

A LC-MS/MS-based quantitative proteomic analysis reveals that expression alterations in the irradiation-selected pancreatic cancer cells result in hyperactivation of the growth factor/cytokine signaling that promotes epithelial-mesenchymal plasticity and enhancement of DNA repair. Functional analysis on one of the most upregulated proteins, CD73, demonstrates that elevated CD73 confers acquired radioresistance by inactivating proapoptotic protein BAD via phosphorylation of BAD at Ser-136 and by maintaining the radioresistant pancreatic cancer cells in a mesenchymal state.
Keywords: Pancreatic cancer, apoptosis, SILAC, tandem mass spectrometry, cancer biology, CD73, radioresistance
Graphical Abstract

Highlights
CD73 is one of the most upregulated proteins in the radioresistant cells.
CD73 upregulation confers radioresistance and irradiation-induced apoptosis.
CD73 confers radioresistance potentially through inactivating protein BAD.
Elevated CD73 is required for maintaining the resistant cells in a mesenchymal state.
Abstract
The molecular mechanisms underlying exceptional radioresistance in pancreatic cancer remain elusive. In the present study, we established a stable radioresistant pancreatic cancer cell line MIA PaCa-2-R by exposing the parental MIA PaCa-2 cells to fractionated ionizing radiation (IR). Systematic proteomics and bioinformatics analysis of protein expression in MIA PaCa-2 and MIA PaCa-2-R cells revealed that several growth factor-/cytokine-mediated pathways, including the OSM/STAT3, PI3K/AKT, and MAPK/ERK pathways, were activated in the radioresistant cells, leading to inhibition of apoptosis and increased epithelial-mesenchymal plasticity. In addition, the radioresistant cells exhibited enhanced capabilities of DNA repair and antioxidant defense compared with the parental cells. We focused functional analysis on one of the most up-regulated proteins in the radioresistant cells, ecto-5′-nucleotidase (CD73), which is a cell surface protein that is overexpressed in different types of cancer. Ectopic overexpression of CD73 in the parental cells resulted in radioresistance and conferred resistance to IR-induced apoptosis. Knockdown of CD73 re-sensitized the radioresistant cells to IR and IR-induced apoptosis. The effect of CD73 on radioresistance and apoptosis is independent of the enzymatic activity of CD73. Further studies demonstrate that CD73 up-regulation promotes Ser-136 phosphorylation of the proapoptotic protein BAD and is required for maintaining the radioresistant cells in a mesenchymal state. Our findings suggest that expression alterations in the IR-selected pancreatic cancer cells result in hyperactivation of the growth factor/cytokine signaling that promotes epithelial-mesenchymal plasticity and enhancement of DNA repair. Our results also suggest that CD73, potentially a novel downstream factor of the enhanced growth factor/cytokine signaling, confers acquired radioresistance by inactivating proapoptotic protein BAD via phosphorylation of BAD at Ser-136 and by maintaining the radioresistant pancreatic cancer cells in a mesenchymal state.
Pancreatic cancer is an exceptionally aggressive type of cancer with the 5-year survival rate of ∼9%, the worst among all the major cancers (1). One of the factors contributing to the high fatality and high rate of recurrence is the poor responses of most pancreatic cancer patients to chemo- or chemoradiotherapy (2). This fact signifies a desperate need for novel therapeutic strategies to treat this fatal disease, a goal that requires a better understanding of the molecular mechanisms underlying the chemo- and chemoradioresistance.
Radiotherapy, which plays an important role in the treatment of nonmetastatic pancreatic cancer patients (3), is limited by development of radioresistance and metastatic progression of surviving cancer cells after radiotherapy (4–7). Radioresistance is caused by the activation of proliferative and pro-survival proteins/pathways (8). On one hand, tumor cells may escape DNA-damage-induced apoptosis by increasing their DNA repair abilities. On the other hand, clinically relevant ionizing radiation (IR)1 can trigger several interconnected cytoplasmic signaling pathways that promote cell proliferation, migration, invasion, and survival (5–7, 9). The ultimate outcome of IR exposure usually is the activation of transcription factors, which lead to expression of the proteins that favor cell proliferation, survival, inflammation, angiogenesis, or other features of cancer cells (7, 10). In the case of acquired radioresistance, those proteins are usually induced by IR over the course of radiotherapy (8). Most pancreatic patients are insensitive to radiotherapy and acquired radioresistance in radio- or chemoradiotherapy is common. The molecular mechanism underlying radioresistance in pancreatic cancer is not well understood.
Ecto-5′-nucleotidase (CD73) is a cell surface enzyme that converts extracellular AMP to adenosine. Because adenosine inhibits antitumor immune responses (11, 12), the immunosuppressive function of CD73 has been the subject of increased study (11, 13, 14). In addition to its role in immunosuppression, CD73 is also a signal and adhesive molecule and regulates apoptosis, growth, motility, and invasion of cancer cells (15, 16). The non-immunological function of CD73 can either be dependent or independent on its enzymatic activity (16–18). Interestingly, CD73 was identified as a key regulator of the epithelial-mesenchymal transition (EMT) program (19). Recently, CD73 expression was found to be associated with the EMT-like “invasive” phenotypes of melanoma (20), to regulate stemness in ovarian carcinoma (21), and to positively correlate with EMT transcription factors in breast cancer (14). In fact, CD73 is a well-established surface marker of mesenchymal stem cells (22). CD73 is overexpressed in multiple types of cancer (11, 23, 24). Importantly, CD73 overexpression on tumor cells correlates with tumor invasion, metastasis, and poor patient survival (12, 23–25).
To explore the potential molecular mechanism underlying the acquired radioresistance of pancreatic cancer in radiotherapy, we generated a stable pancreatic cancer cell line by exposing pancreatic cancer MIA PaCa-2 cells to clinically relevant fractionated irradiation. The resulting cell line showed strong resistance to IR. We then used subcellular fractionation and a SILAC (stable isotope labeling by amino acids in cell culture)-based quantitative proteomic method (26, 27) to identify the proteins that were differentially expressed between the parental cells and the radioresistant cells. The results demonstrated that several growth factor- or cytokine-mediated pathways, including the oncostatin M (OSM)/signal transducer and activator of transcription 3 (STAT3), phosphatidylinositide 3-kinase (PI3K)/α-serine/threonine-protein kinase (AKT), and mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathways, were activated in the radioresistant cells, leading to inhibition of apoptosis and a higher degree of epithelial-mesenchymal plasticity. In addition, the radioresistant cells exhibited enhanced capabilities of DNA repair and antioxidant defense compared with the parental cells. Functional analysis of one of the most up-regulated proteins in the radioresistant cells, CD73, revealed that CD73 up-regulation confers acquired radioresistance in the radioresistant cells and the action is, at least in part, through inactivating the proapoptotic protein BAD by causing BAD phosphorylation at Ser-136 and through maintaining the IR-selected cells in a mesenchymal state.
EXPERIMENTAL PROCEDURES
Cell Culture and Generation of Radioresistant Stable Cell Lines
MIA PaCa-2 cells were routinely maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS and 1% penicillin and streptomycin. Stable radioresistant cells were generated by exposing MIA PaCa-2 cells to a total dose of 60 Gy of γ-radiation over 6 weeks (2 Gy per day for 30 days and 5 times per week). The clones that survived the 60 Gy of IR treatment were expanded and maintained in the same manner as the parental cells. One of the representative radioresistant clones, designated MIA PaCa-2-R, was selected for further analysis.
Experimental Design and Statistical Rationale
For the characterization of the proteomic changes associated with acquired radioresistance in pancreatic cancer cells compared with the parental cells, we used a SILAC-based quantitative proteomic method (26, 27) to systematically compare protein expression between the parental cells and the isogenic, radioresistant cells that were generated through fractionated irradiation. Instead of performing biological replicates in the SILAC proteomic analysis, we fractionated cellular proteins into soluble and membrane fractions and analyzed them independently as an effort to increase the proteome coverage. In fact, protein expression changes in the two fractions exhibited strong correlation and largely validated each other (see Results).
Proteome Labeling and Cell Fractionation
The radioresistant MIA PaCa-2-R cells were cultured in unlabeled DMEM with 10% dialyzed FBS (light medium), and the parental MIA PaCa-2 cells were cultured in labeled DMEM medium containing arginine-13C6 and lysine-13C615N2 (heavy medium) with 10% dialyzed FBS. After isotopic labeling of the proteome, the cells were harvested and washed twice with cold phosphate buffered saline. The cells were then resuspended in 5 packed cell pellet volumes of hypotonic buffer (20 mm Tris, pH 7.5, 5 mm MgCl2, 5 mm CaCl2, 1 mm DTT, 1 mm EDTA, and protease inhibitors), stored on ice for 30 min, and lysed with ∼20 strokes of a tight-fitting pestle (type B) in Dounce homogenizer (Kontes Glass Co., Vineland, NJ) until 95% of cells were ruptured. The lysate was centrifuged at 1000 × g for 15 min at 4 °C. The supernatant was saved, and the pellet was sonicated in the hypotonic buffer on ice with a microtip at 12% power (Branson Digital Sonifier 450; 10 × 0.5 s pulses in each cycle for 3 cycles). The sonicated extract was combined with the supernatant obtained in the previous step, and the mixture was centrifuged at 100,000 × g for 90 min at 4 °C. The resulting supernatant was designated as soluble proteins. The pellet was dissolved in a modified RIPA buffer (50 mm Hepes, pH 7.5, 150 mm NaCl, 1.5 mm MgCl2, 1 mm EGTA, 10% glycerol, 1% Triton X-100, 1% SDS, and protease inhibitors), and the supernatant after a centrifugation was designated as membrane proteins. Equal amounts of soluble proteins and equal amounts of membrane proteins from the two cell populations were mixed, respectively, and fractionated by a 12% SDS-PAGE gel (Bio-Rad Mini gel; 7.2 cm x 8.6 cm) for LC-MS/MS analysis.
LC-MS/MS and Data Analysis
In-gel digestion, database search, and quantification of LC-MS/MS data were performed as described previously (28, 29). Specifically, the entire lane of the Coomassie brilliant blue-stained gel was cut into 10 slices, followed by in-gel digestion with trypsin (Promega, Madison, WI) (30, 31). The resulting peptides were analyzed by LC-MS/MS using a LTQ-Orbitrap XL mass spectrometer (ThermoFisher Scientific, Waltham, MA) operated in a data-dependent mode for tandem MS as described previously (28).
Raw data from LC-MS/MS analysis were processed by MaxQuant (version 1.6.2.10) (32, 33) with the built-in search engine Andromeda (34) and searched against a target-decoy (35) human SwissProt protein database (November 2018; 20,408 entries) retrieved from UniProt (www.uniprot.org). The false discovery rates (FDRs) for peptide and protein identification were both set to 1%. The MS error tolerance was set to 4.5 ppm, and the MS/MS error tolerance was set to 20 ppm. The minimum required peptide length was set to 7 amino acids, and a maximum of 2 missed cleavages was allowed. The variable modifications of acetylation at peptide N terminus and oxidation on methionine, and the fixed modification of cysteine carbamidomethylation were included. SILAC ratios (radioresistant/parental protein expression ratios; light/heavy ratios) were calculated using unique and razor peptides with a minimum ratio count of 2 (33).
The proteins that matched to the reverse database, identified only by site, and common contaminants were removed. The proteins that were identified by single peptide were also discarded. The remaining proteins were analyzed by Perseus (version 1.6.2.2) (36), and the Significance B score was obtained for the quantified proteins. The Significance B score is a significance score for protein SILAC ratios and identifies outliers based on the standard deviation of the protein SILAC ratios of the main distribution and signal intensity (32). A protein was a differentially expressed protein if (1) its ratio was significant by the Significance B score with p < 0.05, and (2) a log2 fold change was larger than 1.5 (representing an actual fold change of 2.82). The significantly changed soluble proteins and membrane proteins were combined and analyzed by the “Core Analysis” module of the software IPA (Ingenuity Pathway Analysis; Ingenuity® Systems, Redwood City, CA), a bioinformatics tool based on information from published literature (37). For the proteins that were identified in both soluble and membrane fractions (the shared proteins), an average of the soluble and membrane SILAC ratios was used for each protein in the IPA analysis (see Results).
Quantitative Real-Time PCR (qRT-PCR)
qRT-PCR was performed as described previously (28, 38). Briefly, total RNA was isolated using RNeasy Mini Kit according to manufacturer's instructions (Qiagen, Valencia, CA). One μg of RNA was reverse transcribed using an iScript cDNA Synthesis Kit following the manufacturer's protocol (Bio-Rad, Hercules, CA), and the resulting cDNA was used for qRT-PCR analysis of the human genes NT5E (protein, CD73), EGFR, OSM, PDGFB, TGFB1 (TGF-β1), TGFB2 (TGF-β2), TGFB3 (TGF-β3), FN1, CDH1 (E-cadherin), ZEB1, and PLAU (urokinase-type plasminogen activator). mRNA abundance was measured using SYBR Green Supermix (ThermoFisher Scientific) from three independent sample preparations. Relative gene expression was calculated according to the traditional 2−ΔΔCt method (39). The primer sequences used for qRT-PCR analysis in this study were listed in supplemental Table S8.
Western Blotting
Western blotting was performed as described previously (40). Specifically, cell lysates were extracted in a modified RIPA buffer (50 mm Hepes, pH7.5, 150 mm NaCl, 1.5 mm MgCl2, 1 mm EGTA, 10% Glycerol, 1% Triton X-100, 1% SDS and protease inhibitors; for detection of BAD phosphorylation, 1 mm sodium orthovanadate, 10 mm sodium fluoride, and 10 mm β-glycerophosphate were included as phosphatase inhibitors). The proteins were separated on a SDS-PAGE (60 μg protein/lane), transferred to a nitrocellulose membrane (Millipore, Billerica, MA), incubated with a primary/secondary antibody, and detected by enhanced chemiluminescence (Clarity Western blotting Substrate, Bio-Rad; or SuperSignal Western Femto Maximum Sensitivity Substrate, ThermoFisher Scientific). Anti-CD73 (Catalogue#: sc-32299), vimentin (sc-6260), and actin (sc-1616) antibodies were from Santa Cruz Biotechnology (Santa Cruz, Dallas, TX); anti-PARP antibody was from BD Biosciences (San Jose, CA, 551025); anti-BAD (phospho-Ser-136) antibody was from Cell Signaling (Danvers, MA, 4366); anti-E-cadherin was from Thermo Fisher Scientific (Waltham, MA, 20874-1-AP); anti-tubulin was from Millipore-Sigma (St. Louis, MO, T-9026).
Kinase Inhibitor Treatment
MIA PaCa-2 cells and MIA PaCa-2-R cells were mock-treated (DMSO) or treated with the PI3K inhibitor LY294002 (AdipoGen Life Sciences; final concentration, 25 μm), dual PI3K/mTOR inhibitor NVP-BEZ235 (Cayman Chemical; 0.2 μm), AKT inhibitor MK-2206 (Cayman Chemical; 1 μm), NVP-BEZ235+MK-2206 (0.2 μm NVP-BEZ235 + 1 μm MK-2206) or MK-2206+LY294002 (1 μm MK-2206 + 25 μm LY294002) for 24 h. After the treatments, lysates of the cells were analyzed by Western blotting with an antibody specifically recognizing phosphorylated BAD at Ser-136.
Stable Overexpression and Knockdown of CD73
For overexpression, the coding sequence of human CD73 was cloned into plasmid pcDNA3.1, and the resulting plasmid was transfected into MIA PaCa-2 cells using calcium phosphate. For knockdown, complementary oligonucleotides containing the cDNA sequences targeting human CD73 (5′-GCCACTAGCATCTCAAATA-3′ and 5′-CGCAACAATGGCACAATT-3′) (41) or the sequence targeting the green fluorescent protein (GFP) (42) were cloned into the retroviral vector pSuppressorRetro (Imgenex). Transfection of one of the two constructs for knockdown of CD73 or the negative control construct (targeting GFP), and selection of stable cells were performed as described previously (43).
Clonogenic Cell Survival Assay
The clonogenic cell survival assay was performed as described previously (43). Briefly, cells in 60-mm dishes were cultured for 6 h and then treated with 0, 2, 4, 6, or 8 Gy of γ-radiation. The cells were then cultured for 21 days with a medium change every 4 days. Colonies with more than 50 cells were counted, and surviving fractions were normalized with plating efficiencies. Triplicate plates were used for each irradiation dose.
Alkaline Comet Assay
The alkaline comet assay was performed according to the protocol described by Singh et al. (1988) (44). Specifically, MIA PaCa-2 cells and MIA PaCa-2-R cells were mock-treated or treated with 2 Gy or 4 Gy of X-rays, and then used for alkaline comet assay as described (44). Images of comets were acquired with Nikon Eclipse Ti2 Inverted Microscope. Fifty randomly selected comet images on each slide (n = 3) were analyzed using the software CASPLab (45).
Cell Invasion Assay
Cell invasion was measured by the invasion of cells through ECM gel-coated transwell inserts. Briefly, 1 × 105 cells in 200 μl serum-free DMEM were added to a 24-well transwell insert (8 μm pore size, VWR International, Radnor, PA) that was coated with 100 μl of 2 mg/ml of ECM gel (Millipore-Sigma, St. Louis, MO), and 600 μl of DMEM containing 10% FBS was added to the lower chamber. After 36 h incubation at 37 °C in 5% CO2, cells in the upper compartment were removed by cotton swab, and cells that had invaded to the lower surface of the insert were fixed with 4% paraformaldehyde, stained with 0.2% crystal violet, and counted under a light microscope.
Immunofluorescence and F-Actin Staining
MIA PaCa-2 and MIA PaCa-2-R cells were mock-irradiated or irradiated with 0.25 Gy of X-rays, stained with an antibody against γ-H2AX (phospho-ser139) (Millipore-Sigma, 05–636) as described by Paull et al. (2000) (46) 15 min after the mock or IR treatment, and images of γ-H2AX staining were acquired with Nikon Eclipse Ti2 Inverted Microscope. Foci of 150 randomly selected cells in 3 groups (50 cells/each) from 2 independent sample preparations for each type of cells were counted using the software ImageJ. F-actin was stained with phalloidin-iFluor488 (Abcam, Cambridge, MA, ab176753) according to the manufacturer's instruction, and images of F-actin staining were also acquired with Nikon Eclipse Ti2 Inverted Microscope.
Reactive Oxygen Species (ROS) Measurement
Cellular ROS concentrations were measured with a FACSAria Fusion flow cytometer (BD Biosciences, San Jose, CA) after incubating cells with the fluorescence probe DCFH-DA (ThermoFisher Scientific) as described previously (43). Briefly, the parental and the radioresistant cells were harvested, washed with cold phosphate-buffered saline, and incubated with 10 μm DCFH-DA for 45 min. Propidium iodide was added to each sample immediately prior to flow cytometry analysis to differentiate dead and live cells. The mean fluorescence intensity of the live cells (propidium iodide negative) was used to represent cellular ROS levels.
Statistical Analysis
The statistical analysis was performed using one-way ANOVA (PSI-PLOT, Pearl River, NY) with data expressed as mean ± S.E. Differences in mean values were considered significant when p was ≤ 0.05 unless otherwise specified.
RESULTS AND DISCUSSION
Identification and Quantification of Differentially Expressed Proteins in the Radioresistant Cells
In order to eliminate the complications caused by genetic background variations in proteomic analysis, we generated radioresistant pancreatic cancer isogenic cell lines for proteomic identification of differentially expressed proteins. We generated the radioresistant cell lines by exposing pancreatic cancer MIA PaCa-2 cells to 60 Gy of fractionated γ-radiation over 6 weeks, a protocol commonly used in clinical radiotherapy. In total 11 clones were recovered, and a representative clone, designated MIA PaCa-2-R, was selected for further investigation. Clonogenic cell survival assay revealed that MIA PaCa-2-R was strongly radioresistant (Fig. 1A). The radioresistance was stable and was retained after more than 30 passages.
Fig. 1.

Radiosensitivity and protein expression changes in the IR-selected pancreatic cancer cells. A, The clonogenic cell survival assay showing that the IR-selected MIA PaCa-2-R cells are strongly radioresistant. Values are the mean ± S.E. of three independent experiments. *p, < 0.05; ***, p < 0.001. B and C, Scatter plots showing the distribution of identified soluble proteins (B) and membrane proteins (C) according to fold change and signal intensity. The significantly up-regulated proteins and down-regulated proteins (p < 0.05 in the Significance B score and log2 fold change > 1.5) are in red and blue, respectively.
We then used a SILAC-based quantitative proteomic method (26, 27) to profile protein expression in the parental MIA PaCa-2 cells and the radioresistant MIA PaCa-2-R cells. To increase the coverage of the proteome being analyzed, we fractionated total cellular protein into soluble fraction and membrane fraction and analyzed them independently. We identified 1896 soluble proteins and 2211 membrane proteins by minimally 1 unique peptide. Of the identified proteins, we could quantify 1850 soluble proteins and 2150 membrane proteins by at least 2 peptides (supplemental Tables S1 and S2). SILAC ratios (L/H ratios: MIA PaCa-2-R/MIA PaCa-2) of the quantified proteins exhibited normal distributions for both soluble and membrane proteins, and the great majority of the log2 fold changes were around 0 (i.e. 1 in actual ratios) (supplemental Fig. S1). Among the quantified proteins, 1070 proteins were shared between the soluble fraction and membrane fraction (supplemental Fig. S2A), and the log2 SILAC ratios of the shared soluble proteins and membrane proteins were positively correlated (supplemental Fig. S2B). The correlation suggests that the soluble portion and membrane portion of a protein are regulated similarly in the radioresistant cells relative to the parental cells. The correlation also suggests that aqueous or membrane subcellular microenvironments do not obviously affect the stability of cellular proteins, at least in the context of the experiment conditions used in this study (i.e. irradiation responsive versus radioresistant pancreatic cancer cells). Cut-offs of p < 0.05 in the Significance B score and a log2 fold change of 1.5 (i.e. 2.82 in actual fold change) in SILAC ratios produced 98 soluble proteins and 130 membrane proteins that were considered differentially expressed between the parental cells and radioresistant cells (Figs. 1B and 1C, and supplemental Table S3). We have verified the expression of multiple selected proteins using Western blotting or qRT-PCR (Figs. 2 and 4, and supplemental Fig. S3). Among the differentially expressed proteins, 23 proteins were identified in both the soluble and membrane fractions. Although 58% of the quantified soluble proteins (1070 out of 1850) were identified in the membrane fraction and 50% of the quantified membrane proteins (1070 out of 2150) were identified in the soluble fraction (supplemental Fig. S2A), the percentage of the shared proteins in the differentially expressed proteins for soluble proteins was 23% (23 out of 98) and for membrane proteins was 18% (23 out of 130). One possible explanation for the lower percentages for the differentially expressed proteins compared with the total quantified proteins is that a portion of the total quantified common proteins that were identified in both fractions were the proteins that “spilled over” from one fraction to another during sample preparation. Because the abundant cellular proteins were more likely to contaminate the other fraction (i.e. from the soluble fraction to membrane fraction or vice versa) than the low abundant proteins, and most of the differentially regulated proteins were not among the abundant cellular proteins (Figs. 1B and 1C), the likelihood of contamination from the other fraction for the differentially regulated proteins was lower than that for the total quantified proteins.
Fig. 2.

The expression of EGFR, OSM, PDGF-BB, TGF-β and FN1 is up-regulated in the radioresistant cells. qRT-PCR analysis of EGFR (A), OSM (B), PDGF-BB (C), TGF-β (D), and FN1 (E) mRNAs in the indicated cells. Fold change was calculated with ΔΔCT method (39) and p was calculated using ΔCT values. Fold change values are the means ± S.E. of three separate sample preparations. **, p < 0.01 and ***, p < 0.001.
Fig. 4.

CD73 is up-regulated in the radioresistant cells. A, A representative mass spectrum (MS1 spectrum) of a lysine-containing peptide (GPLASQISGLYLPYK) derived from protein CD73. B, qRT-PCR analysis of CD73 mRNA in the indicated cells. Values are the mean ± S.E. of three separate sample preparations. *p, < 0.05. C, Western blot analysis of CD73 protein expression in the indicated cells. P, the parental cells; #1–8, different clones of the radioresistant cells (clone #6 was MIA PaCa-2-R). Actin was used as a loading control.
The Expression Changes Enhance the Mitogenic/Cytokine Signaling that Promotes Epithelial-Mesenchymal Plasticity and Inhibits Apoptosis in the Radioresistant Cells
We combined the differentially expressed soluble proteins and membrane proteins and analyzed them with the software IPA (37). For the 23 proteins that were identified in both soluble and membrane fractions, we used the average of soluble and membrane protein SILAC ratios for each protein in the IPA analysis. Because each of the shared proteins was consistently either up-regulated or downregulated in both soluble and membrane fractions in the radioresistant cells, and the differences in the ratios between the soluble proteins and membrane proteins were moderate (supplemental Table S3), using the averages of SILAC ratios for the shared proteins would not affect the outcomes of the IPA analysis. IPA analysis revealed that some well-known mitogenic factors and their related kinases [i.e. platelet-derived growth factor (PDGF-BB), epidermal growth factor receptor 2 (ERBB2), ERK, and AKT1], cytokines (e.g. OSM), transcription factor (e.g. STAT3), and the extracellular matrix protein fibronectin (FN1) that have been shown to promote cancer cell proliferation, stemness, and EMT were among the top activated upstream regulators in the radioresistant cells (Table I). Upstream regulators in IPA are defined as signaling molecules and transcription factors whose activities could explain the observed gene/protein expression changes. The coordinated activations of PDGF-BB, ERBB2, ERK, and AKT1 suggest that two major signaling pathways functioning downstream of the receptor tyrosine kinases, the PI3K/AKT and MAPK/ERK pathways (47), were activated in the radioresistant cells. Consistent with this conclusion, our proteomic analysis revealed that the expression of epidermal growth factor receptor (EGFR), a key upstream receptor tyrosine kinase of the PI3K/AKT and MAPK/ERK signaling, in MIA PaCa-2-R cells was up-regulated compared with the parental cells (supplemental Table S3) and we have confirmed the up-regulation with qRT-PCR (Fig. 2A). The PI3K/AKT pathway is a major pro-survival pathway (48), and activation of the PI3K/AKT and MAPK/ERK pathways is often associated with enhanced cell proliferation, invasion, and EMT (20, 49–51). The cytokine OSM has recently been shown to potently promote epithelial-mesenchymal plasticity, and acquisition of mesenchymal/cancer stem cell properties through activation of STAT3 and the transforming growth factor beta (TGF-β) signaling (52, 53). It is noteworthy that the PI3K/AKT and MAPK/ERK pathways can also function downstream of the OSM-mediated signaling and be activated by Janus kinases (JAKs) (54). FN1 is a known marker of mesenchymal cells (55). Although not being revealed by the IPA as an activated upstream factor, TGF-β, a potent inducer of EMT (14, 56, 57), was also likely activated in the radioresistant cells as the z-score for TGF-β1 in IPA analysis was very close to the cut-off z-score for activation (cut-off z-score for activation in IPA was 2; the z-score for TGF-β1 was 1.914; p value of overlap for TGF-β1 was 5.84E-07). We were not able to detect PDGF-BB, OSM, TGF-β, and FN1 with LC-MS/MS potentially because they are secreted proteins. However, we have checked the expression of these cytokines with qRT-PCR and confirmed that their mRNA levels were significantly up-regulated in the radioresistant cancer cells (Fig. 2). Interestingly, the mRNA levels of TGF-β2 (but not TGF-β1 and TGF-β3) were substantially higher in the radioresistant cancer cells than the parental cells (Fig. 2D). Multiple other upstream regulators were also affected in the radioresistant cells and the changes consistently favored cancer cell proliferation and survival. For example, microRNA Let-7 is known to repress several oncogenes including Ras, MYC and HMGA2 (58), and Let-7 was predicted to be inhibited in the radioresistant cells (Table I). Consistent with the activation or inhibition of these upstream regulators (PDGF-BB, ERBB2, ERK, AKT1, OSM, TGF-β, STAT3, FN1, and Let7), IPA analysis revealed that expression changes in the radioresistant cells would result in inhibition of apoptosis and activation of EMT.
Table I. Activated and inhibited upstream regulators in the radioresistant cells relative to parental cells. The activation states were predicted by Upstream Regulator Analysis in IPA.
| Molecule type | Upstream regulator | Activation state | Activation z-score | p value of overlap |
|---|---|---|---|---|
| Growth factor & kinase | PDGF-BB | Activated | 2.21 | 2.97E-06 |
| ERBB2 | Activated | 2.41 | 2.82E-06 | |
| ERK | Activated | 2.12 | 4.97E-04 | |
| AKT1 | Activated | 2.18 | 1.39E-01 | |
| Cytokine | OSM | Activated | 2.71 | 1.29E-02 |
| IL10 | Activated | 2.04 | 2.64E-01 | |
| CSF3 | Activated | 2.00 | 2.57E-02 | |
| IL1A | Activated | 2.20 | 6.39E-02 | |
| IL15 | Activated | 2.06 | 8.28E-03 | |
| Transcription factor | STAT3 | Activated | 2.21 | 9.83E-02 |
| TCF7L2 | Activated | 2.06 | 3.81E-01 | |
| HSF2 | Inhibited | -2.00 | 4.38E-04 | |
| TP63 | Activated | 2.57 | 3.12E-04 | |
| Estrogen receptor | Inhibited | -2.98 | 9.37E-05 | |
| microRNA | Let-7 | Inhibited | -2.23 | 4.11E-02 |
| miR-122 | Inhibited | -2.23 | 1.62E-03 | |
| Others | CG | Activated | 2.04 | 5.59E-02 |
| FN1 | Activated | 2.54 | 6.91E-07 |
The Expression Changes also Enhance the Capacities of DNA Repair and Antioxidant Defense in the Radioresistant Cells
Besides the cytoplasmic signaling, DNA repair is also known to affect radiosensitivity of tumor cells (5–7, 9). To evaluate whether DNA repair plays any role in the enhanced radioresistance in MIA PaCa-2-R cells, we mock-treated or treated MIA PaCa-2 cells and MIA PaCa-2-R cells with 2 Gy or 4 Gy of X-rays and measured DNA damage 15 min after the exposure using an alkaline comet assay. The results demonstrated that although mock-treated cells did not show significant difference in DNA damage, there were significantly fewer DNA breaks in MIA PaCa-2-R cells than in MIA PaCa-2 cells when the cells were treated with 2 Gy or 4 Gy of X-rays (Fig. 3A), suggesting that the radioresistant cells have developed an enhanced DNA repair capacity compared with the parental cells. To further check the levels of DNA damage in the cells, we measured γ-H2AX foci using immunofluorescence staining after the cells were mock-treated or treated with 0.25 Gy of X-rays. The results showed that the irradiated MIA PaCa-2-R cells contained significantly fewer γ-H2AX foci/cell than the irradiated MIA PaCa-2 (Fig. 3B), confirming the results from the comet assays.
Fig. 3.

DNA repair and anti-ROS defense capabilities are enhanced in the radioresistant cells. A and B, DNA repair capacity is increased in the radioresistant cells. A, MIA PaCa-2 cells and MIA PaCa-2-R cells were mock-treated or treated with 2 or 4 Gy of X-rays, and DNA damage was measured using the alkaline comet assay 15 min after the mock or X-ray exposure. Fifty randomly selected comet images on each slide (n = 3) were analyzed using the software CASPLab. B, The indicated cells were mock-treated or treated with 0.25 Gy of X-rays, and the cells were stained with an antibody against γ-H2AX (phospho-ser139) 15 min after the mock or X-ray exposure. γ-H2AX foci of 150 randomly selected cells in 3 groups (50 cells/each) from 2 independent sample preparations were counted using the software ImageJ. C, The defense against ROS is enhanced in the radioresistance cells. The indicated cells were mock-treated or treated 8 Gy of X-rays, and ROS levels in the cells were measured by monitoring DCFH oxidation with a flow cytometer 30 min after the exposure. Mean fluorescence intensity (MFI) was used to represent ROS levels in the cells. **, p < 0.01 and ***, p < 0.001.
IR is known to kill cancer cells partially through free radicals, ROS in particular (59, 60). To investigate whether ROS is associated with the enhanced DNA repair ability in MIA PaCa-2-R cells, we determined ROS levels in the cells with or without IR treatments. Both untreated and IR-treated MIA PaCa-2-R cells showed significantly lower concentrations of ROS than the counterparts of MIA PaCa-2 cells (Fig. 3C). Furthermore, whereas IR-treated MIA PaCa-2 cells contained significantly higher concentrations of ROS than the untreated cells, IR-treated MIA PaCa-2-R cells showed no significant differences in ROS levels between the untreated and IR-treated cells (Fig. 3C). We also determined ROS levels in other two radioresistant clones (clone #5 and #8, see Fig. 4C) that were selected through fractionated irradiation, and both clones exhibited a similar pattern as MIA PaCa-2-R (Fig. 3C). These results suggest that the radioresistant cells contain an enhanced ROS defense compared with the parental cells. To understand the ROS metabolism in the radioresistant cells, we searched a curated list of genes involved in ROS metabolism (61) against the quantified soluble and membrane proteins (supplemental Tables S1 and S2). The results showed that 11 ROS metabolism-related proteins were quantified, and several important antioxidant proteins [e.g. isocitrate dehydrogenase (IDH1), superoxide dismutase [Mn] (SOD2), major prion protein (PRNP)] (61) were moderately up-regulated (supplemental Table S9). At present, it is not clear whether these moderate changes contributed to the lower ROS levels and enhanced ROS defense in MIA PaCa-2-R cells. In short, the results suggest that the radioresistant cells have developed an enhanced DNA repair capacity compared with the parental cells and the enhancement is potentially the result of an more efficient ROS defense system in the cells. Because lower ROS levels and enhanced ROS defense relative to non-tumorigenic cells are associated with cancer stem cells (61), our results also suggest that the radioresistant cells might have gained properties of cancer stem-like cells.
CD73 is Up-regulated in the Radioresistant Cells
We focused on characterizing one of the top up-regulated proteins, CD73 (Fig. 4A and supplemental Table S3). CD73 is a cell surface enzyme that converts extracellular AMP to adenosine, which inhibits antitumor immune responses (11, 12). CD73 is also characterized by its non-enzymatic activity serving as a signal and adhesive molecule in cell-cell and cell-matrix interactions (15, 16). CD73 exhibits increased expression in different types of cancer (11, 23, 24), and the up-regulation is associated with cancer cell proliferation, migration, invasion (12, 23–25), EMT, and cancer cell stemness (14, 20–22, 24). The roles of CD73 in cancer cell radioresistance and the mechanism by which CD73 enhances cancer cell EMT and stemness remain poorly understood.
We first used qRT-PCR to quantify CD73 mRNA. The results demonstrated that CD73 mRNA level in MIA PaCa-2-R cells was ∼4-fold of that in the parental cells (Fig. 4B). We then used Western blotting to examine CD73 protein expression in the radioresistant clones that were recovered from the 60-Gy γ-radiation. Consistent with the proteomic (Fig. 4A) and qRT-PCR data (Fig. 4B), the results demonstrated that all the tested radioresistant clones exhibited up-regulation of CD73 expression compared with the parental cells (Fig. 4C; clone #6 was MIA PaCa-2-R), suggesting that up-regulation of CD73 might be a common feature of the fractionated irradiation-selected radioresistant pancreatic cancer cells. It has been shown that CD73 expression is induced by TGF-β1 (62) and MAPK signaling (20). Because these two signaling pathways were both activated in the radioresistant cells (Table I and Fig. 2), it is likely that CD73 up-regulation was a consequence of the hyperactivated mitogenic signaling (e.g. MAPK signaling) and/or cytokine signaling (e.g. TGF-β) in the radioresistant cells. Thus, CD73 is likely a downstream factor of those activated signaling pathways.
Manipulation of CD73 Expression Affects Radioresistance in Pancreatic Cancer Cells
To examine whether CD73 up-regulation in MIA PaCa-2-R cells was functionally involved in the radioresistance of the irradiation-selected cells (Fig. 1A), we generated a stable cell line that over-expressed CD73 in the parental MIA PaCa-2 cells (Fig. 5A). We then performed clonogenic cell survival assay to compare long-term survival of the stable cells that over-expressed CD73 (CD73over) and the empty vector transfected stable cells (Vector). The results showed that overexpression of CD73 significantly enhanced the survival of the CD73over cells compared with the control cells at higher doses of γ-radiation (i.e. 4, 6, and 8 Gy) (Fig. 5B), suggesting that overexpression of CD73 confers radioresistance in pancreatic cancer cells.
Fig. 5.
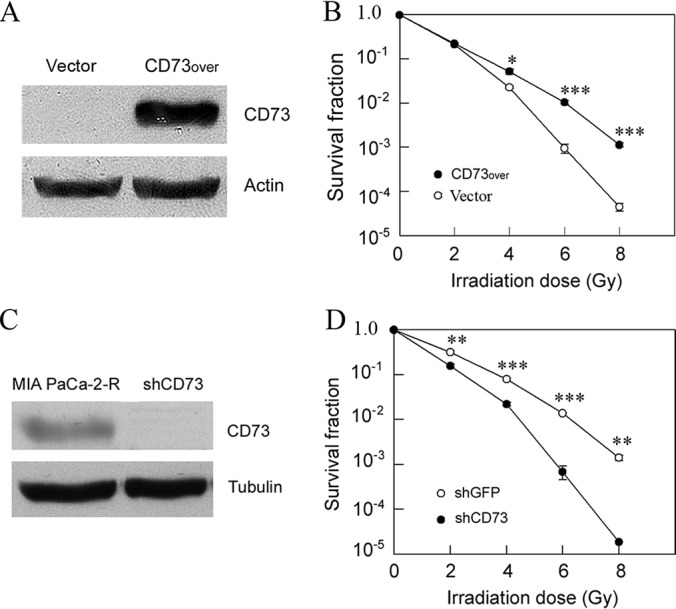
CD73 enhances radioresistance. A, Western blot analysis of CD73 expression in vector-transformed stable cells (Vector) and the stable cells that overexpressed CD73 in MIA PaCa-2 cells (CD73over). B, Clonogenic cell survival assay of the indicated cells. C, Western blot analysis of CD73 expression in MIA PaCa-2-R cells and the cells in which CD73 was knocked down in MIA PaCa-2-R cells (shCD73). D, Clonogenic cell survival assay of control cells (shGFP) and the shCD73 cells. Actin and tubulin were used as loading controls. Values in B and D are the mean ± S.E. of three separate sample preparations. *p, < 0.05; **p, < 0.01; ***p, < 0.001.
To further confirm the role of CD73 in radioresistance, we generated a stable cell line with silenced expression of CD73 in MIA PaCa-2-R cells using shRNA (Fig. 5C). Clonogenic cell survival assay demonstrates that knockdown of CD73 significantly reduced the survival of CD73-knockdown cells (shCD73) compared with control cells (shGFP) (Fig. 5D). The results further support the notion that CD73 up-regulation confers radioresistance in pancreatic cancer cells.
Up-regulation of CD73 Enhances Resistance to IR-Induced Apoptosis and Promotes BAD Phosphorylation at Ser-136
The susceptibility of apoptosis to IR plays an important role in determining radiosensitivity of cancer cells (63, 64). Thus, we assessed apoptosis by determining the cleavage of death substrate PARP after the cells were exposed to IR. Western blot analysis demonstrates that MIA PaCa-2-R cells exhibited decreased cleavage of PARP after the cells were exposed to 5 Gy of γ-radiation compared with the parental cells (Fig. 6A). Overexpression of CD73 in MIA PaCa-2 cells (Fig. 5A) enhanced the resistance of the CD73-overexpressing cells to IR-induced apoptosis (Fig. 6B). Knockdown of CD73 expression in MIA PaCa-2-R cells (Fig. 5C) re-sensitized the radioresistant cells to IR (Fig. 6C). These data demonstrate that elevated CD73 protein confers resistance to IR-induced apoptosis in pancreatic cancer cells.
Fig. 6.

CD73 enhances resistance to IR-induced apoptosis and promotes BAD phosphorylation at Ser-136. A, The radioresistant cells are more resistant to IR-induced apoptosis than the parental cells. B, Ectopic overexpression of CD73 enhances resistance to IR-induced apoptosis. C, Knockdown of CD73 re-sensitizes the radioresistant cells to IR-induced apoptosis. The indicated cells were treated with the indicated doses of γ-radiation. After 48 h of recovery culture, cell lysates were analyzed by Western blotting with the indicated antibodies. D, Western blot analysis of BAD Ser-136 phosphorylation in the indicated cells. shGFP cells and shCD73 cells were derived from MIA PaCa-2-R cells. E, Effect of various kinase inhibitors on BAD Ser-136 phosphorylation. MIA PaCa-2 and MIA PaCa-2-R cells were mock-treated (lanes #1 and 7) or treated with the PI3K inhibitor LY294002 (#2 and 8), dual PI3K/mTOR inhibitor NVP-BEZ235 (#3 and 9), AKT inhibitor MK-2206 (#4 and 10), NVP-BEZ235+MK-2206 (#5 and 11) or MK-2206+LY294002 (#6 and 12) for 24 h, and lysates of the cells were analyzed by Western blotting using the indicated antibodies. Vector, CD73over, shGFP, and shCD73 cells, see Fig. 5 legend. cPARP, 85 kDa fragment of cleaved PARP. Actin and tubulin were used as loading controls.
Pro-apoptotic protein BAD plays an important role in regulating IR-induced apoptosis (65). Phosphorylation of BAD at Ser-136 creates a motif for an interaction with 14–3-3 proteins, which sequester the proapoptotic BAD from antiapoptotic Bcl-2 and Bcl-XL and thus promotes survival (66, 67). We first tested whether BAD Ser-136 phosphorylation was involved in CD73-mediated radioresistance in MIA PaCa-2-R cells. The results demonstrated that MIA PaCa-2-R cells contained significantly higher levels of BAD phosphorylation at Ser-136 than the parental cells (Fig. 6D). BAD Ser-136 could be phosphorylated by AKT (68) or the 70-kDa ribosomal protein S6 kinase (p70S6K) (69). To elucidate which kinase, AKT or p70S6K, plays a major role in phosphorylating BAD at Ser-136, we treated MIA PaCa-2 and MIA PaCa-2-R cells with different kinase inhibitors and determined BAD Ser-136 phosphorylation. The AKT inhibitor MK-2206, PI3K inhibitor LY294002 or a combination of them (MK-2206 + LY294002) had no obvious effect on BAD Ser-136 phosphorylation in MIA PaCa-2-R cells (Fig. 6E, compare lanes 10, 8, and 12 with lane 7), implying that AKT may not be the principal kinase directly responsible for Ser-136 phosphorylation of BAD in MIA PaCa-2-R cells. However, the observation that the dual PI3K/mTOR inhibitor NVP-BEZ235 also showed no significant effect on BAD Ser-136 phosphorylation (Fig. 6E, compare lane 9 with lane 7) and that a combination of NVP-BEZ235 and MK-2206 significantly decreased the phosphorylation (Fig. 6E, compare lane 11 with lane 7) suggests that AKT is involved in BAD Ser-136 phosphorylation. Given the complex interplays of multiple factors in this signaling system (70), further specific studies will be needed to understand the signaling pathway(s) and specific kinase(s) that are directly responsible for the inactivation of BAD in the radioresistant MIA PaCa-2-R cells. The effect of these kinase inhibitors on BAD Ser-136 phosphorylation in the parental MIA PaCa-2 cells could not be accurately assessed because of weak Western blotting signals (Fig. 6E, lanes 1–6). Strikingly, knockdown of CD73 (shCD73) in MIA PaCa-2-R cells reduced BAD Ser-136 phosphorylation to the level comparable to that in the parental cells (Fig. 6D, compare lane 4 with lanes 3, 2, and 1), demonstrating that BAD Ser-136 phosphorylation in the radioresistant MIA PaCa-2-R cells depends on the up-regulated CD73. In short, these results suggest that elevated CD73 enhances the resistance of the radioresistant cancer cells to IR-induced apoptosis, at least in part, through promoting BAD phosphorylation at Ser-136.
CD73-Mediated Radioresistance is Independent of the Enzymatic Activity of CD73
CD73 can affect different aspects of cancer cells through enzyme-dependent or enzyme-independent mechanisms (16–18). To examine whether the enzymatic activity of CD73 is important for its effect on radioresistance in pancreatic cancer cells, we generated a stable cell line that over-expressed an enzyme-inactive mutant human CD73 in MIA PaCa-2 cells. The mutant CD73 was generated through substituting histidine residues 92 and 194 with alanine via site-directed mutagenesis (71). Clonogenic cell survival assay demonstrates that overexpression of wild-type CD73 (CD73over) or the enzyme-inactive mutant CD73 (mCD73over) (Fig. 7A) both significantly increased the survival fractions when the cells were treated at higher doses of γ-radiation (i.e. 6 and 8 Gy). However, there was no significant difference between the CD73over cells and the mCD73over cells (Fig. 7B), suggesting that the enzymatic activity of CD73 is not involved in conferring radioresistance in pancreatic cancer cells. To further test this notion, we treated the control cells, the CD73over cells, and the mCD73over cells with 5 Gy of γ-radiation and measured the PARP cleavage. Consistent with the results from clonogenic cell survival assays (Fig. 7B), both CD73over cells and the mCD73over cells exhibited elevated apoptotic resistance to γ-radiation compared with the control cells (Vector), but there was no difference in susceptibility to IR-induced apoptosis between the two types of cells (Fig. 7C). We also determined BAD Ser-136 phosphorylation in the cells, and no obvious difference in BAD Ser-136 phosphorylation between CD73over cells and the mCD73over cells was observed (Fig. 7D). These results suggest that CD73-mediated radioresistance and the resistance to IR-induced apoptosis in the pancreatic cancer cells are independent of the enzymatic activity of CD73.
Fig. 7.
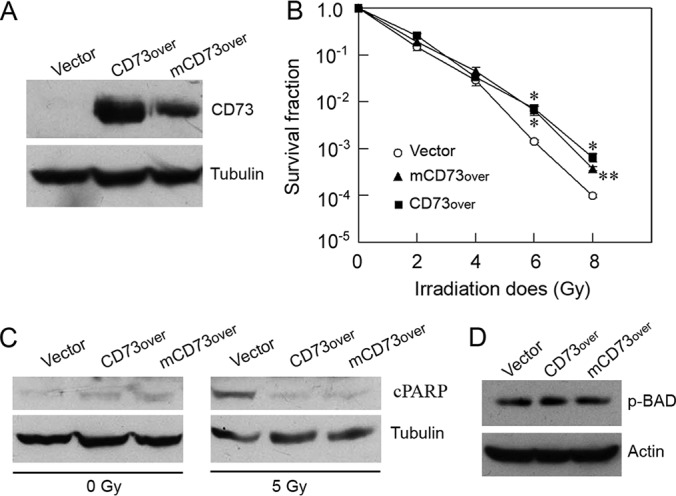
CD73-mediated radioresistance and apoptosis resistance are independent of the enzymatic activity of CD73. A, Western blot analysis of CD73 expression in vector-transformed stable cells (Vector), the stable cells that overexpressed wild-type CD73 (CD73over) or an enzyme-inactive mutant CD73 (mCD73over). B, Clonogenic cell survival assay of the indicated cells. Values are the mean ± S.E. of three separate sample preparations. * and ** indicate the statistical differences between the Vector cells and the CD73over cells or between the Vector cells and the mCD73over cells. *p, < 0.05; **p, < 0.01. C, Western blot analysis of PARP cleavage (cPARP) in the indicated cells, which were treated as described in Fig. 6 legend. D, Western blot analysis of BAD Ser-136 phosphorylation in the indicated cells. Actin and tubulin were used as loading controls.
CD73 Up-regulation is Required for Maintaining the Radioresistant Cells in a Mesenchymal State
We observed dramatic differences in the morphology of the parental MIA PaCa-2 cells, radioresistant MIA PaCa-2-R cells, and CD73-knockdown cells (CD73 was silenced in MIA PaCa-2-R cells). Most MIA PaCa-2 cells firmly attached to the bottom of tissue culture dish and did not form large clumps of cells even when the cells reached full confluency (Fig. 8A, left panel). Although most MIA PaCa-2-R cells initially attached to the bottom of culture dish, the cells increasingly aggregated to form large clumps of cells even though the attached cells have not reached confluency; the attached MIA PaCa-2-R cells had a mesenchymal spindle-shaped morphology, whereas the cells in clumps were rounded (Fig. 8A, middle panel). The cells in large clumps were not dead cells because they could not be stained by trypan blue (data not shown). In fact, when the cells in clumps were isolated and plated in a new culture dish, they could divide, with some of them regaining the ability to attach to the bottom of the culture dish (supplemental Fig. S4A). When CD73 was silenced, CD73-knockdown cells developed a more cobblestone-like morphology, which is typical for epithelial cells, and did not form clumps of cells even after they reached confluency (Fig. 8A, right panel). The images of F-actin staining of the cells with fluorescence-labeled phalloidin confirmed that the rounded MIA PaCa-2-R cells were prone to formation of cell clumps and the cells with CD73 knockdown have a more cuboidal, epithelial morphology (Fig. 8B). We have checked additional radioresistant clones (clones #5 and #8, see Fig. 4C) and they showed similar F-actin staining as MIA PaCa-2-R cells (supplemental Fig. S4B). In contrast to knockdown of CD73, overexpression of CD73 in MIA PaCa-2 cells did not change the morphology of the cells in cell culture (data not shown).
Fig. 8.

Role of CD73 in maintaining the radioresistant cells in a mesenchymal state. A, Morphology of the indicated cells. Images of live cells in culture were taken by Nikon Eclipse Ti2 Inverted Microscope. B, Staining of F-actin in the indicated cells with fluorescence-labeled phalloidin. The indicated cells in coverslips were fixed, stained with phalloidin-iFluor488 (green) and DAPI (blue), and images of the stained cells were acquired by Nikon Eclipse Ti2 Inverted Microscope. C, D and G, qRT-PCR (C and G) or Western blotting (D) analysis of E-cadherin mRNA/protein in the indicated cells. E, Western blot analysis of vimentin in the indicated cells. F, qRT-PCR analysis of ZEB1 mRNA in the indicated cells. H, Invasion assay for the indicated cells. Values in C, F, and G are the mean ± S.E. of three separate sample preparations. Values in H are the mean ± S.E. of six separate sample preparations. shCD73 and shGFP cells, see Fig. 5 legend. Tubulin and actin in D and E were loading controls. **p, < 0.01, and ***p, < 0.001. Note: Results in E were from the same Western blot analysis as shown in Fig. 6D; the same Western blotting membrane was stripped and reprobed with an anti-vimentin antibody.
MIA PaCa-2 cells are known to exhibit characteristics of both epithelial cells and mesenchymal cells (72, 73). The morphological changes in MIA PaCa-2-R cells compared with MIA PaCa-2 cells implied that the radioresistant cells might have shed some of their epithelial features and gained more mesenchymal properties. To test this possibility, we determined the expression of the epithelial cell marker E-cadherin, mesenchymal cell marker vimentin (56, 57), and a key EMT transcription factor, ZEB1, that appears to play a crucial role in EMT activation and stemness maintenance in pancreatic cancer cell (53, 55, 74, 75), by qRT-PCR and/or Western blotting. The mRNA level of E-cadherin was significantly lower in MIA PaCa-2-R cells than in MIA PaCa-2 cells (Fig. 8C). Consistent with this, we detected a truncated E-cadherin at ∼80 kDa in MIA PaCa-2 cells but not in MIA PaCa-2-R cells using Western blotting (Fig. 8D, compare lane 2 with lane 1), suggesting that the expression of E-cadherin is decreased in MIA PaCa-2-R cells compared with the parental cells. The protein expression of vimentin was increased in MIA PaCa-2-R cells compared with the parental cells (Fig. 8E, compare lane 2 with lane 1). ZEB1 is crucial for both EMT activation and stemness maintenance in pancreatic cancer cells (74), and may link EMT and stemness maintenance in pancreatic cancer (55, 74). In addition, it has recently been shown that ZEB1 plays a unique role in the OSM-induced EMT in pancreatic cancer (53). The ZEB1 mRNA levels were significantly higher in the radioresistant cells than in the parental cells (Fig. 8F). Furthermore, our IPA analysis and proteomic data have respectively shown that the mesenchymal cell marker FN1 (55) and EMT-regulating protein urokinase-type plasminogen activator (gene name, PLAU) (7) were up-regulated in the radioresistant cells compared with the parental cells (Table I and supplemental Table S3). We have verified the expression of both genes with qPCR (Fig. 2 and supplemental Fig. S3). These results suggest that the IR-selected radioresistant MIA PaCa-2-R cells have shed epithelial features and gained the properties that are normally associated with mesenchymal/cancer stem cells. E-cadherin and ZEB1 proteins were not detected in our proteomic analysis potentially because of low levels of expression (Fig. 8D) (76). Vimentin protein was detected in both the soluble and membrane fractions, and quantification of the LC-MS/MS data showed that protein expression of vimentin in MIA PaCa-2-R cells was moderately higher than in MIA PaCa-2 cells (1.5-fold in the soluble fraction and 1.6-fold in the membrane fraction) (supplemental Tables S1 and S2).
Knockdown of CD73 in MIA PaCa-2-R cells resulted in a >700-fold increase in E-cadherin mRNA levels (Fig. 8G) and a substantial increase in E-cadherin protein levels (Fig. 8D, compare lane 4 with lanes 2 and 3). Strikingly, CD73 knockdown caused disappearance of an E-cadherin protein band at ∼40 kDa in Western blotting, suggesting that CD73 knockdown prevented truncation of E-cadherin in the cells. This conclusion was supported by the observation that the great majority of the E-cadherin protein in the CD73-silenced cells was the full-length version of the protein at ∼120 kDa (Fig. 8D). These results demonstrate that CD73 can modulate E-cadherin protein levels in pancreatic cancer cells through affecting both E-cadherin mRNA transcription/mRNA stability and posttranslational modification of E-cadherin protein. Concomitantly, knockdown of CD73 decreased protein expression of vimentin in the cells (Fig. 8E, compare lane 4 with lanes 2 and 3). In contrast to knockdown, overexpression of CD73 in MIA PaCa-2 cells had no effect on the expression of E-cadherin and vimentin in the cells (data not shown), consistent with the observation that overexpression of CD73 in MIA PaCa-2 cells did not change the morphology of the cells. The expression results coincide with the morphological changes in CD73-knockdown cells and MIA PaCa-2-R cells (Fig. 8A and 8B) as well as the situation for the CD73-overexpressed cells. To further assess the role of CD73 in the maintenance of a mesenchymal phenotype of the radioresistant cells, we examined cell invasion, a crucial trait of the mesenchymal-like cells developed through EMT, using a cell invasion assay. The results demonstrated that knockdown of CD73 significantly reduced the number of the radioresistant cells that invaded through the ECM gel (Fig. 8H). Collectively, these results strongly suggest that the elevated CD73 in the radioresistant cells is required for maintaining the cells in a mesenchymal state. The role of CD73 in maintaining the radioresistant cells in a mesenchymal/invasive state is relevant to its role in conferring radioresistance because it has been well established that the mesenchymal cells developed through EMT and the cancer cells with cancer stem-like cell properties usually are resistant to chemo- or radiotherapy (7, 77–79).
CD73 is a surface marker of mesenchymal stem cells (22), and its expression has been associated with the invasive phenotypes of melanoma (20) and stemness in ovarian carcinoma (21). In the present study, we demonstrate that CD73 up-regulation was necessary and enough to maintain the radioresistant cells in a mesenchymal state. However, the observations that forced overexpression of CD73 in MIA PaCa-2 cells did not alter the expression of E-cadherin and vimentin and the phenotype of the cells demonstrate that CD73 up-regulation alone is not sufficient to drive pancreatic cancer cells toward a more mesenchymal state. It could be that the development of a more invasive/mesenchymal state in pancreatic cancer cells over the course of fractionated irradiation requires changes in at least two critical proteins/pathways and a single change is not enough to trigger the change. After a more invasive/mesenchymal state is developed, perhaps some of those changes are required for continued maintenance of the invasive/mesenchymal state in the cells. The results from this study demonstrate that up-regulation of CD73 expression may be one of the critical changes that drives the development of an invasive/mesenchymal state over the course of fractionated irradiation and is one of the determining factors that maintain the invasive/mesenchymal state in the pancreatic cancer cells that have acquired radioresistance. It is also possible that CD73 is not involved in driving the development of an invasive/mesenchymal state during fractionated irradiation but evolves to become one of the pillars that support cellular programs maintaining the radioresistant cells in the invasive/mesenchymal state.
In summary, by using a quantitative proteomic approach coupled with bioinformatics analysis, we have systematically compared protein expression between a parental pancreatic cancer cell line and a radioresistant cell line that was generated by fractionated irradiation. Our results suggest that protein expression alterations in the IR-selected radioresistant cells relative to the parental cells cause hyperactivation of several interconnected signaling pathways involving OSM/STAT3, PI3K/AKT, MAPK/ERK and TGF-β, and enhancement of DNA repair capability in the radioresistant cells. Functional analysis of CD73, a likely downstream factor of the hyperactivated signaling pathways, demonstrates that the up-regulated CD73 confers acquired radioresistance through inhibiting IR-induced apoptosis and maintaining the radioresistant pancreatic cancer cells in a mesenchymal/invasive state.
DATA AVAILABILTY
The MS proteomic data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRoteomics IDEntifications (PRIDE) partner repository with the data set identifier PXD012922.
Supplementary Material
Footnotes
* This work was supported in part by NIH grant R03 CA169692 and a grant from the Arkansas Biosciences Institute (to Du). We thank the University of Arkansas Honors College Research grant (to Nguyen and Sonney) and State of Arkansas Undergraduate Research Fellowship (to Nguyen) for providing funding for undergraduate research. Sicairos is a recipient of the University of Arkansas Doctoral Academy Fellowship. The license for the IPA software was purchased through the Chancellor's Innovation and Collaboration Fund at the University of Arkansas, and the Nikon Eclipse Ti2 Inverted Microscope used in this study was partially funded by the Arkansas Biosciences Institute. The authors declare that they have no conflicts of interest with the contents of this article.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- IR
- ionizing radiation
- AKT
- α-serine/threonine-protein kinase
- BAD
- Bcl-2 antagonist of cell death
- CD73/NT5E
- 5′-nucleotidase/ecto-5′-nucleotidase
- EGFR
- epidermal growth factor receptor
- EMT
- epithelial-mesenchymal transition
- ERK
- extracellular signal-regulated kinase
- FN1
- fibronectin
- OSM
- oncostatin M
- MAPK
- mitogen-activated protein kinase
- PDGF
- platelet-derived growth factor
- PI3K
- phosphatidylinositide 3-kinase
- qRT-PCR
- quantitative real-time PCR
- ROS
- reactive oxygen species
- STAT3
- signal transducer and activator of transcription 3
- TGF-β
- transforming growth factor beta.
REFERENCES
- 1. Siegel R. L., Miller K. D., and Jemal A. (2019) Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34 [DOI] [PubMed] [Google Scholar]
- 2. Lohr J. M. (2007) Medical treatment of pancreatic cancer. Expert Rev. Anticancer Ther. 7, 533–544 [DOI] [PubMed] [Google Scholar]
- 3. Tsai J. Y., Iannitti D. A., and Safran H. (2003) Combined modality therapy for pancreatic cancer. Semin. Oncol. 30, 71–79 [DOI] [PubMed] [Google Scholar]
- 4. Marie-Egyptienne D. T., Lohse I., and Hill R. P. (2013) Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: potential role of hypoxia. Cancer Lett. 341, 63–72 [DOI] [PubMed] [Google Scholar]
- 5. Ahmed K. M., and Li J. J. (2008) NF-kappa B-mediated adaptive resistance to ionizing radiation. Free Radic. Biol. Med. 44, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boss M. K., Bristow R., and Dewhirst M. W. (2014) Linking the history of radiation biology to the hallmarks of cancer. Radiat. Res. 181, 561–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee S. Y., Jeong E. K., Ju M. K., Jeon H. M., Kim M. Y., Kim C. H., Park H. G., Han S. I., and Kang H. S. (2017) Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 16, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deorukhkar A., and Krishnan S. (2010) Targeting inflammatory pathways for tumor radiosensitization. Biochem. Pharmacol. 80, 1904–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hidalgo M. (2012) New insights into pancreatic cancer biology. Ann. Oncol. 23, 135–138 [DOI] [PubMed] [Google Scholar]
- 10. Criswell T., Leskov K., Miyamoto S., Luo G., and Boothman D. A. (2003) Transcription factors activated in mammalian cells after clinically relevant doses of ionizing radiation. Oncogene 22, 5813–5827 [DOI] [PubMed] [Google Scholar]
- 11. Jin D., Fan J., Wang L., Thompson L. F., Liu A., Daniel B. J., Shin T., Curiel T. J., and Zhang B. (2010) CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 70, 2245–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stagg J., and Smyth M. J. (2010) Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 29, 5346–5358 [DOI] [PubMed] [Google Scholar]
- 13. Stagg J., Divisekera U., McLaughlin N., Sharkey J., Pommey S., Denoyer D., Dwyer K. M., and Smyth M. J. (2010) Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. U.S.A. 107, 1547–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Turcotte M., Allard D., Mittal D., Bareche Y., Buisseret L., Jose V., Pommey S., Delisle V., Loi S., Joensuu H., Kellokumpu-Lehtinen P. L., Sotiriou C., Smyth M. J., and Stagg J. (2017) CD73 promotes resistance to HER2/ErbB2 antibody therapy. Cancer Res. 77, 5652–5663 [DOI] [PubMed] [Google Scholar]
- 15. Zhi X., Chen S., Zhou P., Shao Z., Wang L., Ou Z., and Yin L. (2007) RNA interference of ecto-5′-nucleotidase (CD73) inhibits human breast cancer cell growth and invasion. Clin. Exp. Metastasis 24, 439–448 [DOI] [PubMed] [Google Scholar]
- 16. Mikhailov A., Sokolovskaya A., Yegutkin G. G., Amdahl H., West A., Yagita H., Lahesmaa R., Thompson L. F., Jalkanen S., Blokhin D., and Eriksson J. E. (2008) CD73 participates in cellular multiresistance program and protects against TRAIL-induced apoptosis. J. Immunol. 181, 464–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sadej R., and Skladanowski A. C. (2012) Dual, enzymatic and non-enzymatic, function of ecto-5′-nucleotidase (eN, CD73) in migration and invasion of A375 melanoma cells. Acta Biochim. Pol. 59, 647–652 [PubMed] [Google Scholar]
- 18. Wu R., Chen Y., Li F., Li W., Zhou H., Yang Y., and Pei Z. (2016) Effects of CD73 on human colorectal cancer cell growth in vivo and in vitro. Oncol. Rep. 35, 1750–1756 [DOI] [PubMed] [Google Scholar]
- 19. Xiong L., Wen Y., Miao X., and Yang Z. (2014) NT5E and FcGBP as key regulators of TGF-1-induced epithelial-mesenchymal transition (EMT) are associated with tumor progression and survival of patients with gallbladder cancer. Cell Tissue Res. 355, 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reinhardt J., Landsberg J., Schmid-Burgk J. L., Ramis B. B., Bald T., Glodde N., Lopez-Ramos D., Young A., Ngiow S. F., Nettersheim D., Schorle H., Quast T., Kolanus W., Schadendorf D., Long G. V., Madore J., Scolyer R. A., Ribas A., Smyth M. J., Tumeh P. C., Tuting T., and Holzel M. (2017) MAPK signaling and inflammation link melanoma phenotype switching to induction of CD73 during immunotherapy. Cancer Res. 77, 4697–4709 [DOI] [PubMed] [Google Scholar]
- 21. Lupia M., Angiolini F., Bertalot G., Freddi S., Sachsenmeier K. F., Chisci E., Kutryb-Zajac B., Confalonieri S., Smolenski R. T., Giovannoni R., Colombo N., Bianchi F., and Cavallaro U. (2018) CD73 regulates stemness and epithelial-mesenchymal transition in ovarian cancer-initiating cells. Stem Cell Reports 10, 1412–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Calloni R., Cordero E. A., Henriques J. A., and Bonatto D. (2013) Reviewing and updating the major molecular markers for stem cells. Stem Cells Dev. 22, 1455–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sadej R., Spychala J., and Skladanowski A. C. (2006) Expression of ecto-5′-nucleotidase (eN, CD73) in cell lines from various stages of human melanoma. Melanoma Res. 16, 213–222 [DOI] [PubMed] [Google Scholar]
- 24. Katsuta E., Tanaka S., Mogushi K., Shimada S., Akiyama Y., Aihara A., Matsumura S., Mitsunori Y., Ban D., Ochiai T., Kudo A., Fukamachi H., Tanaka H., Nakayama K., Arii S., and Tanabe M. (2016) CD73 as a therapeutic target for pancreatic neuroendocrine tumor stem cells. Int. J. Oncol. 48, 657–669 [DOI] [PubMed] [Google Scholar]
- 25. Wang L., Zhou X., Zhou T., Ma D., Chen S., Zhi X., Yin L., Shao Z., Ou Z., and Zhou P. (2008) Ecto-5′-nucleotidase promotes invasion, migration and adhesion of human breast cancer cells. J. Cancer Res. Clin. Oncol. 134, 365–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu H., Pan S., Gu S., Bradbury E. M., and Chen X. (2002) Amino acid residue specific stable isotope labeling for quantitative proteomics. Rapid Commun. Mass Spectrom. 16, 2115–2123 [DOI] [PubMed] [Google Scholar]
- 27. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., and Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 28. Liu L., Zhou J., Wang Y., Mason R. J., Funk C. J., and Du Y. (2012) Proteome alterations in primary human alveolar macrophages in response to influenza A virus infection. J. Proteome Res. 11, 4091–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dhamad A. E., Zhou Z., Zhou J., and Du Y. (2016) Systematic proteomic identification of the heat shock proteins (Hsp) that interact with estrogen receptor alpha (ERalpha) and biochemical characterization of the ERalpha-Hsp70 interaction. PLoS ONE 11, e0160312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Du Y., Zhou J., Fan J., Shen Z., and Chen X. (2009) Streamline proteomic approach for characterizing protein-protein interaction network in a RAD52 protein complex. J. Proteome Res. 8, 2211–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Du Y. C., Gu S., Zhou J., Wang T., Cai H., Macinnes M. A., Bradbury E. M., and Chen X. (2006) The dynamic alterations of H2AX complex during DNA repair detected by a proteomic approach reveal the critical roles of Ca(2+)/calmodulin in the ionizing radiation-induced cell cycle arrest. Mol. Cell. Proteomics 5, 1033–1044 [DOI] [PubMed] [Google Scholar]
- 32. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 33. Tyanova S., Temu T., and Cox J. (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 [DOI] [PubMed] [Google Scholar]
- 34. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 35. Elias J. E., and Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 36. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 [DOI] [PubMed] [Google Scholar]
- 37. Thomas S., and Bonchev D. (2010) A survey of current software for network analysis in molecular biology. Hum. Genomics 4, 353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Y., Zhou J., and Du Y. (2014) hnRNP A2/B1 interacts with influenza A viral protein NS1 and inhibits virus replication potentially through suppressing NS1 RNA/protein levels and NS1 mRNA nuclear export. Virology 449, 53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 40. Zhou Z., Zhou J., and Du Y. (2012) Estrogen receptor alpha interacts with mitochondrial protein HADHB and affects beta-oxidation activity. Mol. Cell. Proteomics 11, M111 011056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhi X., Wang Y., Zhou X., Yu J., Jian R., Tang S., Yin L., and Zhou P. (2010) RNAi-mediated CD73 suppression induces apoptosis and cell-cycle arrest in human breast cancer cells. Cancer Sci. 101, 2561–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jeffery J. M., Urquhart A. J., Subramaniam V. N., Parton R. G., and Khanna K. K. (2010) Centrobin regulates the assembly of functional mitotic spindles. Oncogene 29, 2649–2658 [DOI] [PubMed] [Google Scholar]
- 43. Zhou J., and Du Y. (2012) Acquisition of resistance of pancreatic cancer cells to 2-methoxyestradiol is associated with the upregulation of manganese superoxide dismutase. Mol Cancer Res. 10, 768–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Singh N. P., McCoy M. T., Tice R. R., and Schneider E. L. (1988) A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 175, 184–191 [DOI] [PubMed] [Google Scholar]
- 45. Konca K., Lankoff A., Banasik A., Lisowska H., Kuszewski T., Gozdz S., Koza Z., and Wojcik A. (2003) A cross-platform public domain PC image-analysis program for the comet assay. Mutat. Res. 534, 15–20 [DOI] [PubMed] [Google Scholar]
- 46. Paull T. T., Rogakou E. P., Yamazaki V., Kirchgessner C. U., Gellert M., and Bonner W. M. (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10, 886–895 [DOI] [PubMed] [Google Scholar]
- 47. Sharma S. V., Bell D. W., Settleman J., and Haber D. A. (2007) Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 7, 169–181 [DOI] [PubMed] [Google Scholar]
- 48. Manning B. D., and Toker A. (2017) AKT/PKB signaling: navigating the network. Cell 169, 381–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xie L., Law B. K., Chytil A. M., Brown K. A., Aakre M. E., and Moses H. L. (2004) Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 6, 603–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen B., Zeng X., He Y., Wang X., Liang Z., Liu J., Zhang P., Zhu H., Xu N., and Liang S. (2016) STC2 promotes the epithelial-mesenchymal transition of colorectal cancer cells through AKT-ERK signaling pathways. Oncotarget 7, 71400–71416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou Q., Chen J., Feng J., Xu Y., Zheng W., and Wang J. (2017) SOSTDC1 inhibits follicular thyroid cancer cell proliferation, migration, and EMT via suppressing PI3K/Akt and MAPK/Erk signaling pathways. Mol. Cell Biochem. 435, 87–95 [DOI] [PubMed] [Google Scholar]
- 52. Junk D. J., Bryson B. L., Smigiel J. M., Parameswaran N., Bartel C. A., and Jackson M. W. (2017) Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 36, 4001–4013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smigiel J. M., Parameswaran N., and Jackson M. W. (2017) Potent EMT and CSC phenotypes are induced by oncostatin-M in pancreatic cancer. Mol Cancer Res. 15, 478–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Heinrich P. C., Behrmann I., Haan S., Hermanns H. M., Muller-Newen G., and Schaper F. (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou P., Li B., Liu F., Zhang M., Wang Q., Liu Y., Yao Y., and Li D. (2017) The epithelial to mesenchymal transition (EMT) and cancer stem cells: implication for treatment resistance in pancreatic cancer. Mol. Cancer 16, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Polyak K., and Weinberg R. A. (2009) Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer 9, 265–273 [DOI] [PubMed] [Google Scholar]
- 57. Weinberg R. A. (2014) the Biology of Cancer, 2nd Ed., Garland Science, Taylor & Francis Group, LLC, New York, NY [Google Scholar]
- 58. Barh D., Malhotra R., Ravi B., and Sindhurani P. (2010) MicroRNA let-7: an emerging next-generation cancer therapeutic. Curr. Oncol. 17, 70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Riley P. A. (1994) Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 65, 27–33 [DOI] [PubMed] [Google Scholar]
- 60. Kong Q., and Lillehei K. O. (1998) Antioxidant inhibitors for cancer therapy. Med. Hypotheses 51, 405–409 [DOI] [PubMed] [Google Scholar]
- 61. Diehn M., Cho R. W., Lobo N. A., Kalisky T., Dorie M. J., Kulp A. N., Qian D., Lam J. S., Ailles L. E., Wong M., Joshua B., Kaplan M. J., Wapnir I., Dirbas F. M., Somlo G., Garberoglio C., Paz B., Shen J., Lau S. K., Quake S. R., Brown J. M., Weissman I. L., and Clarke M. F. (2009) Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Avila-Ibarra L. R., Mora-Garcia M. L., Garcia-Rocha R., Hernandez-Montes J., Weiss-Steider B., Montesinos J. J., Lizano-Soberon M., Garcia-Lopez P., Don-Lopez C. A., Torres-Pineda D. B., Chacon-Salinas R., Vallejo Castillo L., Perez-Tapia S. M., and Monroy-Garcia A. (2019) Mesenchymal stromal cells derived from normal cervix and cervical cancer tumors increase CD73 expression in cervical cancer cells through TGF-beta1 production. Stem Cells Dev. 28, 477–488 [DOI] [PubMed] [Google Scholar]
- 63. Dewey W. C., Ling C. C., and Meyn R. E. (1995) Radiation-induced apoptosis: relevance to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 33, 781–796 [DOI] [PubMed] [Google Scholar]
- 64. Meyn R. E., Stephens L. C., and Milas L. (1996) Programmed cell death and radioresistance. Cancer Metastasis Rev. 15, 119–131 [DOI] [PubMed] [Google Scholar]
- 65. Maier P., Hartmann L., Wenz F., and Herskind C. (2016) Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int. J. Mol. Sci. 17, pii: E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Datta S. R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., and Greenberg M. E. (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241 [DOI] [PubMed] [Google Scholar]
- 67. Zha J., Harada H., Yang E., Jockel J., and Korsmeyer S. J. (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14–3-3 not BCL-X(L). Cell 87, 619–628 [DOI] [PubMed] [Google Scholar]
- 68. Downward J. (1999) How BAD phosphorylation is good for survival. Nat. Cell Biol. 1, E33–E35 [DOI] [PubMed] [Google Scholar]
- 69. Harada H., Andersen J. S., Mann M., Terada N., and Korsmeyer S. J. (2001) p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. U.S.A. 98, 9666–9670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sathe A., Chalaud G., Oppolzer I., Wong K. Y., von Busch M., Schmid S. C., Tong Z., Retz M., Gschwend J. E., Schulz W. A., and Nawroth R. (2018) Parallel PI3K, AKT and mTOR inhibition is required to control feedback loops that limit tumor therapy. PLoS ONE 13, e0190854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gutensohn W., Resta R., Misumi Y., Ikehara Y., and Thompson L. F. (1995) Ecto-5′-nucleotidase activity is not required for T cell activation through CD73. Cell. Immunol. 161, 213–217 [DOI] [PubMed] [Google Scholar]
- 72. Deer E. L., Gonzalez-Hernandez J., Coursen J. D., Shea J. E., Ngatia J., Scaife C. L., Firpo M. A., and Mulvihill S. J. (2010) Phenotype and genotype of pancreatic cancer cell lines. Pancreas 39, 425–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gradiz R., Silva H. C., Carvalho L., Botelho M. F., and Mota-Pinto A. (2016) MIA PaCa-2 and PANC-1 - pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci. Rep. 6, 21648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wellner U., Schubert J., Burk U. C., Schmalhofer O., Zhu F., Sonntag A., Waldvogel B., Vannier C., Darling D., zur Hausen A., Brunton V. G., Morton J., Sansom O., Schuler J., Stemmler M. P., Herzberger C., Hopt U., Keck T., Brabletz S., and Brabletz T. (2009) The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 11, 1487–1495 [DOI] [PubMed] [Google Scholar]
- 75. Krebs A. M., Mitschke J., Lasierra Losada M., Schmalhofer O., Boerries M., Busch H., Boettcher M., Mougiakakos D., Reichardt W., Bronsert P., Brunton V. G., Pilarsky C., Winkler T. H., Brabletz S., Stemmler M. P., and Brabletz T. (2017) The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 19, 518–529 [DOI] [PubMed] [Google Scholar]
- 76. Palma Cde S., Grassi M. L., Thome C. H., Ferreira G. A., Albuquerque D., Pinto M. T., Ferreira Melo F. U., Kashima S., Covas D. T., Pitteri S. J., and Faca V. M. (2016) Proteomic analysis of epithelial to mesenchymal transition (EMT) reveals cross-talk between SNAIL and HDAC1 proteins in breast cancer cells. Mol. Cell. Proteomics 15, 906–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Goldman A., Majumder B., Dhawan A., Ravi S., Goldman D., Kohandel M., Majumder P. K., and Sengupta S. (2015) Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat. Commun. 6, 6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sun L., Yao Y., Liu B., Lin Z., Lin L., Yang M., Zhang W., Chen W., Pan C., Liu Q., Song E., and Li J. (2012) MiR-200b and miR-15b regulate chemotherapy-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene 31, 432–445 [DOI] [PubMed] [Google Scholar]
- 79. Singh A., and Settleman J. (2010) EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29, 4741–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.