energy 27.0, activation time of 15 ms and enabled dynamic exclusion.
Peptide identification was conducted with de novo sequencing-assisted database search using PEAKS DB software version 8 (32). The number of entries in the database searched were 95,244 with no enzyme specificity and 100 missed cleavages permitted. Carbamidomethylation of cysteines was set as a fixed modification. Oxidation of methionine and protein N-terminal acetylation were set as variable modifications. Parent mass error tolerance and fragment mass tolerance were set to 15.0 ppm and 0.5 Da respectively. Threshold score for accepting individual spectra was set to 0.01. The identified peptide sequences from H. sapiens (UP000005640, release version 2017_05) and P. falciparum (UP000001450, release version 2016_04) proteome databases detected in multiple samples were shortlisted. The mass-to-charge ratio and retention time of 123 shortlisted peptides were imported into TraceFinder (ThermoFisher). The peak intensity was obtained by manually adjusting the integration and accounting for retention time drift, when required. Log-transformed fold-difference and student t test were calculated in Microsoft Excel.
β-hematin Polymerization
A propionate buffer-based assay was conducted to assess the role of JPC-3210 in β-hematin polymerization (33). CQ, MQ, JPC-3210 (HCl salt) and DMSO were tested in duplicate in deep 96-well sample collection plates (800 μl round well, Waters) on three separate occasions. AQm, PYN, and JPC-3210 (base) were tested once. Test compounds (1 mm) were diluted in 1 m propionate buffer pH 5.2 containing 20% DMSO, then 360 μl of each test compound was added to deep 96-well sample collection plates (Waters) and serially diluted in 180 μl propionate buffer. For positive and negative controls, propionate buffer alone was used. Freshly prepared hemin solution (10 μl at 2 mm diluted in 0.1 m NaOH) was added to all wells, and 10 μl of the lipid catalyst phosphatidylcholine (0.5 mg/ml in propionate buffer) was also added to all wells except for the negative control. Each well was mixed thoroughly by pipetting, before incubating for 16 h at 37 °C with gentle agitation to allow β-hematin crystal polymerization. The following day, 100 μl of 7.5% sodium dodecyl sulfate (SDS) was added to each well to terminate crystal formation and mixed thoroughly by pipetting. Plates were incubated at room temperature for 10 min with gentle agitation, followed by centrifugation at 400 × g for 3 min to pellet any crystals formed. The supernatant (50 μl) in each well was then transferred to a new 96-well flat bottom plate (Costar) containing 200 μl of 2.5% SDS. A blank control of propionate buffer was also included, 250 μl/well. The absorbance was detected at 405 nm on a Perkin Elmer Ensight Plate Reader. The mean absorbance value of blank control was subtracted from test compounds, followed by normalization to the positive and negative controls. Values were graphed against log concentration using GraphPad Prism and the error expressed as standard deviation. IC50 values were obtained from three separate replicates unless otherwise stated, with error expressed as standard deviation.
Additional β-hematin studies were conducted using an acetate buffer-based method described in da Silva et al. (34). Briefly, 100 μl of a fresh 6.5 mm solution of β-hematin (Sigma) in 0.2 m NaOH was mixed with 200 μl of 3 m sodium acetate, 25 μl of 17.4 m acetic acid and 25 μl of the tested drugs in triplicate, or the solvent alone as the negative control. The final concentration of drugs was 2 mm. After 48 h of incubation with gentle agitation at 37 °C, samples were centrifuged for 15 min at 3300 × g, the supernatant discarded, and the pellet was washed with 200 μl DMSO. This latter step was repeated once more and the pellet was finally washed with water. The pellet was then dissolved in 200 μl of 0.1 m NaOH. After a further 1:8 dilution in 0.1 m NaOH, absorption at 405 nm was measured using a Tecan Infinite M200 microplate reader. Experiments were carried out on two separate occasions. Results are expressed as percentage of inhibition of β-hematin formation compared with the negative control, as calculated with the following equation; % Inhibition = 100*(1 − (OD drug)/(OD solvent).
Puromycin Labeling Assay
Synchronized trophozoite-stage parasites (30 h post invasion) at 5% parasitaemia and 2% hematocrit were incubated with PYN, CQ, MQ, AQm, JPC-3210 (1 μm), and DMSO control for 5 h. Puromycin (400 nm) (Sigma) was added for the last 15 min of incubation to pulse label translation products as previously described (35). Following puromycin labeling, parasites were harvested, and proteins isolated as previously described (36). Briefly, parasite cultures were centrifuged at 3000 × g for 5 min, supernatant removed and iRBCs lysed with 0.01% saponin in 1 × PBS. Released parasites were centrifuged 3000 × g for 5 min, washed in PBS three times and the pellets frozen at −80 °C.
Samples were thawed in RIPA lysis buffer (4% SDS, 0.5% Triton X-114 in 1 × PBS) supplemented with Calbiochem protease inhibitor mixture and total protein concentration was measured using Pierce BCA protein assay kit. Protein concentrations were adjusted to 1 μg/μl and 10 μg was resolved on 4–12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto a nitrocellulose membrane. The membrane was blocked using 5% skim milk powder in PBS/0.05% Tween20 for 1 h prior to overnight incubation in anti-puromycin monoclonal antibody (Merck, Mouse Monoclonal, Clone 12D10, 1:1000).
Membranes were washed and incubated in anti-mouse Ig HRP conjugate (1:2,000) (Sigma) for 1 h, washed extensively and chemiluminescent detection (Thermo SuperSignal West Dura) signal detected using a Gel Logic 1500 Imaging System (Carestream Health Inc.). Quantitation of total signal intensity of each lane was measured with Image-J software (NIH) and expressed as a fraction of Control (DMSO treated parasite cultures).
Hemoglobin Fractionation
The hemoglobin fractionation assay was adapted from (37). Aliquots of 6.5 ml of 30–32 h post invasion parasite cultures were adjusted to 8% parasitaemia and 2% hematocrit and then incubated with MQ, CQ, and JPC-3210 1 μm or a DMSO control for either 1 or 5 h. Following incubation, the media was aspirated off and the culture was incubated with 2.3 ml of 0.1% saponin in 1 × PBS with protease inhibitors (complete mini protease inhibitor mixture (Roche)) for 10 min at 4 °C to lyse the iRBCs. The parasites were washed three times with PBS and stored at −80 °C.
For the hemoglobin fractionation, lysed parasites were resuspended in 50 μl of Milli-Q water and sonicated for 5 min in a water bath sonicator. Following sonication, 50 μl of 0.2 m HEPES (pH 7.5) was added and the samples were centrifuged at 4000 rpm for 20 min. The supernatant containing the hemoglobin fraction was carefully transferred to new tubes and 50 μl of 4% of SDS was added before the samples were incubated at 95 °C for 5 min. Following heating, 50 μl of 0.3 m NaCl and 50 μl of 25% (v/v) pyridine (Sigma) in 0.2 m HEPES was added, the samples were vortexed and transferred to a 96-well plate. This sample contained the hemoglobin fraction.
The pellets were treated with 50 μl of MilliQ water and 50 μl of 4% of SDS and resuspended before being sonicated for 5 min and incubated at 95 °C for 5 min to solubilize the free heme. Following incubation, 50 μl of 0.2 m HEPES, 0.3 m NaCl and 25% pyridine were added to the samples. The samples were then subsequently centrifuged at 4000 rpm for 20 min. The supernatant was transferred to the 96-well plate, corresponding to the free heme fraction.
The remaining pellet containing the hemozoin fraction was solubilized by resuspending with 50 μl of MilliQ water and 50 μl of 0.3 m NaOH. The samples were sonicated for 15 min before 50 μl of 0.2 m HEPES, 0.3 m HCl and 25% pyridine was added. The samples were then transferred to the 96-well plate, corresponding to the hemozoin fraction. The absorbance of each fraction was measured at a 405 nm wavelength using a Perkin Elmer Ensight Plate Reader. The samples were normalized via a paired analysis to the DMSO control and graphed as their fold change versus DMSO ± S.E. All fractions had >4 replicates from >2 independent experiments apart from the 5 h CQ hemozoin fraction for which there was only 3 replicates from one experiment.
RESULTS
Parasite Viability Drug Pulse Assay
To explore the onset of action of JPC-3210 and other test antimalarials, a pulse assay was performed to assess parasite killing kinetics. Following a 1 h drug pulse against trophozoite-stage parasites, JPC-3210, MQ, and CQ did not kill parasites at values as high as 1 μm (Fig. 1A). After a 5 h drug pulse against trophozoite-stage parasites, all these compounds lead to parasite death (as measured by parasite survival after one full life-cycle following the drug pulse), with JPC-3210 being the most potent (IC50 of 15 ± 5 nm) compared with MQ (IC50 of 103 ± 14 nm) and CQ (IC50 of 215 ± 28 nm) (Fig. 1A). However, when visually inspected via light microscopy, parasites treated with JPC-3210, MQ or CQ appeared normal and were comparable to the DMSO control in morphology (Fig. 1B). They also showed significant morphological differences to the DHA positive control, which appeared pyknotic, consistent with parasite death that has been previously reported for high doses of DHA (38). Overall, the low IC50 value of 15 nm demonstrates that JPC-3210 has rapid parasite killing kinetics compared with MQ and CQ, irreversibly damaging the parasite within 5 h. Therefore, as no significant parasite killing occurred within the first hour, and no effect on cellular integrity was observed within the first 5 h, these time points of drug exposure were selected to investigate the MoA of JPC-3210.
Fig. 1.

Parasite killing kinetics of JPC-3210, MQ and CQ against Plasmodium falciparum (3D7) parasites after 1 and 5 h incubations. A, Representative curves of antimalarial potency 48 h after either a 1 or 5 h pulse incubation with JPC-3210 (blue) MQ (red) or CQ (green) plotted as the % survival versus the log (concentration). IC50 values were generated from three independent experiments, performed in duplicate and expressed as the mean of the three replicates ± standard deviation. B, A panel of representative giemsa stained parasites treated with 1 μm of JPC-3210, MQ, CQ, a DMSO negative control or a 1 μm DHA positive control for parasite death after 5 h.
Untargeted Metabolomics Analysis of JPC-3210-treated P. falciparum Trophozoite-stage Parasites
Tightly synchronized trophozoite-stage parasites were incubated with 1 μm of a panel of antimalarials for 1 h to identify the initial drug-induced changes in metabolite abundance and to compare changes seen in JPC-3210 to current antimalarials with known MoA. Upon performing untargeted metabolomics, 536 metabolites were putatively identified (supplemental Table S1) based on accurate mass and predicted RT using the IDEOM database (26). The mean relative abundances of each metabolite in JPC-3210 and comparator antimalarial drug treated samples were expressed as fold-change relative to the untreated (DMSO) control to provide metabolic profiles for each test compound (Fig. 2A). The heat map shows that the metabolic profiles induced by JPC-3210, MQ, and DHA were similar, as all three treatments significantly depleted short-chain peptides. This observation was supported by sPLS-DA analysis where JPC-3210, MQ, and DHA clustered closely, and separated from the untreated control, based predominantly on the differential abundance of short peptides (Fig. 2B, 2D).
Fig. 2.

Metabolomics analysis of Plasmodium falciparum (3D7)-infected red blood cells following treatment with JPC-3210, AQm, ATQ, CQ, DHA, LF, MQ, PYN, TQ, and DMSO. A, Heat map of all metabolite (y axis) abundances expressed as mean fold change versus DMSO control in all sample groups (x axis). All compounds (1 μm for 1 h) were tested with at least three independent biological replicates. Yellow indicates no change while red and blue indicates increased and decreased abundances, respectively. B, C, Sparse partial least square-discriminant analysis (sPLS-DA) of all samples, showing scores plot for components one and two (B) or components one and three (C). These sPLS-DA plots were generated using the top 10 metabolites for each component. Points represent individual sample replicates while the 95% confidence interval is represented by the shaded region. D–F, Loadings plots listing the 10 metabolites contributing to the variance across components one to three respectively. Heat maps represent the relative abundance of each metabolite with red and green representing high and low levels, respectively.
This profile was distinct from the 4-aminoquinolines, PYN, AQm and TQ where 11 unique short peptides were increased following treatment, leading to a separation from the untreated control and JPC-3210 across component two in sPLS-DA analysis (Fig. 2B, 2E). The remaining compounds; CQ, LF and ATQ do not share this distinct metabolic profile and clustered closely with the untreated control in sPLS-DA analysis (Fig. 2B). This suggests that the short incubation time (1 h) for this experiment did not allow time for drug-induced metabolic changes to accumulate to a detectable level.
A distinct metabolic profile was also observed for ATQ, with a significant increase in dihydroorotate and N-carbamoyl-l-aspartate, which lead to the separation of ATQ from the untreated control across component three of the sPLS-DA analysis (Fig. 2C, 2F). This is consistent with the known MoA of ATQ as a cytochrome bc1 inhibitor, which prevents re-oxidation of the ubiquinone cofactor and therefore indirectly inhibits dihydroorotate dehydrogenase, resulting in inhibition of de novo pyrimidine synthesis and accumulation of the enzyme substrate, dihydroorotate, and it's precursor, N-carbamoyl-l-aspartate (39). While cytochrome bc1 is involved in mitochondrial function, ATQ monotherapy has little impact on the mitochondrial membrane potential compared with the positive control mitochondrial uncoupler CCCP (carbonyl cyanide m-chlorophenyl hydrazone) (40). Unsurprisingly, JPC-3210 also showed no significant inhibition of mitochondrial activity (p < 0.05; supplemental Fig. S1).
Overall, the untargeted metabolomics analysis demonstrated that the impact of JPC-3210 on P. falciparum metabolism was limited to peptide metabolism. Further, a number of the short peptides that were significantly depleted following treatment with JPC-3210, DHA, and MQ could be mapped to hemoglobin α- and β-chains (Fig. 3). This is consistent with these peptides originating from hemoglobin and their depletion demonstrates that hemoglobin digestion is affected by JPC-3210 treatment.
Fig. 3.

Metabolomics data for the di-, tri- and tetra-peptides originating from Plasmodium falciparum (3D7)-infected red blood cells following treatment with JPC-3210, AQm, ATQ, CQ, DHA, LF, MQ, PYN, TQ, and DMSO. A, Heat map of all di-, tri- and tetra peptides detected (y axis) from all compounds (1 μm for 1 h) expressed as the mean fold change versus DMSO control from at least three biological replicates. Yellow indicates no change, while red and blue indicates increased and decreased abundances respectively. Peptides colored blue in the DMSO control were not detected in those samples. B, Hemoglobin α (Hb α) and β (Hb β) chain sequences. Blue bars indicate the peptides that could be mapped to the hemoglobin sequence, which were significantly depleted (p value < 0.05) following JPC-3210 treatment compared with the DMSO control.
Peptidomics Analysis of JPC-3210 Treated Trophozoite-stage Parasites
A limitation of the metabolomics approach is that only peptides up to four amino acids in length can be identified with this workflow, and to identify longer-chain hemoglobin-derived peptides, a peptidomics approach was undertaken. The peptidomics approach provides a semi-quantitative analysis of endogenous peptides (up to ∼10 kDa) present in drug-treated parasites.
Peptidomics analysis of trophozoite-stage parasites treated with JPC-3210 (1 μm), CQ (1 μm), MQ (1 μm), and DHA (10 nm) for 5 h identified a total of 79 endogenous H. sapien and 43 P. falciparum peptides. Parasites treated with JPC-3210 had 28 significantly perturbed peptides (p value < 0.05) (supplemental Table S2), 19 of which originated from hemoglobin α and β chains (Fig. 4). The abundance of these hemoglobin-derived peptides was reduced in JPC-3210 treated samples compared with DMSO control (Fig. 4). DHA and MQ treatment also significantly reduced the abundance of 27 and 17 peptides from hemoglobin respectively (Fig. 4). In comparison, CQ treatment significantly increased the abundance of 11 peptides from hemoglobin (Fig. 4). This demonstrated that hemoglobin digestion is significantly inhibited in JPC-3210-, DHA-, and MQ-treated parasites, but not for CQ-treated parasites where an accumulation of long chain peptides was observed. This suggested that the JPC-3210 MoA may be like other fast acting antimalarials such as MQ and DHA, particularly in regard to its effect on hemoglobin digestion.
Fig. 4.

Peptidomics analysis of Plasmodium falciparum (3D7) parasites treated with JPC-3210, CQ, MQ, DHA and DMSO control. A, Sequence coverage and relative abundance of endogenous peptides originating from hemoglobin α (Hb α) and hemoglobin β (Hb β). Peptide abundances are the average fold change following 5 h of drug treatment, expressed relative to the untreated control (DMSO) from at least three biological replicates. Bars represent significant changes in peptide abundance relative to the untreated control (p value < 0.05). B, Mirror chromatograms from all detected Hb α and β peptides; JPC-3210 treated versus DMSO, MQ treated versus DMSO, DHA treated versus DMSO, and CQ treated versus DMSO. The chromatograms demonstrate an overall decrease in hemoglobin derived peptides in JPC-3210, MQ, and DHA treated (top panel) compared with control, while for CQ treated (top panel), the chromatogram demonstrated an overall increase in hemoglobin derived peptides compared with control.
Hemoglobin Fractionation Analysis of JPC-3210 Treated Trophozoite-Stage Parasites
The untargeted metabolomics and peptidomics analyses indicated that JPC-3210 treatment results in a reduction in peptides originating from hemoglobin. To further explore the effects of JPC-3210 on the hemoglobin digestion pathway, the levels of hemoglobin, free heme and hemozoin were measured using a hemoglobin fractionation approach adapted from (31). Following a 5 h incubation with JPC-3210, levels of intraparasitic hemoglobin, free heme and hemozoin were all decreased when compared with the DMSO control. MQ showed a similar profile to JPC-3210, with hemoglobin, free heme and hemozoin levels reduced when compared with the DMSO control (Fig. 5). CQ treatment significantly increased hemoglobin levels, and decreased hemozoin levels, while free heme levels remained unchanged compared with the DMSO control (Fig. 5).
Fig. 5.

Hemoglobin fractionation of JPC-3210-, MQ- and CQ-treated Plasmodium falciparum (3D7) parasites compared with a DMSO control. Bar charts representing the relative levels of hemoglobin, free heme and hemozoin in trophozoite-stage parasites following either a 1 or 5 h incubation with 1 μm JPC-3210, MQ or CQ expressed as the fold change when compared with a DMSO control. Bars represent the mean of >4 paired replicates from at least two independent experiments (apart from 5 h CQ hemozoin, 3 replicates from one experiment) with the error bars expressed as S.E.
A shorter, 1 h, incubation with JPC-3210, and MQ also resulted in decreased levels of free heme and hemoglobin. Hemozoin levels in JPC-3210 and MQ treated parasites remained unchanged after the 1 h incubation. Parasites treated with CQ had a slight decrease in hemoglobin levels, while the levels of free heme and hemozoin remained unchanged, consistent with the minimal impact observed in the metabolomics studies for CQ after 1 h incubation (Fig. 5).
JPC-3210 Is An Intermediate Inhibitor of β-hematin Polymerization
The untargeted metabolomics analysis showed JPC-3210 to have a metabolic profile similar to MQ, a drug known to inhibit β-hematin polymerization (41). Therefore, the ability of JPC-3210 to inhibit β-hematin polymerization was screened in vitro using an acetate buffer system. This assay showed inhibition of β-hematin polymerization by JPC-3210 at a concentration of 2 mm (Fig. 6A), similar to 4-aminoquinolines, whose primary mode of antimalarial action is widely accepted to be mediated by inhibition of β-hematin polymerization (42). However, this assay uses very high concentrations of inhibitor and therefore is limited in its sensitivity. To confirm the observations of the acetate buffer assay, a more sensitive propionate buffer system was employed, which uses lower drug concentrations and a shorter incubation time than the acetate buffer system, in part due to the incorporation of a lipid catalyst (43).
Fig. 6.
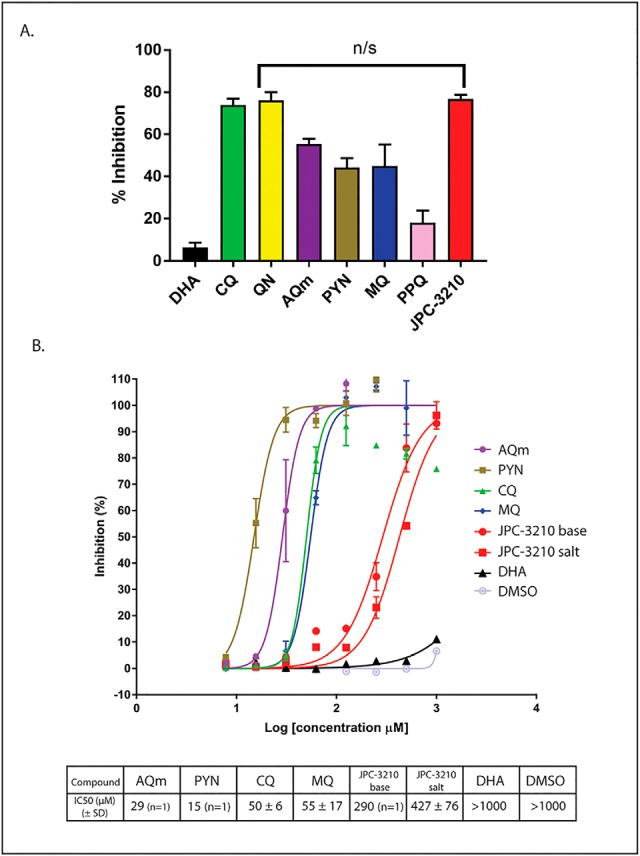
β-hematin polymerization assays to explore the effects of JPC-3210 on hemazoin formation. A, Bar chart showing percent inhibition of hematin polymerization in an acetate buffer by DHA (black), MB (light blue), CQ (green), QN (yellow), AQm (purple), PYN (gold), MQ (dark blue), PPQ (pink) and JPC-3210 (red) at 2 mm final concentration from two independent repeats. Error bars are expressed as standard deviation. B, Percentage inhibition of β-hematin polymerization in a propionate buffer versus the log10 (concentration of compound) for AQm (purple), PYN (gold), CQ (green), MQ (blue), JPC-3210 base (red circles), JPC-3210 HCl (red squares), DHA (black) and DMSO (silver). Assays were performed with at least two technical replicates. IC50 concentrations were determined from at least three independent replicates unless otherwise stated.
The propionate buffer assay was performed using serially diluted concentrations of several antimalarials, allowing IC50 values to be determined (Fig. 6B). The positive control β-hematin polymerization inhibitors AQm, PYN, MQ, and CQ all gave IC50 values in the low range of the assay (15 to 55 μm). The untreated and negative controls, DMSO and DHA did not show any inhibition (IC50 >1000 μm). JPC-3210 showed some level of inhibition (IC50 290 μm), in between the positive control, CQ or MQ, and the negative control, DMSO. Given the improved sensitivity of the propionate buffer assay it can be concluded that JPC-3210 is an intermediate inhibitor of β-hematin polymerization, demonstrating ∼10-fold weaker activity than the quinoline antimalarials.
Quantitative Proteomics Analysis of JPC-3210 Treated P. falciparum Trophozoite-stage Parasites
Quantitative proteomics analysis was performed on trophozoite-stage parasites treated with JPC-3210, MQ, DHA and CQ for 5 h to identify proteins that were differentially regulated following drug exposure. A total of 2014 proteins were identified with at least two unique peptides in a minimum of three independent biological replicates across all treatment conditions (supplemental Table S3). Similar proteins were depleted in abundance following treatment with JPC-3210 and MQ (Fig. 7C), which was demonstrated by the hierarchical clustering analysis. sPLS-DA analysis also showed close clustering of JPC-3210 with MQ, while this model also showed DHA-treated parasites trending in the same direction (component 1), albeit to a lesser extent (Fig. 7A). Analysis of the features that allowed separation of JPC-3210 from other antimalarials and DMSO control demonstrated that the primary impact of JPC-3210 on the P. falciparum proteome is like that of MQ (Fig. 7B).
Fig. 7.

Proteomics analysis of Plasmodium falciparum (3D7) parasites treated with JPC-3210, CQ, MQ, DHA, and DMSO control. A, Sparse partial least squares - discriminant analysis (sPLS-DA) of the proteomics data for parasites treated with CQ (red), DHA (green), JPC-3210 (blue), MQ (pink), and DMSO control (purple). The sPLS-DA plot was generated using the top 40 proteins for each component. Points represent individual sample replicates while the 95% confidence interval is represented by the shaded region. B, Variable importance in projection (VIP) scores of the 40 proteins that contributed to the variance in component one. Heat maps represent the relative abundance of each significant protein with red and green representing high and low respectively. C, Hierarchical clustering of the different sample groups, treated with CQ (red), DHA (green), JPC-3210 (blue), MQ (pink), and DMSO control (purple). Vertical clustering displays similarities between sample groups, while horizontal clusters reveal the relative abundances of the 60 most significantly different proteins. All compounds were tested with at least three independent biological replicates. White indicates no change, while red and blue indicates increased and decreased abundances respectively. Ward's minimum variance method algorithm was used to generate the hierarchical cluster analysis.
However, while most of the significantly perturbed proteins following MQ treatment were down-regulated, JPC-3210 treatment also resulted in significant up-regulation of many proteins (Fig. 8A, 8B). Analysis of proteins significantly up-regulated following JPC-3210 treatment revealed that JPC-3210 treatment has a unique proteomic signature characterized by significant enrichment of proteins involved in regulation of translation. This set of translation-associated proteins was not affected by MQ treatment (Fig. 8C, 8D).
Fig. 8.

Proteomics analysis of dysregulated protein abundances following JPC-3210, and MQ treatment. A–B, Volcano plot of differential protein abundance following treatment. Proteins identified in at least three independent experiments following treatment with 1 μm (A) JPC-3210 (2014 proteins, indicated by red color dots) or (B) MQ (2014 proteins, blue dots) for 5 h. Proteins above the significance threshold (p value < 0.05) and outside the gray shaded area (fold change ≥ 1.5) were considered significant. C–D, Network analysis of trophozoite-stage parasite proteins perturbed following treatment with (C) JPC-3210, and (D) MQ. The network analysis was built using the STRINGdb interaction network analysis output (connectivity was based on experimental, database and co-expression evidence with a minimum interaction score of 0.7) in Cytoscape 3.6 with the ClusterONE algorithm. Node size represents p value and node color represents fold-change from at least three independent replicates. E, Puromycin labeling of trophozoite stage 3D7 parasites treated with 1 μm JPC-3210 (red), CQ (green), AQm (purple), PYN (gold) or a DMSO control for 5 h. Bars represent the ratio of labeled puromycin for the respective compounds relative to the DMSO control ± the S.E. from three independent replicates.
Inhibition of Protein Translation
To assess the effect of JPC-3210 on parasite protein translation, synchronized 3D7 parasites were treated for 5 h with drug prior to being pulse-labeled with puromycin (35). Puromycin-bound newly synthesized proteins were detected by immunoblotting with antipuromycin antibody. The intensity of the total signal of each lane was measured with Image-J software. The results showed that JPC-3210 inhibited protein translation by 40% compared with DMSO-treated parasites (Fig. 8E). However, protein translation was also decreased for each of the other tested compounds after 5 h incubation, suggesting that inhibition of protein translation may be a nonspecific consequence of antimalarial treatment of parasites under these conditions.
DISCUSSION
Combining multiple “omics” approaches provides a comprehensive analysis of biochemical changes following drug treatments and is therefore an effective way of investigating the MoA of previously unexplored compounds. The conditions for this analysis involving relatively short durations of exposure at high doses (1 μm for all compounds apart from DHA in proteomics and peptidomics) were chosen to ensure that a robust response was observed that was not due to nonspecific secondary parasite death responses. For example, previous metabolomic studies have observed only minor metabolic changes after CQ treatment at lower doses (100 nm in Cobbold et al. (18), and 2× IC50 in Allman et al. (19)) and therefore a high dose was used for all compounds to ensure a measurable response within this time period. Further, as demonstrated by the pulse assay (Fig. 1B), the parasites appeared visually healthy and there were no discernible differences between JPC-3210, MQ, CQ or the DMSO control parasites, in contrast to the parasite death phenotype observed in the DHA-treated parasites following a 5 h treatment. Therefore, the metabolomics study was conducted after 1 h incubation with the test compounds to allow detection of the metabolic pathways that are initially targeted, and the proteomics samples were collected after a 5 h incubation to allow time for parasite responses to be translated to detectable differences in protein levels.
In this study, untargeted metabolomics following a short incubation with JPC-3210 revealed a significant depletion of short peptides originating from hemoglobin, suggesting that JPC-3210 affects hemoglobin metabolism (Fig. 2). This was supported by the depletion of longer hemoglobin-derived peptides, detected using a peptidomics approach (Fig. 3). Further, a similar peptide signature was observed for parasites treated with MQ and DHA and both compounds are known to inhibit hemoglobin digestion, albeit by distinct mechanisms (44, 45). DHA is activated by free heme within the parasite (46), resulting in the production of free radical species which covalently bind to a number of targets within the parasite's digestive vacuole including multiple hemoglobin-digesting proteases, such as the plasmepsins (47). MQ, like all quinoline antimalarials, is thought to have pleiotropic effects, with some studies demonstrating an ability for MQ to inhibit hemozoin formation in the digestive vacuole (48), which is likely to compromise digestive vacuole function.
Inhibition of the hemoglobin digestion pathway was further explored through the hemoglobin fractionation assay (Fig. 5). Hemoglobin, heme and hemozoin species were all depleted following a 5 h treatment with both JPC-3210 and MQ, while hemoglobin and heme were also reduced after a 1 h incubation. The changes in hemoglobin, heme and hemozoin levels supports the changes observed in hemoglobin-derived peptides using the metabolomics and peptidomics approach, and the similarity in profiles suggests that these two compounds may be acting to quickly inhibit hemoglobin digestion by the parasite via a similar mechanism.
Hemoglobin digestion is a crucial process for parasite survival and occurs within the specialized food vacuole of the parasite (49). It is mediated by several aspartic and cysteine proteases, which produce peptides that are further digested by aminopeptidases to produce free amino acids necessary for parasite growth and development (50). The depletion of both long and short chain hemoglobin-derived peptides, and hemoglobin itself, suggested that JPC-3210 may inhibit an early step in the digestion pathway, by inhibiting hemoglobin uptake or more generally interfering with digestive vacuole formation and/or function (51). For example; the actin-perturbing agent, jasplakinolide inhibits the actin-dependent process of hemoglobin uptake into the food vacuole, and alterations to digestive vacuole acidic pH has been shown to also prevent normal functioning of the vacuole (52, 53). Further, MQ has previously been proposed to act via inhibition of hemoglobin endocytosis (54). Inhibition of hemoglobin uptake or alteration in food vacuole function would presumably result in inhibition of hemoglobin digestion and subsequently the depletion of hemoglobin and hemoglobin-derived peptides observed here.
The six other antimalarial drugs in this study induced significantly different metabolomics, peptidomics and hemoglobin fractionation profiles compared with JPC-3210. ATQ treatment induced specific metabolomics perturbations to intermediates of the de novo pyrimidine synthesis pathway that correspond directly to the known MoA for this drug, consistent with previous reports (17–19). CQ treatment resulted in increased abundance of long chain hemoglobin-derived peptides and increased levels of hemoglobin itself as measured by peptidomics and hemoglobin fractionation analysis (Fig. 3). Treatment with CQ was also characterized by the reduction in hemozoin and no change in the upstream free heme following 5 h treatment (Fig. 5). This is consistent with previous reports detailing an increase in undigested hemoglobin levels in parasites treated with CQ (37, 54) and is also consistent with the proposed MoA of CQ as an inhibitor of hemozoin formation. In contrast to our observations, Combrinck et al. (37) also reported an increase in free heme following CQ treatment, however, this could potentially be explained by the shorter incubation time (5 h compared with 32 h) and later stage parasites (trophozoite-stage compared with early ring-stage parasites) used in our assay. A significant difference in the short chain hemoglobin-derived peptides using metabolomics was not observed for CQ, likely due to the short (1 h) incubation time for the metabolomics study. Interestingly, the related 4-aminoquinolines, PYN and AQm induced significant increases in several short peptides, consistent with a previous study of CQ after a 5 h drug incubation (17). Sequence analysis of peptides affected as a result of treatment with PYN and AQm indicated that these short peptides did not arise from hemoglobin, and likely indicates an impact on some other protein turnover pathway within the parasite. Therefore, inhibition of the hemoglobin digestion pathway by JPC-3210 indicates a MoA that is distinct from the 4-aminoquinolines.
A β-hematin polymerization inhibition assay was performed to confirm that JPC-3210 is acting via a different mechanism to the 4-aminoquinolines (Fig. 6). Inhibition of hemazoin crystal formation is generally accepted as the primary MoA of 4-aminoquinolines such as, CQ, AQm, and PYN (42). Atomic force microscopy has recently been used to reveal how CQ inhibits hematin crystallization, which is by binding to the planar hematin crystal surfaces, inhibiting crystallization (55) and leading to the accumulation of toxic free heme. In addition, Dorn et al. (56) found that other quinoline antimalarial drugs, AQm, CQ, HF, MQ, PYN, QN, and quinacrine are all able to inhibit β-hematin polymerization, and the inhibition directly correlates with their ability to inhibit P. falciparum growth.
Using two different in vitro β-hematin polymerization inhibition assays (Fig. 6), we showed that JPC-3210 does possess some β-hematin polymerization inhibiting activity. However, the IC50 was an order of magnitude higher than all the tested quinoline antimalarials, despite comparable antiparasitic activity levels, suggesting that JPC-3210 does not act primarily by inhibition of β-hematin polymerization like the quinoline antimalarials. Interestingly, MQ showed inhibition of β-hematin polymerization similar to that of CQ, but induced very different metabolomics, peptidomics, and fractionation profiles, suggesting that the inhibition of β-hematin polymerization may not be the primary MoA of MQ.
Metabolomics and peptidomics analysis, as well as the hemoglobin fractionation assay, revealed that JPC-3210 inhibits hemoglobin digestion. However, it is not clear whether this represents the primary drug target, or a secondary parasite response. Interestingly, JPC-3210 clustered with DHA and MQ, which have been shown to impact multiple aspects of parasite biochemistry. The artemisinins (e.g. DHA) have been shown to dysregulate processes involved in glycolysis, hemoglobin degradation, antioxidant defense, and protein turnover pathways (47) while several potential drug targets have been proposed for MQ, including inhibition of hemoglobin digestion, protein translation (57), and purine salvage (58). Our proteomics analysis of JPC-3210-treated parasites revealed additional pathways involved in the JPC-3210 MoA. Quantitative proteomics analysis showed that both JPC-3210 and MQ resulted in decreased abundance of proteins involved in DNA replication and synthesis (Fig. 8C–8D). Unlike MQ, JPC-3210 also reduced a small cluster of parasite surface proteins and induced an accumulation of a large cluster of proteins involved in regulation of protein translation (Fig. 8C). This suggested that the JPC-3210 MoA may involve dysregulation of protein translation mechanisms, and importantly, suggests some difference in the MoA compared with MQ. However, the difference in potency between the two compounds after 5 h should be considered, as the rapid action of JPC-3210 could provide some explanation for the more extensive changes observed following JPC-3210 treatment.
The inhibition of parasite protein translation by JPC-3210 was demonstrated using puromycin labeling, but surprisingly, MQ, AQm, CQ and PYN also inhibited protein translation to a similar extent. This suggests that this analysis of cellular protein synthesis provides little mechanistic insight into a compound's MoA, and that the up-regulation of translation regulation machinery observed for JPC-3210 is not a conserved response to overall inhibition of protein translation. Some inhibition of protein translation by CQ and AQm has previously been demonstrated using the S35 labeling assay (59), confirming the nonspecific nature of this type of assay. It has been suggested that a recently developed in vitro translation assay would be necessary to investigate drug mechanisms that involve translation inhibition in P. falciparum (59).
While this study has revealed many similarities in the MoA's of JPC-3210 and MQ, one significant difference between these two compounds is the potent activity of JPC-3210 against the MQ-resistant parasite strains, TM90-C2B and TM91-C235 (6). MQ resistance is primarily mediated by increased expression of the efflux transporter Pfmdr1, rather than modification of the drug target (60), and the lack of cross-resistance indicates that JPC-3210 is not a substrate for this transporter. Therefore, we propose that while the overlapping aspects of the MoA of JPC-3210 and MQ might have implications for the selection of combination therapies, it should not limit the activity of JPC-3210 in MQ-resistant parasites.
A particularly promising feature of JPC-3210 is the rapid onset of action that was discovered using the drug pulse assays. While not as fast-acting as the artemisinins, the 15 nm IC50 from a 5 h pulse corresponds to a 7-fold, and 14-fold, improvement in activity during this initial exposure phase compared with mefloquine and chloroquine, respectively. It is anticipated that this rapid killing effect will contribute to desirable pharmacodynamics in vivo and minimize the opportunity for parasites to develop resistance de novo.
In conclusion, our biochemical assays combined with a multi-omics analysis using high resolution orbitrap mass spectrometry has provided evidence that JPC-3210 possesses a MoA like MQ, involving inhibition of the hemoglobin digestion pathway and dysregulation of DNA replication. However, compared with MQ, JPC-3210 demonstrated additional dysregulation of translation proteins, rapid parasite killing kinetics, and possesses excellent potency against MQ resistant strains. These findings provide additional impetus for the clinical appraisal of JPC-3210 by the Medicines for Malaria Venture within its pipeline of drug development.
Data availability
Proteomics and Peptidomics spectrometry data and search results have been deposited to the ProteomeXchange Consortium via the PRIDE (61) partner repository with the dataset identifier PXD013539 and project name 'Multi-omics analysis demonstrates unique mode of action of a potent new antimalarial compound, JPC-3210, against Plasmodium falciparum'.
Metabolomics spectrometry data and search results have been deposited to the Metabolomics Workbench Consortium with project name 'Multi-omics analysis demonstrates unique mode of action of a potent new antimalarial compound, JPC-3210, against Plasmodium falciparum' and Data track identification (1528/DataTrackID1713).
Supplementary Material
Acknowledgments
We thank the Australian Red Cross Blood Service for the provision of O-positive human blood. We thank the Monash Proteomics and Metabolomics Facility for infrastructure and technical support. The opinions expressed are those of the authors and do not reflect those of the Australian Defense Force Joint Health Command or any extant Australian Defense Force policy.
Footnotes
* This research was funded by the Australian Defense Organisation and the NHMRC through fellowship (APP1148700) and project (APP1163235) grants. G.D.H and D.P.J. who work for Jacobus Pharmaceutical Company Inc., declare a financial interest in the development of JPC-3210 and thus declare a conflict of interest. All other authors have no conflicts of interest to declare.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- MoA
- mode of action
- ACT
- artemisinin combination therapies
- ACN
- Acetonitrile
- ART
- artemisinin
- AQm
- N-desethylamodiaquine
- ATQ
- atovaquone
- CCCP
- carbonyl cyanide m-chlorophenyl hydrazone
- CQ
- chloroquine
- DDA
- data dependent analysis
- DIA
- data independent analysis
- DHA
- dihydroartemsinin
- DMSO
- dimethylsulfoxide
- FA
- formic acid
- FDR
- false discover rate
- HF
- halofantrine
- h
- hour
- IC50
- 50% inhibition concentration
- iRT
- indexed retention time
- iRBCs
- infected red blood cells
- JPC
- Jacobus Pharmaceutical Company
- LC-MS
- liquid chromatography-mass spectrometry
- LF
- lumefantrine
- m/z
- mass to charge ratio
- MQ
- mefloquine
- μL
- microlitre
- μM
- micromolar
- mM
- millimolar
- PBS
- phosphate-buffered saline
- PPQ
- piperaquine
- P. falciparum
- Plasmodium falciparum
- PCA
- principal-component analysis
- PYN
- pyronaridine
- QN
- quinine
- RBCs
- red blood cells
- NaOH
- sodium hydroxide
- sPLS-DA
- sparse partial least squares - discriminant analysis
- TQ
- tafenoquine.
REFERENCES
- 1. Organisation WH. World malaria report 2018. World Health Organisation. 2018; http://www.who.int/iris/handle/10665/275867
- 2. Dondorp A. M., Nosten F., Yi P., Das D., Phyo A. P., Tarning J., Lwin K. M., Ariey F., Hanpithakpong W., Lee S. J., Ringwald P., Silamut K., Imwong M., Chotivanich K., Lim P., Herdman T., An S. S., Yeung S., Singhasivanon P., Day N. P. J, Lindegardh N., Socheat D., and White N. J. (2009) Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361, 455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parobek C. M., Parr J. B., Brazeau N. F., Lon C., Chaorattanakawee S., Gosi P., Barnett E. J., Norris L. D., Meshnick S. R., Spring M. D., Lanteri C. A., Bailey J. A., Saunders D. L., Lin J. T., and Juliano J. J. (2017) Partner-drug resistance and population substructuring of artemisinin-resistant Plasmodium falciparum in Cambodia. Genome Biol. Evol. 9, 1673–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Martin R. E., Shafik S. H., and Richards S. N. (2018) Mechanisms of resistance to the partner drugs of artemisinin in the malaria parasite. Curr. Opin. Pharmacol. 42, 71–80 [DOI] [PubMed] [Google Scholar]
- 5. Birrell G. W., Heffernan G. D., Schiehser G. A., Anderson J., Ager A. L., Morales P., MacKenzie D., van Breda K., Chavchich M., Jacobus L. R., Shanks G. D., Jacobus D. P., and Edstein M. D. (2018) Characterization of the preclinical pharmacology of the new 2-aminomethylphenol, JPC-3210, for malaria treatment and prevention. Antimicrob Agents Chemother. 62, pii: e01335–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chavchich M., Birrell G. W., Ager A. L., MacKenzie D. O., Heffernan G. D., Schiehser G. A., Jacobus L. R., Shanks G. D., Jacobus D. P., and Edstein M. D. (2016) Lead selection of a new aminomethylphenol, JPC-3210, for malaria treatment and prevention. Antimicrob. Agents Chemother. 60, 3115–3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heffernan DPJ Gavin D., Krasucki Peter E., Saionz Kurt W., Schiehser Guy A., Shieh Hong-Ming, Terpinski Jacek, Zhao Wenyi, Ager Arba L., Chavchich Marina, Birrell Geoffrey W., Shanks G. Dennis, Edstein Michael D. (2015) Identification of 2-aminomethylphenol antimalarials with potent in vitro and in vivo activity against plasmodium blood stages. 250th American Chemical Society National Meeting & Expositio, 16–20 August, 2015, Boston, MA [Google Scholar]
- 8. Muller I. B., and Hyde J. E. (2010) Antimalarial drugs: modes of action and mechanisms of parasite resistance. Future Microbiol. 5, 1857–1873 [DOI] [PubMed] [Google Scholar]
- 9. Fichera M. E., and Roos D. S. (1997) A plastid organelle as a drug target in apicomplexan parasites. Nature. 390, 407–409 [DOI] [PubMed] [Google Scholar]
- 10. Mc G. I., and Smith D. A. (1952) Daraprim in treatment of malaria; a study of its effects in falciparum and quartan infections in West Africa. Br. Med. J. 1, 730–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bopp S. E., Manary M. J., Bright A. T., Johnston G. L., Dharia N. V., Luna F. L., McCormack S., Plouffe D., McNamara C. W., Walker J. R., Fidock D. A., Denchi E. L., and Winzeler E. A. (2013) Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet. 9, e1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thomas J. A., Tan M. S. Y., Bisson C., Borg A., Umrekar T. R., Hackett F., Hale V. L., Vizcay-Barrena G., Fleck R. A., Snijders A. P., Saibil H. R., and Blackman M. J. (2018) A protease cascade regulates release of the human malaria parasite Plasmodium falciparum from host red blood cells. Nat. Microbiol. 3, 447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goodman C. D., Buchanan H. D., and McFadden G. I. (2017) Is the mitochondrion a good malaria drug target? Trends Parasitol. 33, 185–193 [DOI] [PubMed] [Google Scholar]
- 14. Clyde D. F., Miller R. M., DuPont H. L., and Hornick R. B. (1971) Antimalarial effects of tetracyclines in man. J. Trop. Med. Hyg. 74, 238–242 [PubMed] [Google Scholar]
- 15. Klonis N., Xie S. C., McCaw J. M., Crespo-Ortiz M. P., Zaloumis S. G., Simpson J. A., and Tilley L. (2013) Altered temporal response of malaria parasites determines differential sensitivity to artemisinin. Proc. Natl. Acad. Sci. U.S.A. 110, 5157–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bridgford J. L., Xie S. C., Cobbold S. A., Pasaje C. F. A., Herrmann S., Yang T., Gillett D. L., Dick L. R., and Ralph S. A., Dogovski C, Spillman NJ, Tilley L. (2018) Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat. Commun. 9, 3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Creek D. J., Chua H. H., Cobbold S. A., Nijagal B., MacRae J. I., Dickerman B. K., Gilson P. R., Ralph S. A., and McConville M. J. (2016) Metabolomics-based screening of the malaria box reveals both novel and established mechanisms of action. Antimicrob. Agents Chemother. 60, 6650–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cobbold S. A., Chua H. H., Nijagal B., Creek D. J., Ralph S. A., and McConville M. J. (2016) Metabolic dysregulation induced in Plasmodium falciparum by dihydroartemisinin and other front-line antimalarial drugs. J. Infect. Dis. 213, 276–286 [DOI] [PubMed] [Google Scholar]
- 19. Allman E. L., Painter H. J., Samra J., Carrasquilla M., and Llinas M. (2016) Metabolomic profiling of the malaria box reveals antimalarial target pathways. Antimicrob. Agents Chemother. 60, 6635–6649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trager W. (1994) Cultivation of malaria parasites. Methods Cell Biol. 45, 7–26 [DOI] [PubMed] [Google Scholar]
- 21. Giannangelo C., Stingelin L., Yang T., Tilley L., Charman S. A., and Creek D. J. (2018) Parasite-mediated degradation of synthetic ozonide antimalarials impacts in vitro antimalarial activity. Antimicrob. Agents Chemother. 62, e01566–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smilkstein M., Sriwilaijaroen N., Kelly J. X., Wilairat P., and Riscoe M. (2004) Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 48, 1803–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chong J., Soufan O., Li C., Caraus I., Li S., Bourque G., Wishart D. S., and Xia J. (2018) MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46, W486–W94, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N. T., Roth A., Bork P., Jensen L. J., and von Mering C. (2017) The STRING database in : quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Creek D. J., Jankevics A., Burgess K. E., Breitling R., and Barrett M. P. (2012) IDEOM: an Excel interface for analysis of LC-MS-based metabolomics data. Bioinformatics. 28, 1048–1049 [DOI] [PubMed] [Google Scholar]
- 27. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., Baker T. A., Brusniak M. Y., Paulse C., Creasy D., Flashner L., Kani K., Moulding C., Seymour S. L., Nuwaysir L. M., Lefebvre B., Kuhlmann F., Roark J., Rainer P., Detlev S., Hemenway T., Huhmer A., Langridge J., Connolly B., Chadick T., Holly K., Eckels J., Deutsch E. W., Moritz R. L., Katz J. E., Agus D. B., MacCoss M., Tabb D. L., and Mallick P. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Siddiqui G., Srivastava A., Russell A. S., and Creek D. J. (2017) Multi-omics based identification of specific biochemical changes associated with PfKelch13-mutant artemisinin-resistant Plasmodium falciparum. J. Infect. Dis. 215, 1435–1444 [DOI] [PubMed] [Google Scholar]
- 29. Tjhin E. T., Spry C., Sewell A. L., Hoegl A., Barnard L., Sexton A. E., Siddiqui G., Howieson V. M., Maier A. G., Creek D. J., Strauss E., Marquez R., Auclair K., and Saliba K. J. (2018) Mutations in the pantothenate kinase of Plasmodium falciparum confer diverse sensitivity profiles to antiplasmodial pantothenate analogues. PLoS Pathog. 14, e1006918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rappsilber J., Ishihama Y., and Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 31. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 32. Zhang J., Xin L., Shan B., Chen W., Xie M., Yuen D., Zhang W., Zhang Z., Lajoie G. A., and Ma B. (2012) PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 11, M111.010587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baelmans R., Deharo E., Munoz V., Sauvain M., and Ginsburg H. (2000) Experimental conditions for testing the inhibitory activity of chloroquine on the formation of beta-hematin. Exp. Parasitol. 96, 243–248 [DOI] [PubMed] [Google Scholar]
- 34. da Silva G. N., Maria N. R., Schuck D. C., Cruz L. N., de Moraes M. S., Nakabashi M., Graebin C., Gosmann G., Garcia C. R., and Gnoatto S. C. (2013) Two series of new semisynthetic triterpene derivatives: differences in anti-malarial activity, cytotoxicity and mechanism of action. Malar J. 12, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schmidt E. K., Clavarino G., Ceppi M., and Pierre P. (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods. 6, 275–277 [DOI] [PubMed] [Google Scholar]
- 36. Fontaine S. D., Spangler B., Gut J., Lauterwasser E. M., Rosenthal P. J., and Renslo A. R. (2015) Drug delivery to the malaria parasite using an arterolane-like scaffold. ChemMedChem. 10, 47–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Combrinck J. M., Fong K. Y., Gibhard L., Smith P. J., Wright D. W., and Egan T. J. (2015) Optimization of a multi-well colorimetric assay to determine haem species in Plasmodium falciparum in the presence of anti-malarials. Malar J. 14, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dogovski C., Xie S. C., Burgio G., Bridgford J., Mok S., McCaw J. M., Chotivanich K., Kenny S., Gnädig N., Straimer J., Bozdech Z., Fidock D. A., Simpson J. A., Dondorp A. M., Foote S., Klonis N., and Tilley L. (2015) Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol. 13, e1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fry M., and Pudney M. (1992) Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharmacol. 43, 1545–1553 [DOI] [PubMed] [Google Scholar]
- 40. Srivastava I. K., Rottenberg H., and Vaidya A. B. (1997) Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J. Biol. Chem. 272, 3961–3966 [DOI] [PubMed] [Google Scholar]
- 41. Zhang J., Krugliak M., and Ginsburg H. (1999) The fate of ferriprotorphyrin IX in malaria infected erythrocytes in conjunction with the mode of action of antimalarial drugs. Mol. Biochem. Parasitol. 99, 129–141 [DOI] [PubMed] [Google Scholar]
- 42. Sullivan D. J., Gluzman I. Y., Russell D. G., and Goldberg D. E. (1996) On the molecular mechanism of chloroquine's antimalarial action. Proc. Natl. Acad. Sci. U.S.A. 93, 11865–11870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gorka A. P., Alumasa J. N., Sherlach K. S., Jacobs L. M., Nickley K. B., Brower J. P., de Dios A. C., and Roepe P. D. (2013) Cytostatic versus cytocidal activities of chloroquine analogues and inhibition of hemozoin crystal growth. Antimicrob. Agents Chemother. 57, 356–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Klonis N., Crespo-Ortiz M. P., Bottova I., Abu-Bakar N., Kenny S., Rosenthal P. J., and Tilley L. (2011) Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc. Natl. Acad. Sci. U.S.A. 108, 11405–11410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gabay T., Krugliak M., Shalmiev G., and Ginsburg H. (1994) Inhibition by anti-malarial drugs of haemoglobin denaturation and iron release in acidified red blood cell lysates–a possible mechanism of their anti-malarial effect? Parasitology. 108, 371–81 [DOI] [PubMed] [Google Scholar]
- 46. Meshnick S. R., Yang Y. Z., Lima V., Kuypers F., Kamchonwongpaisan S., and Yuthavong Y. (1993) Iron-dependent free radical generation from the antimalarial agent artemisinin (qinghaosu). Antimicrob. Agents Chemother. 37, 1108–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ismail H. M., Barton V., Phanchana M., Charoensutthivarakul S., Wong M. H. L., Hemingway J., Biagini G. A., O'Neill P. M., and Ward S. A. (2016) Artemisinin activity-based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proc. Natl. Acad. Sci. U.S.A. 113, 2080–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Olafson K. N., Nguyen T. Q., Rimer J. D., and Vekilov P. G. (2017) Antimalarials inhibit hematin crystallization by unique drug–surface site interactions. Proc. Natl. Acad. Sci. U.S.A. 114, 7531–7536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Francis S. E., Sullivan D. J. Jr, and Goldberg D. E. (1997) Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu. Rev. Microbiol. 51, 97–123 [DOI] [PubMed] [Google Scholar]
- 50. Gluzman I. Y., Francis S. E., Oksman A., Smith C. E., Duffin K. L., and Goldberg D. E. (1994) Order and specificity of the Plasmodium falciparum hemoglobin degradation pathway. J. Clin. Invest. 93, 1602–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kolakovich K. A., Gluzman I. Y., Duffin K. L., and Goldberg D. E. (1997) Generation of hemoglobin peptides in the acidic digestive vacuole of Plasmodium falciparum implicates peptide transport in amino acid production. Mol. Biochem. Parasitol. 87, 123–135 [DOI] [PubMed] [Google Scholar]
- 52. Lazarus M. D., Schneider T. G., and Taraschi T. F. (2008) A new model for hemoglobin ingestion and transport by the human malaria parasite Plasmodium falciparum. J. Cell Sci. 121, 1937–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. van Schalkwyk D. A., Saliba K. J., Biagini G. A., Bray P. G., and Kirk K. (2013) Loss of pH control in Plasmodium falciparum parasites subjected to oxidative stress. PLoS ONE 8, e58933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Famin O., and Ginsburg H. (2002) Differential effects of 4-aminoquinoline-containing antimalarial drugs on hemoglobin digestion in Plasmodium falciparum-infected erythrocytes. Biochem. Pharmacol. 63, 393–398 [DOI] [PubMed] [Google Scholar]
- 55. Olafson K. N., Ketchum M. A., Rimer J. D., and Vekilov P. G. (2015) Mechanisms of hematin crystallization and inhibition by the antimalarial drug chloroquine. Proc. Natl. Acad. Sci. U.S.A. 112, 4946–4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dorn A., Vippagunta S. R., Matile H., Bubendorf A., Vennerstrom J. L., and Ridley R. G. (1998) A comparison and analysis of several ways to promote haematin (haem) polymerisation and an assessment of its initiation in vitro. Biochem. Pharmacol. 55, 737–747 [DOI] [PubMed] [Google Scholar]
- 57. Wong W., Bai X. C., Sleebs B. E., Triglia T., Brown A., Thompson J. K., Jackson K. E., Hanssen E., Marapana D. S., Fernandez I. S., Ralph S. A., Cowman A. F., Scheres S. H. W., and Baum J. (2017) Mefloquine targets the Plasmodium falciparum 80S ribosome to inhibit protein synthesis. Nat. Microbiol. 2, 17031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dziekan J. M., Yu H., Chen D., Dai L., Wirjanata G., Larsson A., Prabhu N., Sobota R. M., Bozdech Z., and Nordlund P. (2019) Identifying purine nucleoside phosphorylase as the target of quinine using cellular thermal shift assay. Sci. Transl. Med. 11, pii: eaau3174. [DOI] [PubMed] [Google Scholar]
- 59. Sheridan C. M., Garciam V. E., Ahyong V., and DeRisi J. L. (2018) The Plasmodium falciparum cytoplasmic translation apparatus: a promising therapeutic target not yet exploited by clinically approved anti-malarials. Malar. J. 17, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Price R. N., Uhlemann A. C., Brockman A., McGready R., Ashley E., Phaipun L., Patel R., Laing K., Looareesuwan S., White N. J., Nosten F., and Krishna S. (2004) Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 364, 438–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., Tiwary S., Cox J., Audain E., Walzer M., Jarnuczak A. F., Ternent T., Brazma A., and Vizcaíno J. A. (2019) The PRIDE database and related tools and resources in: improving support for quantification data. Nucleic Acids Res. 47, D442–D500 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics and Peptidomics spectrometry data and search results have been deposited to the ProteomeXchange Consortium via the PRIDE (61) partner repository with the dataset identifier PXD013539 and project name 'Multi-omics analysis demonstrates unique mode of action of a potent new antimalarial compound, JPC-3210, against Plasmodium falciparum'.
Metabolomics spectrometry data and search results have been deposited to the Metabolomics Workbench Consortium with project name 'Multi-omics analysis demonstrates unique mode of action of a potent new antimalarial compound, JPC-3210, against Plasmodium falciparum' and Data track identification (1528/DataTrackID1713).