

We benchmarked ultraviolet photodissociation (UVPD) based on 213 nm photons for the characterization of proteoforms <30 kDa from three different samples (Pseudomonas aeruginosa, mouse hearts and human fibroblasts). Product ion abundance analysis of 213 nm UVPD data sets was used for improving the database search ultimately leading to increased identification rates. 213 nm UVPD outperformed HCD in terms of proteoform characterization (defined by the C-score). Finally, we described UVPD fragmentation propensities and the observation of a 213 nm UVPD-specific product ion.
Keywords: Fourier transform MS, mass spectrometry, post-translational modifications, protein identification, tandem mass spectrometry, FDR, product ion, proteoform, top-down proteomics, UVPD
Graphical Abstract

Highlights
Analysis of product ions produced by 213 nm UVPD is used to refine database search.
A product ion at the N-terminus of Pro, y-2, is observed in 213 nm UVPD spectra.
213 nm UVPD provides more complete proteoform characterization than HCD.
HCD and 213 nm UVPD are complementary fragmentation methods for proteoforms <30 kDa.
Abstract
Top-down proteomics studies intact proteoform mixtures and offers important advantages over more common bottom-up proteomics technologies, as it avoids the protein inference problem. However, achieving complete molecular characterization of investigated proteoforms using existing technologies remains a fundamental challenge for top-down proteomics. Here, we benchmark the performance of ultraviolet photodissociation (UVPD) using 213 nm photons generated by a solid-state laser applied to the study of intact proteoforms from three organisms. Notably, the described UVPD setup applies multiple laser pulses to induce ion dissociation, and this feature can be used to optimize the fragmentation outcome based on the molecular weight of the analyzed biomolecule. When applied to complex proteoform mixtures in high-throughput top-down proteomics, 213 nm UVPD demonstrated a high degree of complementarity with the most employed fragmentation method in proteomics studies, higher-energy collisional dissociation (HCD). UVPD at 213 nm offered higher average proteoform sequence coverage and degree of proteoform characterization (including localization of post-translational modifications) than HCD. However, previous studies have shown limitations in applying database search strategies developed for HCD fragmentation to UVPD spectra which contains up to nine fragment ion types. We therefore performed an analysis of the different UVPD product ion type frequencies. From these data, we developed an ad hoc fragment matching strategy and determined the influence of each possible ion type on search outcomes. By paring down the number of ion types considered in high-throughput UVPD searches from all types down to the four most abundant, we were ultimately able to achieve deeper proteome characterization with UVPD. Lastly, our detailed product ion analysis also revealed UVPD cleavage propensities and determined the presence of a product ion produced specifically by 213 nm photons. All together, these observations could be used to better elucidate UVPD dissociation mechanisms and improve the utility of the technique for proteomic applications.
Contemporary systems biologists are faced with the grand challenge of elucidating sophisticated cellular processes arising from the physical and temporal dynamics of thousands of different biomolecules. In this context, the analysis of proteomes, or proteomics, is of primary importance given the role played by proteins as effector molecules within a cell (1). Generally, two main approaches are prevalent in proteomics. First and most commonly, proteins are analyzed after proteolysis by methodologies referred to as bottom-up proteomics (BUP)1 (2), extended bottom-up proteomics (eBUP) (3, 4) or middle-down proteomics (MDP) (5, 6), depending on the average mass of the resulting proteolytic peptides (about 1–3, 3–5 and >5 kDa, respectively). Alternatively, intact proteoforms, gene products with their specific set of genetic and chemical modifications (7), are characterized using the approach known as top-down proteomics (TDP) (8, 9). As the only mass-spectrometry (MS)-based proteomic technology that can characterize all sources of variability simultaneously present on an intact protein molecule (10), such as post-translational modifications (PTMs) or single nucleotide polymorphisms (SNPs), TDP offers unique advantages when compared with proteolysis-based proteomics. Importantly, TDP is not limited by the 'protein inference' problem that complicates analysis of BUP, eBUP, and MDP data and negatively impacts the specificity of these latter approaches (11). TDP ultimately delivers highly specific information about biological events otherwise impossible to study in a high-throughput manner, including endogenous protein processing and PTM cross-talk (12), an important phenomenon in epigenetics (13) as well as in cancer biology (14).
To achieve its full potential, TDP should fully characterize the proteoforms present in a sample. Although high mass resolution and mass accuracy MS, particularly Fourier transform MS (FTMS) (15), can lead to the accurate determination of a proteoform's molecular weight, the precise identification and localization of the sources of chemical and genetic variation present in a proteoform require tandem MS (MS2) capable of producing a variety of fragment ions distributed throughout the entire primary structure. Traditional ion activation technologies cause extensive fragmentation of smaller proteins, but present challenges when analyzing larger proteins. The most used ion dissociation strategies are vibrational excitation techniques (also known as “energy-threshold” fragmentations), such as low energy collision-induced dissociation (CID) and beam-style CID, including higher-energy collisional dissociation (HCD) (16). All vibrational excitation technologies primarily lead to the detection of relatively small terminal fragments when applied to large biomolecules, leading to incomplete characterization of the amino acid sequence. Additionally, activation by vibrational excitation also has the potential to produce a large number of internal fragments (i.e. fragment ions produced by at least two consecutive fragmentation events that do not include either of the proteoform termini), so that large canonical N- and C-terminal ions (typically, b- and y-type ions) might be reduced to smaller fragments, decreasing their spectral signal-to-noise ratio (SNR) (17).
An alternative to vibrational excitation methods, radical-driven fragmentation is based on the interaction between analyte ions and electrons (18). The most popular electron-based ion activation techniques for intact protein analysis are electron capture dissociation (ECD) (19) and electron transfer dissociation (ETD) (20). ECD and ETD can produce extensive fragmentation (primarily through c- and z-ions) throughout the entirety of the proteoform molecule, making them ideal for targeted top-down MS experiments, even of very large proteins (21–24). At the same time, the lack of specificity in cleavage sites results in low SNR for a vast number of potential fragment ions which can be compensated primarily by transient averaging, and furthermore both ECD and ETD require more time to implement than vibrational excitation, (25); these two features limited so far the implementation of radical driven fragmentation to large-scale TDP, with a few notable exceptions (26, 27). Recent studies by Coon and co-workers demonstrated the potential offered by activating precursor ions using IR photons during ETD experiments (the so-called AI-ETD), and this strategy proved particularly useful for increasing analysis throughput in TDP runs using both liquid chromatography (LC) and capillary zone electrophoresis for proteoform separation (28, 29). However, this promising instrument setup is still not commercially available.
Another fragmentation technique has existed for more than three decades but is recently getting renewed attention by a broad audience in the proteomic field. This type of fragmentation relies on the interaction of ions with photons, where the photon energy dictates the physical mechanism underlying the dissociation of the polypeptide ion (30). Specifically, the absorption of low-energy (∼0.1 eV/photon) infrared photons induces a progressive increase of ion internal energy, and ultimately leads to the cleavage of the peptide bond. This process, known as infrared multiphoton dissociation (IRMPD), generates a series of b- and y-ions like other vibrational excitation fragmentation methods (31). Conversely, the fast energy deposition mechanism that characterizes the absorption of high-energy UV photons (∼5–10 eV/photon) produces a completely different fragmentation pattern, either through the interconversion to vibrational energy of the initial electronic excitation (major pathway leading to bond rupture) or via direct dissociation (cleavage occurring on the femtosecond time-scale because of the generation of an excited-state electron that occupies a dissociative orbital) (32, 33). This process, called ultraviolet photodissociation (UVPD), was introduced in the 1980s (34, 35) and has undergone a resurgence with further development given its unique properties. Particularly, UVPD at 157 nm induced the complete backbone fragmentation of singly-charged peptides with the formation of the three main series of a/x, b/y and c/z-ions (some of which were present in their radical form), as well as v and w side chain-loss ions and immonium ions (36). Furthermore, irradiation with 157 nm photons was also shown to cleave inter- and intra-molecular disulfide bonds (37). UVPD with 193 nm photons generated via an ArF excimer laser produces the same fragment ion types (38), and when applied to intact proteins resulted in nearly full sequence coverage of 29 kDa carbonic anhydrase (39). 193 nm UVPD also demonstrated very low dependence on the precursor charge state, unlike ETD or HCD. This feature underlies the recently reported application of UVPD for the dissociation of protein complexes under native MS conditions, where precursor cations are characterized by low charge density (40, 41). In consideration of these appealing premises, Cannon et al. applied 193 nm UVPD to the characterization of intact proteoforms on the LC time scale. This pioneering TDP study led to the identification of 46 UniProt entries from purified ribosomes of Escherichia coli, versus 44 identified by HCD (42). More recently, Cleland et al. used the same ArF 193 nm laser in combination with a Tribrid Orbitrap mass spectrometer to characterize proteoforms in HeLa cells, demonstrating that 193 nm UVPD produced a higher average sequence coverage than HCD (43). Finally, a recent study also demonstrated the possibilities offered by UVPD performed using 355 nm photons on polypeptide ions generated with either electrospray or matrix-assisted laser desorption ionization (44).
Here, we describe the implementation of UVPD based on 213 nm photons generated by the 5th harmonic of a solid-state Nd:YAG laser. Previous studies in which 213 nm UVPD was coupled to a quadrupole-Orbitrap mass spectrometer showed that photons at this wavelength induced polypeptide fragmentation similar to that achieved with 193 nm photons (45–49). Notably, all previous proteomics studies based on 193 nm UVPD were carried out at a laser power of ∼1–2 mJ/pulse, whereas the 213 nm laser beam power is much lower at 25 μJ/pulse in this study. This difference is counterbalanced by a higher pulsing frequency of the solid-state device. Importantly, the Nd:YAG laser is considerably more economical than an excimer 193 nm laser. In our study, we first benchmarked the performance of 213 nm UVPD on a quadrupole-Orbitrap-linear ion trap mass spectrometer using standard proteins with masses ranging from 8.5 to 29 kDa. We subsequently performed the high-throughput analysis of proteoforms <30 kDa from one prokaryote, Pseudomonas aeruginosa, and two eukaryotic organisms, Mus musculus and Homo sapiens. To fully assess the potential offered by this newly introduced ion activation method, 213 nm UVPD was compared with traditional HCD-based TDP not only in terms of the total number of unique proteoforms and UniProt entries identified, but more importantly in regard to the quality of proteoform characterization as measured by sequence coverage and proteoform characterization score (C-score) (50). Finally, we analyzed the 213 nm UVPD data with a focus on the propensity to form different types of product ions, in order to improve database searching. In summary, with the present study we provide a comprehensive guide to efficiently perform TDP using 213 nm UVPD.
EXPERIMENTAL PROCEDURES
Chemicals
Water, acetonitrile (ACN), chloroform, methanol, acetic acid, and formic acid (FA) were purchased in LC-MS purity grade (Fisher Scientific, Rockford, IL). Dithiothreitol (DTT), phosphate buffered saline (PBS), penicillin-streptomycin, and HALT protease and phosphatase inhibitor mixture were obtained from Thermo Fisher Scientific. Sodium butyrate, sodium dodecyl sulfate (SDS), Dulbecco's Modified Eagle's Medium, lyophilized ubiquitin (U6253), trypsinogen (T1143), and myoglobin (M5696) were purchased from Sigma Aldrich (St. Louis, MO).
Preparation of Samples for High-throughput Proteomics
Pseudomonas aeruginosa (strain PAO1) cells were cultured in rich media and harvested at mid-log phase. Mouse cardiac myocytes were obtained from adult mice as previously reported (51). Briefly, mouse hearts were prepared for protein extraction by cryopulverization using a Retsch Mixer Mill MM400 (frequency 30 Hz, 2 min cycle), keeping tissue samples in liquid nitrogen between two consecutive milling sessions. Primary IMR90 human fibroblasts were cultured as previously described (52). Briefly, cells were grown adherently in Dulbecco's Modified Eagle's Medium, supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. At 80% confluence, cells were pelleted by centrifugation and washed with PBS. For all samples (either cell pellets or tissue powder), cell lysis was carried out with a buffer composed of 4% SDS (w/v), 1 mm DTT, 10 mm Tris-HCl pH = 7.8, 10 mm sodium butyrate, and HALT protease and phosphatase inhibitor mixture diluted according to the manufacturer's recommendation. Resuspended cell pellets were boiled in lysis buffer for 10 min under shaking (600 rpm). Proteins in the cell lysate were purified by acetone-precipitation at −20 °C overnight, and the resulting protein pellets were re-solubilized using 1% SDS (w/v). Protein quantification was performed by BCA assay (Pierce BCA Protein Assay Kit, Thermo Scientific). Prior to online liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, proteins were subjected to molecular weight (MW) fractionation with a GELFrEE 8100 Fractionation System (Expedeon, Harston, Cambridgeshire, UK) using a 10% T cartridge (53). One or two lanes were loaded with 350–500 μg of protein each, and the first 5 or 6 fractions (which include the proteins <30 kDa) were collected. GELFrEE fractions were analyzed by SDS-PAGE (10 μl loaded/lane) and visualized by silver staining (supplemental Fig. S1). Each fraction was purified by MeOH/CHCl3/H2O precipitation according to a previously described protocol (54). Pellets were resuspended in Solution A (vide infra). For P. aeruginosa, a single lane was used. For the mouse hearts, corresponding fractions from two different lanes (e.g. fraction 1 of each lane) were pooled in order to obtain enough homogeneous material for multiple LC injections, as previously described (55). For the IMR90 proteins, GELFrEE fractions from a single lane were analyzed in a specific set of experiments without pooling, and 2 lanes in total were used (one for HCD and one for UVPD).
Liquid Chromatography
Proteoforms from each GELFrEE fraction were subjected to nanocapillary reversed-phase liquid chromatography performed on a Dionex Ultimate 3000 chromatographic system (Thermo Scientific, Sunnyvale, CA). Two main setups, previously described in detail, were used (55, 56). P. aeruginosa and mouse heart samples were separated using the combination of an in-house prepared trap column (150 μm i.d., 25 mm length) and analytical column (75 μm i.d., 250 mm length) packed with PLRP-S resin (5 μm particle size, Agilent, Santa Clara, CA). Separations were carried out at 300 nL/min. IMR90-derived proteins were separated using monolithic trap (PepSwift, 200 μm i.d., 20 mm length, Thermo Scientific) and analytical columns (RP-4H, 100 μm i.d., 500 mm length, Thermo Scientific) at a 1 μl/min flow rate. Samples loaded on the trap column in 100% Solution A were separated using a gradient of Solution B from 5 to 15% in 2 min, followed by a ramp to 50% in 50 min. Columns were washed using 95% B for 5 min followed by re-equilibration at 5% B for 15 min. Solution A was composed of 4.8% ACN in H2O with 0.2% FA, whereas Solution B consisted of 4.8% H2O in ACN and 0.2% FA. The column temperature was set to 55 °C and 35 °C for PLRP-S and RP-4H, respectively. The column outlet was coupled to a nanoelectrospray ionization (nanoESI) source built in-house. This source employed a 15 μm i.d. electrospray emitter (New Objective, Woburn, MA) packed at the tip with ∼5 mm of PLRP-S resin to prevent outgassing.
Mass Spectrometry
All MS experiments were carried out on an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific, San Jose, CA) operated in “protein mode” with 2 mTorr N2 in the HCD cell. The instrument was controlled through Tune v. 3.0 (Thermo Scientific). For all measurements the high capacity transfer capillary temperature was set at 320 °C, the ion funnel RF was set at 30%, and a 15 V offset between SRIG and MP00 was used to improve desolvation and declustering. Top-down MS acquisition parameters were set as follows (unless otherwise stated): broadband MS (MS1) used 120,000 resolving power (at m/z 200), with an automatic gain control (AGC) target value of 2e5. MS2 spectra were acquired at 60,000 resolving power (at m/z 200), with target AGC values of 2e5 or 5e5. Four microscans were averaged for both MS1 and MS2. The UVPD setup is like that described previously (47, 48) (supplemental Fig. S2).
For direct infusion experiments, proteins were resuspended at 1 μm concentration in a mix of H2O (49.9%), ACN (49.9%) and acetic acid (0.2%). Each single protein was infused at 5 μl/min (via syringe pump) to an IonMax electrospray source (Thermo Scientific) operating at 3.5 kV. Acquisition was performed via Tune, and each acquisition method applied either a single normalized collision energy (NCE) value for HCD or a specific number of laser pulses for UVPD, and sequentially fragmented different precursor charge states (four charge states for ubiquitin and five charge states for both myoglobin and carbonic anhydrase) selected in the quadrupole using a 5 m/z isolation window.
For LC-MS/MS experiments using the nanoESI source, a 2.1 kV potential was applied through a PEEK high voltage union interconnecting the LC column and spray emitter. Precursor ions were quadrupole selected using a 3 m/z isolation window. The dynamic exclusion option was used with a 60 s duration. Data acquisition was performed by applying a data-dependent top-2 method, with the two most abundant precursors selected for fragmentation.
Experimental Design, Data Analysis and Statistical Rationale
In direct infusion experiments, each targeted precursor was fragmented for 30 s, corresponding to ∼50 spectra. Each spectrum was analyzed separately, and mean and standard deviation values for sequence coverage and ratio of matched versus unmatched fragment ions were calculated. All LC-MS/MS experiments for a single GELFrEE fraction and a selected ion activation method (i.e. HCD or UVPD) were repeated in triplicate. In the case of HCD, the Normalized Collision Energy (NCE) was set at 23%, 24%, and 25% for the first, second and third technical replicate, respectively. For UVPD we applied a differing number of pulses that were increased in steps of 5 pulses across the technical replicates (from a minimum of 30 to a maximum of 65, depending on the GELFrEE fraction) following an order specified in supplemental Table S1. RAW files from LC-MS/MS experiments were analyzed through TDPortal 1.0 (http://nrtdp.northwestern.edu/tdportal-request/), according to a previously described workflow (26, 55). Briefly, a cRAWler algorithm grouped deconvoluted MS2 spectra and corresponding precursor neutral masses. Deconvolution was carried out with Xtract (Thermo Scientific). The resulting combinations of precursor and fragment masses were searched against a database of candidate proteoforms generated from a UniProt formatted text file for P. aeruginosa (Taxon ID: 208964, Proteome ID: UP000002438), Mus musculus (Taxon ID: 10090, Proteome ID: UP000000589) or Homo sapiens (Taxon ID: 9606, Proteome ID: UP000005640). The proteoform search space was generated in silico, allowing up to 11 PTMs or sequence variations per candidate proteoform. Three different database searches were run in parallel for the same set of masses, including: (1) an Absolute Mass search with precursor tolerance of 2.2 Da; (2) a Biomarker search with 10 ppm precursor tolerance; (3) an Absolute Mass search with 200 Da precursor tolerance applying the Δm mode to account for unexpected modifications. The tolerance applied to fragments was 10 ppm. Each forward hit consisted of a proteoform with associated p-score (57), E-value (58) and C-score (50). An instantaneous q-value was also associated with each identified proteoform, as a result of searching a decoy database and using a false-discovery rate (FDR) estimation (for which a scrambled version of the UniProt database is used) (59). Distinct q-values were obtained separately for each of the three searches. After single FDR cutoffs were determined for each search, a global FDR estimation was finally generated according to the procedure described by Higdon et al. (60), using only the hit with the best q-value obtained from the three searches. Identical FDR calculations were applied at the protein isoform and UniProt entry (protein) level. All reported proteoforms and entries derived from a 1% FDR cutoff. A unique ProteoForm Record number (PFR) was assigned to confidently identified proteoforms. Identified proteoforms with a C-score >40 were uploaded to an online repository hosted by the Consortium for Top Down Proteomics (http://repository.topdownproteomics.org/) and cross-referenced by UniProt. Graphical fragmentation maps were obtained using ProSight Lite (available at http://prosightlite.northwestern.edu/) (61, 62). Heatmaps representing the propensities of HCD and UVPD to cleave between specific pairs of amino acid residues were generated according to the procedure described by Haverland et al. (63).
RESULTS
Performance of 213 nm UVPD on Standard Proteins
For the initial evaluation of UVPD performance, three standard proteins of different molecular weight (MW) were used: ubiquitin (8.5 kDa), myoglobin (17 kDa), and carbonic anhydrase (29 kDa). Similar to other dissociation techniques capable of producing high protein sequence coverage—namely ETD and ECD—in 213 nm UVPD experiments a large part of the precursor current is not directly transformed into sequence-informative, dissociated product ions, probably because of poor photon absorption—although, unlike ECD and ETD, UVPD does not generate charge-reduced species. As mentioned previously, with 213 nm UVPD it is possible to compensate for the low power of the solid-state Nd:YAG laser by using multiple laser pulses to activate and dissociate protein cations. For standard proteins directly infused into the ESI source of the Tribrid mass spectrometer, the decrease in precursor peak area and simultaneous increase in fragment ion area as a function of applied 213 nm laser shots can be easily calculated (supplemental Fig. S3). A precursor “half-life” (t1/2) can be estimated by fitting the obtained data points. This half-life is roughly inversely proportional to the collisional cross-section of the protein (which typically increases with the mass of a denatured protein cation). Supplemental Fig. S4 shows the sequence coverage obtained for myoglobin and carbonic anhydrase as a function of the number of used laser pulses. Empirically, it was possible to determine that the maximum sequence coverage for each standard protein was obtained at several pulses corresponding to approximately three times t1/2 when the spectra were collected at 120,000 resolution (at 200 m/z) using 100 microscans. However, such conditions do not reflect the actual parameters used in proteomic experiments, where the elution of proteins from an on-line coupled column dictates rapid fragmentation and spectral acquisition. Therefore, we recorded spectra with 4 microscans/spectrum, and compared 213 nm UVPD with HCD to provide a direct comparison to the most used ion activation technique in TDP. Initially, both HCD and UVPD main parameters (NCE for HCD and the number of laser pulses for UVPD) were varied to determine whether optimal values could be identified to generate maximized sequence coverage and fraction of matched product ions. For HCD, NCE was increased in steps of 2% from 14% to 30%. The number of pulses in UVPD was increased in steps of 5, from 5 to 60. Four different precursors were isolated and subsequently fragmented in the case of ubiquitin, namely the 8+, 10+, 11+ and 13+ charge states, whereas five were selected for both myoglobin (14+, 16+, 20+, 24+, 25+) and carbonic anhydrase (29+, 32+, 35+, 38+, 43+). Fig. 1A and 1B show the sequence coverage obtained for the three proteins as a function of the fragmentation parameters for HCD and UVPD. Although these graphs average the results obtained for all the analyzed charged states, it is apparent (particularly in the case of ubiquitin) that under these experimental conditions the selection of the correct NCE in HCD is fundamental to obtain the best sequence coverage possible, whereas in UVPD the variation of the number of pulses does not substantially alter the final sequence coverage.
Fig. 1.

Sequence coverage and ratio of matched/total fragment ions as a function of ion activation parameters. Fig. 1A and 1B show the sequence coverage for the three standard proteins ubiquitin (8.5 kDa), myoglobin (17 kDa) and carbonic anhydrase (29 kDa) as a function of normalized collision energy and number of laser pulses for HCD and UVPD, respectively. Fig. 1C and 1D show the ratio between matched and total fragment ions as a function of the same parameters for HCD and UVPD, respectively. For each protein, multiple charge states (listed in the Experimental section) were fragmented, and plotted results were obtained by averaging the values relative to each interrogated charge state using a certain fragmentation energy/number of laser pulses. Error bars correspond to standard deviation.
When the ratio between fragments matched to the protein sequence relative to the total number of detected fragments is considered, HCD and UVPD again exhibit different behavior. Fig. 1C shows that in HCD the best ratio is obtained around narrow windows of NCE values which are centered on different values for proteins of different masses. Specifically, ubiquitin has the highest matching fragment ratio at low NCE values (14–18%), whereas the opposite happens for carbonic anhydrase, which reaches the best ratio at high NCE values (26–30%). Myoglobin is in between, requiring low NCE values, but in a less pronounced fashion than ubiquitin. Conversely, UVPD shows very little variation in the matched/total fragment ratio as the number of laser pulses increases: all three proteins reach their best ratio at the lowest number of pulses. However, the decrease in that ratio observed from 5 to 60 pulses is very limited for ubiquitin and myoglobin, and slightly higher for carbonic anhydrase (Fig. 1D). When sequence coverage and ratio of matched/total fragment ions is analyzed as a function of the protein charge density (calculated as number of charges of the precursor per kDa of protein average mass), HCD and UVPD demonstrate further differences. In HCD, at least one of the proteins, myoglobin, exhibits a clear trend toward lower charge density precursors to produce the best sequence coverage, whereas a less obvious trend toward higher charge density precursors is shown by ubiquitin. Interestingly, the sequence coverage of carbonic anhydrase stays constant throughout the entire charge density range investigated (Fig. 2A). In UVPD, both myoglobin and carbonic anhydrase reach the same level of sequencing independently of the charge state of the isolated precursor, and ubiquitin shows variations that do not indicate any clear trend, which might potentially be related to variations in the protein conformation rather than the charge density itself (Fig. 2B). Regarding the matched/total fragment ratio, in HCD, only myoglobin shows a clear propensity to return higher ratios when low charge state precursors are selected for fragmentation, whereas the other two proteins do not display any clear tendency (Fig. 2C). Conversely, proteins fragmented by UVPD show moderate but consistent variations along the charge density axis, with ubiquitin and myoglobin having their related matched/total fragment ratios increased with charge density whereas carbonic anhydrase exhibits an opposite trend. However, charge density-based variations are minor in UVPD (ubiquitin and myoglobin increase the ratio from a minimum of 0.3 and 0.14 to a maximum of 0.39 and 0.25, respectively, whereas for carbonic anhydrase the ratio decreases from 0.22 to 0.13), whereas in HCD the changes are more apparent with the ratio for myoglobin passing from a maximum of 0.27 to a minimum of 0.06 (Fig. 2D). Importantly, we also used these MS2 spectra recorded under typical proteomic conditions to determine the propensity of 213 nm UVPD to generate each of the 9 types of termini-containing product ions first attributed to 193 nm UVPD in 2013 (39) (i.e. a, a+, b, c, x, x+, y, y−, z•, where “+” and “−” respectively indicate the presence or absence of an extra hydrogen compared with the standard ion form) (supplemental Fig. S5) (64). Like for 193 nm UVPD, the most abundant class of N-terminus containing ions was a/a+-type ions (39). In terms of C-terminus containing ions, 193 nm UVPD produces mainly z•-type ions whereas UVPD at 213 nm primarily generated x/x+-type ions in the case of ubiquitin and myoglobin, and y/y− for carbonic anhydrase.
Fig. 2.

Sequence coverage and ratio of matched/total fragment ions as a function of charge state density of protein precursors in HCD and UVPD. The charge density is expressed as number of charges per kDa of protein average mass. Fig. 2A and 2B show the variation in sequence coverage for the three standard proteins for HCD and UVPD, respectively. Fig. 2C and 2D display the dependence of the fraction of matched ions over charge state density for HCD and UVPD, respectively. In these plots, all the applied NCE values (for HCD) or number of laser pulses (for UVPD) are averaged together. Error bars correspond to standard deviation.
213 nm UVPD Top-down Proteomics and Standard Database Search
Proteoforms <30 kDa from P. aeruginosa, M. musculus and H. sapiens were analyzed by LC-MS/MS, with each single GELFrEE fraction characterized both by HCD and 213 nm UVPD. For the initial database search, two ion types (i.e. b- and y-ions) were considered for HCD, whereas all nine canonical termini-containing ions were included for UVPD data. A summary of the results is reported in supplemental Fig. S6. HCD fragmentation returned a larger number of both UniProt entries and proteoforms identified at 1% FDR (an average increase of +32% and +111% at the protein and proteoform level, respectively). Notably, the majority of reported UniProt entries were identified by both HCD and UVPD (specifically, 68.6%, 56.9%, and 57.1% of the total for bacterial, mouse and human samples, respectively). However, the situation is radically different at the proteoform level, where fewer than a third of the total identified proteoforms are identified by both HCD and UVPD (22.7%, 18.9%, and 28.3%, respectively). The lower identification numbers obtained by UVPD are counterbalanced by the greater characterization provided by this type of ion activation compared with HCD. Supplemental Fig. S7 shows an increase in average proteoform sequence coverage by UVPD fragmentation over HCD for each analyzed sample (a 48%, +27%, and +27% improvement for P. aeruginosa, M. musculus and H. sapiens, respectively). Additionally, UVPD outperformed HCD in terms of the fraction of identified proteoforms that are fully characterized (i.e. with an associated C-score >40). The right panels of supplemental Fig. S7 show the C-score distributions for the three proteoform datasets, and the fully characterized fraction of the datasets by UVPD is equivalent to 100, 77.3 and 86.3% of the total for P. aeruginosa, M. musculus and H. sapiens, respectively. For the human fibroblasts, HCD returned 410 fully characterized proteoforms versus 392 for UVPD, despite the total number of proteoforms identified by UVPD being only ∼57% of those identified by HCD. Interestingly, the advantage in characterization offered by UVPD seems to not be limited to a specific mass range. Supplemental Fig. S8 displays the C-score distribution for the proteoforms identified in the HCD and UVPD data sets as a function of MW, and with the exception of very small proteoforms (average mass <5 kDa), the fraction of proteoforms with C-score >40 is always larger for UVPD than for HCD. Notably, the analysis of the proteoforms identified by both HCD and UVPD in mouse and human samples (supplemental Fig. S9 and S10, respectively) shows that 213 nm UVPD returns +16% and +12% proteoforms with C-score >40 compared with HCD for the two data sets, respectively. Although the average sequence coverage obtained by the two fragmentation techniques for this sub-group of proteoforms is comparable (for instance, in the case of human proteoforms the average sequence coverage yielded by HCD is 30% whereas for UVPD it is 31%), 213 nm UVPD can often provide better localization of PTMs, single amino acid substitutions or other modifications than HCD (Fig. 3).
Fig. 3.

Comparison between HCD and 213 nm UVPD fragmentation of proteoforms on the LC time scale. Tandem mass spectra for proteoform PFR 13328 (from UniProt entry P60468) are shown: Fig. 3A displays the HCD MS2 spectrum whereas Fig. 3B represents the UVPD spectrum. Note that whereas the base peak in Fig. 3A is represented by the product ion b738+ (m/z 916), the UVPD MS2 spectrum is largely dominated by the undissociated precursor (m/z 764), as it is often the case for UVPD, and product ion intensities have been magnified 5-fold. Despite similar sequence coverage and number of matched fragments, only 213 nm UVPD could precisely localize the phosphorylation at Ser16; in the case of HCD, Ser12 and Ser13 - annotated in UniProt as potential phosphorylation sites - cannot be ruled out. The complementarity between backbone cleavages induced by HCD (covering only the C-terminus) and UVPD (fragmenting primarily in the middle of the sequence, but also in part of the N-terminus) is apparent.
Analysis of 213 nm UVPD Product Ions and Consequences on Database Search
The analysis of the population of matched product ions for standard proteins (supplemental Fig. S5) or monoclonal antibodies characterized in previous studies (48) suggested that not all fragment ion types might be produced with equal frequency by 213 nm UVPD (or, possibly, that their long-term stability is not identical). We performed a product ion analysis considering the nine fragment ion types for each proteoform spectral match (PrSM) (59) from the three available data sets (i.e. bacterial, mouse, human). The results, summarized in Fig. 4, clearly show that two fragment types alone, a and a+, account for more than 50% of all matched ions in each proteome context studied here. Notably, the relative abundance associated with each ion type remains highly consistent from one data set to the other. Considering these results, we performed new database searches while progressively removing ions one-by-one in inverse order of abundance, until we searched only one ion type (a+, the most abundant). We then reported the variation in identified UniProt accession numbers, unique proteoforms (both calculated at 1% FDR), and average sequence coverage as a function of ion types (Fig. 5A–5C and supplemental Fig. S11). The graphs for all three proteomes share the same trend, and specifically they reach maxima in the number of entry and proteoform identifications when the four most abundant ion types (a+, a, x+ and y−) are used (with the only exception of the unique proteoform in the human data set, reaching the maximum when only one ion type is considered, Fig. 5B). Removing the five least abundant product ion types results in losing 5–6% of total backbone cleavages previously matched when using all nine canonical fragments (Fig. 5C and supplemental Fig. S11C). However, the C-score distribution is not substantially affected by the sequence coverage reduction, as displayed in Fig. 5D. Detrimental effects on proteoform characterization are instead observed when only a+-ions are searched (Fig. 5D gray histogram). Database search results obtained by including only the four most abundant product ion types are summarized in Fig. 6. Compared with the Venn diagrams derived from the canonical search results (supplemental Fig. S6), the new plots show not only an increase in the total number of identifications for UVPD (on average of +14% for UniProt entries and +31% for proteoforms), but also that most of the additional identifications correspond to entries or proteoforms previously identified by HCD (i.e. shared between HCD and UVPD). Remarkably, not only did the adoption of optimized search parameters reduce the identification gap between HCD and UVPD, but for the first time a higher number of UniProt accession entries were identified in the same sample using UVPD over HCD (human data set, 245 versus 265 identified entries for HCD and UVPD, respectively).
Fig. 4.
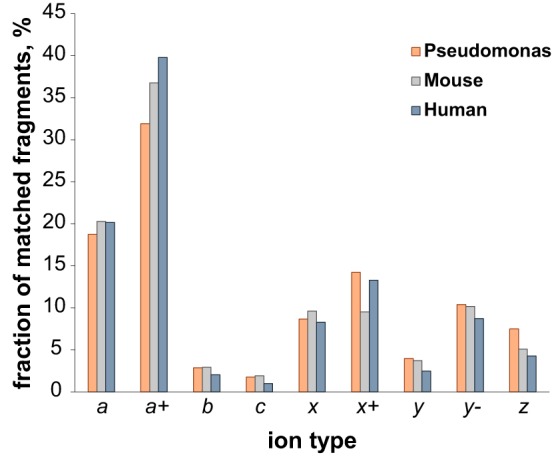
Breakdown of matched fragment ions by type derived from 213 nm UVPD experiments. The histogram shows the relative frequency of each ion type within the total population of matched fragment ions generated by 213 nm UVPD. Color coding distinguishes between the bacterial (orange), mouse (gray) and human (blue) data sets. All nine canonical ions produced by UVPD are considered.
Fig. 5.

Variation of database search results for UVPD experiments as a function of considered product ion pool. Fig. 5A and 5B show the variation in the number of unique UniProt accession and proteoforms identified at 1% FDR, respectively, as a function of the considered number of ions. Fig. 5C shows the decrease in sequence coverage produced by the reduction in the pool of product ions considered in the database search. Fig. 5D displays the C-score distribution for pools of nine, four, and a single product ion. All graphs refer to the H. sapiens data set. Product ions were removed from the canonical list of nine starting from the least abundant type, according to the relative frequency analysis summarized in Fig. 4.
Fig. 6.
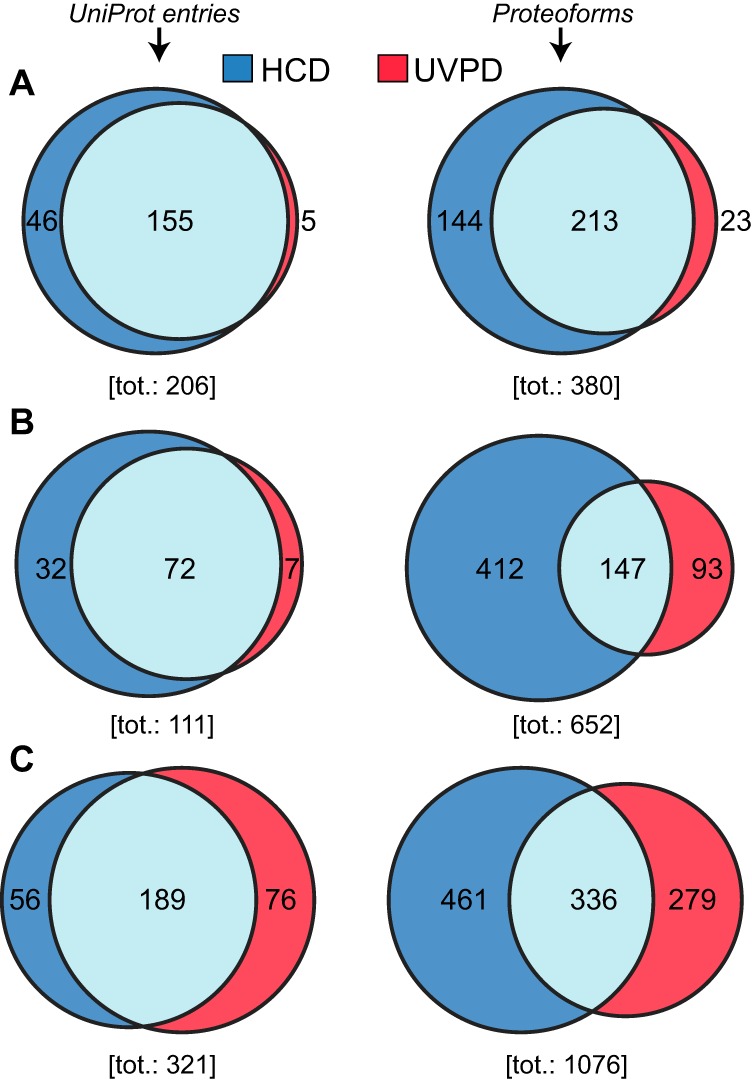
Comparison of HCD results versus UVPD considering the four most abundant ion types. A, B, and C, display Venn diagrams for P. aeruginosa, M. musculus and H. sapiens, respectively. The left column refers to UniProt accession whereas the right column refers to unique proteoforms.
A Product Ion Type Specific for Photodissociation with 213 nm Photons
The manual validation of tandem mass spectra of standard proteins suggested that a product ion type, x+x+, not included in the canonical list of UVPD product ions (which was originally based on studies with 193 nm photons) could be generated by 213 nm UVPD at the N-terminus of Pro. This finding was confirmed by performing the database search of the UVPD human data set including the newly discovered ion in the product ion list. The heatmap in supplemental Fig. S12 shows strong evidence that y-2-ions are produced by UVPD, almost exclusively at the N-terminal side of proline residues. Oftentimes these ions are among the most abundant in 213 nm UVPD spectra, as shown in supplemental Fig. S13 and S14.
DISCUSSION
Applying 213 nm UVPD to Targeted or High-throughput Top-down MS
The primary goal of this study was to evaluate the performance of the newly introduced 213 nm UVPD setup combined with high resolution Orbitrap FTMS for high-throughput TDP. This type of application demands highly specific instrument settings, particularly for tandem MS, as a fine balance between acquisition rate/duty cycle optimization and spectral quality must be achieved. Specifically, with the increase of MW, the dissociation of a polypeptide produces progressively more complex spectra with a larger number of different fragment ions that are often multiply-charged. With the precursor ion current split into a multitude of fragmentation channels, ion statistics becomes a fundamental parameter to finely control. To improve spectral signal-to-noise ratio (SNR) for low abundant fragment ions, the most employed strategy in FTMS is the acquisition of multiple time-domain transients (i.e. microscans) that are summed prior to Fourier transformation. However, the number of microscans that can be averaged in high-throughput experiments is limited, as the instrument cycle time cannot be extended over the limits that are imposed by the chromatographic time scale. Moreover, activations based on vibrational excitation such as CID and HCD are highly efficient in converting the precursor ion current into fragment ion current usable for obtaining sequence information—without the need of supplemental activation—when compared with other activations, including UVPD (Fig. 3) (65, 66). Ultimately, these features allowed HCD to become the standard ion activation technology for TDP, and at the same time limited the application of other activation techniques such as ECD/ETD (18) primarily to targeted top-down MS experiments. In these experiments, where a single purified protein is directly infused for a prolonged time (67) or analyzed in a series of LC-MS/MS runs from which time-domain transients are collected and summed (68, 69), high spectral SNR can be achieved for many more product ions. An example clearly illustrating the differences between targeted and high-throughput top-down experiments is given by recent applications of activated ion ETD: Riley et al. used multiple Orbitrap fills (three, with that increasing ∼3 times the final spectral SNR) and averaged 3 microscans/spectrum for characterizing AI-ETD in targeted mode (which also took advantage of extremely high resolving power, 240,000 at m/z 400, for MS2 spectra) (70). Conversely, the same research group applied conditions similar to those of our study (SNR improvement obtained averaging 4 microscans/spectrum, and MS2 spectra recorded at resolving power 60,000 at m/z 200) when using AI-ETD in high-throughput mode (29). Because of the substantial difference in the data acquisition parameters, in the present study we sought to characterize 213 nm UVPD by separately performing targeted and large-scale top-down experiments. When used for targeted top-down MS, 213 nm UVPD shows sequencing capabilities in line with those of 193 nm UVPD (supplemental Fig. S15). Furthermore, in targeted mode it is possible to determine an optimal number of laser pulses to achieve the maximum sequence coverage for proteins of different MW (supplemental Fig. S4). The finding that ∼3x the 't1/2' yields near optimal sequence coverage can be optionally used by the data acquisition software to automatically determine the number of laser shots based on the precursor mass, when this is calculated on-the-fly (primarily in the case of peptides or small proteins). However, when averaged microscans are reduced to 4/spectrum, as in LC-MS/MS TDP experiments, the maximum sequence coverage obtained via HCD or UVPD on standard proteins was similar (Fig. 1 and 2). Nevertheless, although HCD showed a substantial dependence on NCE and charge state density of the selected precursor, UVPD was less dependent on equivalent parameters (i.e. number of laser pulses and precursor charge density) and led to a relatively constant level of protein sequencing. This characteristic can be fundamental in TDP, where the selection of precursor ions for tandem MS is a stochastic process. Furthermore, exposing protein cations to multiple laser shots does not seem to significantly alter the matched/total fragment ion ratio (Fig. 1D), an important feature when the statistical confidence of identifying a specific proteoform in high-throughput TDP experiments is based on a p-score (vide infra). Finally, although we clearly demonstrated that proteomics-like conditions differ dramatically from those used in targeted top-down MS experiments, we took advantage of the MW-based pre-fractionation of extracted proteins facilitated by GELFrEE to estimate an optimal number of laser pulses for each fraction to analyze by LC-MS/MS. The number of laser pulses, reported in supplemental Table S1, was progressively reduced for higher MW fractions. Applying this tuning strategy would be difficult for the more powerful 193 nm excimer lasers, where 1 or 2 laser pulses are used with limited possibility of power dampening (71).
A New Set of Database Search Parameters for UVPD
The latest technological advances in MS (in particular the development and application of hybrid tandem MS technologies) (72) are being successfully applied to top-down proteomics, necessitating the investigation of the performance of 213 nm UVPD on different proteomes with various degrees of complexity. We calculated the average number of annotated PTMs per gene product (i.e. UniProt entry) being equal to only 0.087 for P. aeruginosa, whereas this number dramatically grows to 2.76 and 2.55 for mouse and human, respectively. This difference could be explained considering that P. aeruginosa expresses fewer protein-modifying enzymes and lacks histones, but we believe that another contributing factor is that mouse and human proteomes are among those annotated more in detail because of a high number of proteomic laboratories focused on these two organisms. Regardless, the different levels of complexity of the samples is reflected in supplemental Table S2, which reports the absolute numbers and relative percentages of post-translationally modified proteoforms identified for the three sample types. For the two mammalian proteomes, the fraction of identified proteoforms exhibiting PTMs exceeds 70% of the total. 213 nm UVPD has proven to be capable of fully characterizing proteoforms (i.e. C-score >40) more often than HCD. However, previous comparative studies (43) have shown that UVPD exhibits a gap in identification performance compared with HCD. Through the analysis of the relative frequencies of occurrence of the nine fragment ions produced by UVPD, we have determined that including in the database search a subset restricted to the four product ion types produced with the highest propensities leads to improved identification results. The conclusion that searching for four ion types represents the optimal choice for UVPD data is not only reached considering the increased identification numbers (both at the protein entry and proteoform levels), but is also supported by the observation that the use of this subset of product ions primarily increases the number of UVPD identifications that are shared with HCD. In other words, it seems that the new search parameters improved the recovery of proteoforms and protein entries present within the sample. Importantly, the increase in identifications is obtained for both bacterial and mammalian proteomes, although the relative increase in identifications is more similar between the mammalian data sets. Supplemental Table S2 also indicates that the increase in proteoform identification was constant for both unmodified and modified proteoforms. To explain these results, we need to consider that the FDR calculation is based on q-values derived from p-scores. The p-score is based on a Poisson distribution (Eq. 1) (73):
| (1) |
Where P(n,f) is the probability of a random proteoform match, f is the total number of detected ions, and n the number of matching ions. A key variable is x, which corresponds to the mean probability of matching one ion by chance, Eq. 2:
| (2) |
with 111.1 being the mass of averagine (74) and Ma the defined mass accuracy (tolerance) for fragment ion match; nPossFrags is instead a parameter that specifies the number of potential fragment types that can be used to assign a backbone cleavage. The p-score originally used nPossFrags = 2, as it was developed for beam-style CID, where only b- and y-ions are typically produced. This number needs to be dramatically increased for UVPD (nPossFrags = 9) if all product ions are considered (and this is what was done for the standard search in this study and in the past (42, 43)). However, this leads to an overestimation of x, as normally most of these 9 product ions are not actually produced by UVPD, or their relative abundance is low enough to prevent their detection unless the spectral SNR is increased by extensive transient/spectrum averaging. Including all the 9 canonical ion types would make sense only if they were produced with similar frequency, as increasing the number of ion types considered generally decreases the p-score obtained for a given number of matched ions: in other words, if 9 ion types are considered, reaching good p-score values (for passing the FDR cutoff) requires a higher number of matched ions in a spectrum (supplemental Fig. S16). This, combined with the fact that to offset increased false-positive ion match rates the FDR cutoff shifts toward stricter p-score requirements as the number of considered ion types increases, explains the reduced identification rates when 9 product ion types are used for the database search. Considering that the number of identified proteoforms increased by 35 and 39% (for human and mouse data sets, respectively) when setting nPossFrags = 4, we resolved to include only a+, a, x+, and y- product ions in future database searches of UVPD LC-MS/MS top-down data.
Why 213 nm UVPD Can Characterize Proteoforms Better than HCD
An additional observation supporting the move from nine to four product ions for UVPD searches is that the C-score distribution does not change when considering only the most frequently matched fragment ion types, as shown in Fig. 5D. This is true despite a non-negligible decrease in sequence coverage when the five least abundant fragment ion types are removed (Fig. 5C and supplemental Fig. S11C). Based on a Bayesian model, the Characterization score (50) is used here as in previous large-scale top-down studies (43, 55) to distinguish three categories of identified proteoforms: those with C-score <3 that are identified without possibility of knowing the exact position of the modifications they carry, those with C-score comprised between 3 and 40, where the localization of modification can be narrowed down but not unambiguously assigned, and those with C-score >40 where modifications are assumed precisely localized based on the matched product ions. Achieving complete molecular characterization of the proteoforms identified with high statistical confidence is of primary importance to take full advantage of the theoretical superiority of the top-down approach over the reductionist bottom-up one, especially in the case of proteoforms carrying combinations of multiple genetic/chemical modifications, as discussed also in a recent piece by the Consortium for Top-Down Proteomics (75). Although HCD outperforms UVPD with regard to the numbers of proteoforms identified at 1% FDR, our study shows that the C-score distributions generated by accounting for either canonical or reduced groups of UVPD product ion types are substantially unbalanced in favor of fully characterized proteoforms, whereas in HCD experiments the fraction of proteoforms with C-score >40 is about one half or one third of the total in the case of human and mouse proteomes, respectively (supplemental Fig. S7). Remarkably, these results derived from LC-MS/MS experiments with limited spectral averaging applied to MS2 spectra (4 microscans), a condition that should have favored the vibrational excitation ion activation because of its higher efficiency of conversion of precursor ion current into product ion current. Further, supplemental Fig. S9 and S10 clearly show that for proteoforms identified by both HCD and UVPD, with overall similar sequence coverage, UVPD normally characterizes the proteoforms to a higher degree than HCD. With all this considered, it is apparent that the average sequence coverage alone cannot explain UVPD's higher efficiency in proteoform characterization. It is therefore necessary to move the focus from sequence coverage to product ions and their features in order to fully clarify this phenomenon. We ultimately determined two main reasons that explain the observed differences between HCD and UVPD. First, a recent study by Lyon et al. based on TDP experiments (fully comparable to those performed here (76)) suggests that UVPD generates a lower fraction of internal fragments than HCD. Although tools for matching internal fragments and manually validating their presence in top-down MS spectra exist (48), such product ions are not used in database searches and therefore cannot contribute to proteoform characterization in high-throughput studies (and for the same reason they have a negative impact on the p-score calculation). A secondary consequence of multiple collisional fragmentation events (leading to the production of internal fragments) is also that termini-containing large product ions are reduced to smaller ones. Supplemental Fig. S17 displays the mass distribution of matched product ions from HCD and 213 nm UVPD data sets, and for each of the three analyzed proteomes UVPD-generated fragments are on average larger than HCD fragments. This might also suggest that UVPD cleaves more often in the central portion of large polypeptide chains than HCD, perhaps because its cleavage mechanism is less dependent on the position of protonation sites compared with vibrational excitation activation (77, 78). A recent study based on the fragmentation of low-charged proteins by HCD and UVPD supports this assertion (79). This hypothesis leads to the second reason that explains the improved characterization achieved by UVPD, which is its lower cleavage specificity compared with HCD. A comparison of several graphical fragmentation maps of the same proteoform obtained by HCD and UVPD, like those reported in supplemental Fig. S10, suggest that the primary, secondary or even tertiary structure of a proteoform cation in the gas phase might play a more important role in directing the fragmentation of a polypeptide backbone for HCD than for UVPD. Supplemental Fig. S18 reports the frequency of consecutive backbone cleavages produced by HCD and UVPD. The histogram indicates a similar general behavior between the two activation types, as most of the backbone cleavages they produce are located alone (i.e. followed and preceded by unassigned backbone cleavage sites) or in short series of two or three consecutive cleavages along the protein sequence. However, the very short series are more common for UVPD, whereas the long series (i.e. four or more consecutive assigned cleavages) are primarily present in HCD spectra. The logical implication is that HCD might result in complete sequence coverage of certain portions of a protein sequence with a concurrent lack of characterization of other sequence portions—potentially those where PTMs or other sources of variation might occur, which is precisely what is observed in supplemental Fig. S10C and S10D. The randomness of UVPD backbone cleavages, likely because of direct dissociation (where electronic excitation leads to fast backbone cleavage without energy redistribution through vibrational states), is further supported by the analysis of cleavage propensity maps. The cleavage propensity of a product ion can be defined as the relative frequency with which this product ion cleaves between a specific amino acid pair. Fig. 7A and 7B display the heatmaps that summarize the cleavage propensities of b- and y-ions generated by HCD (considering the H. sapiens data set containing a few thousand identified PrSMs). In full agreement with the available literature regarding collisional activation of polypeptides (80–85), HCD product ions are frequently produced at the N-terminus of Pro and at the C-terminus of Asp (and, to a lower extent, of Glu). When we then consider the four most abundant product ions in 213 nm UVPD (the N-terminal containing a+- and a-ions, and the C-terminal containing x+- and y–ions), some fragmentation hotspots are apparent (Fig. 7C and 7D, and supplemental Fig. S19); however, the favorable residue pairs differ substantially passing from one type of product ion to the other. Although related a+- and a-ions show similar cleavage propensities (Fig. 7C and supplemental Fig. S19A, respectively), it is also noticeable that the latter ion type seems to appear at the C-terminal side of Cys with slightly higher frequency. More surprisingly, the complementary x+-ion (Fig. 7D) shows different cleavage frequencies, and it seems to occur frequently at the N-terminus of proline and, to a lesser extent, of cysteine. Importantly, all 213 nm UVPD product ions seem to be frequently produced next to phenylalanine and tyrosine (and on their C-terminal side), in agreement with reports based on 193 nm UVPD (79). The frequency of cleavage at the C-terminus of these aromatic residues is also the main difference between the cleavage propensities of HCD-produced y-ions and UVPD produced y–ions (that also show a pronounced “proline effect”).
Fig. 7.

Heatmap of fragmentation propensities for HCD and UVPD. Matched backbone cleavages are indicated by residue pairs. For all panels, X X′ (columns) refers to fragmentation occurring C-terminal to the amino acid residue, whereas X X′ (rows) refers to fragmentation occurring N-terminal to the amino acid residue. A and B, display propensities for matched b- and y-ions generated by HCD, respectively. C and D, display propensities for matched a+- and x+-ions generated by UVPD, respectively The H. sapiens data set was considered to generate the heatmaps. White color indicates average cleavage frequency.
Proline Effect in Hydrogen-depleted y-ion Series
The propensity of cleavage at the N-terminal side of Pro is characteristic not only of y–ions but, in an even more pronounced fashion, also of doubly hydrogen-deficient y-2-ions. First reports on the generation of y-2-ions by UVPD linked their production specifically to the use of 213 nm photons for inducing photodissociation of the polypeptide backbone (45, 86). Although our study is the first to systematically investigate the cleavage propensity of y-2-ions across a large dataset based on the fragmentation of intact proteins, the previous studies, based on the analysis of shorter polypeptides, also reported the generation of y-2-ions at the N-terminus of Pro. Furthermore, in agreement with the above-mentioned peptide-based studies, y-2-ions were never reported for UVPD top-down experiments using 193 nm lasers. Finally, y-2-ions are not uniquely produced by 213 nm UVPD. Their generation has been reported also in electron ionization dissociation (EID) experiments aimed at the characterization of proteins under native MS conditions (87). Although some similarities between EID and 193 nm UVPD have been proposed with respect to their use in native MS experiments, further studies will be needed to understand if the proline effect observed for y-2-ions in 213 nm UVPD is found also in EID.
Conclusions
In this study we have evaluated the use of 213 nm UVPD for top-down proteomics. We also reported on the frequent generation of a product ion type, y-2, that seems specific for UVPD using 213 nm photons and that should be considered in future iterations of software for the analysis of targeted UVPD tandem mass spectra. Importantly, an in-depth analysis of UVPD-derived product ions led to improved outcomes of database searches for large-scale TDP. Although at the current state HCD is still capable of returning more proteoform identifications than UVPD, we envision that our findings will assist UVPD to play a more prominent role in TDP applications in disease biology research and clinical studies, where its improved capability for proteoform characterization could be used in discovery and validation studies by a larger number of proteomic laboratories.
DATA AVAILABILITY
All the RAW files referring to LC-MS experiments have been deposited into MassIVE (http://massive.ucsd.edu/) with the identifier MSV000083972.
Supplementary Material
Acknowledgments
We thank Thomas M. Vondriska (UCLA) for providing mouse cardiac myocytes and Michael W. Senko (Thermo Scientific) for fruitful discussion.
Footnotes
* This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Grant No. P41 GM108569 for the National Resource for Translational and Developmental Proteomics (NRTDP). Several authors are involved in software commercialization and Thermo Fisher Scientific is an Industrial Collaborator of the NRTDP. The authors have declared a conflict of interest.
 This article contains supplemental Figures and Tables.
This article contains supplemental Figures and Tables.
1 The abbreviations used are:
- BUP
- bottom-up proteomics
- UVPD
- ultraviolet photodissociation
- HCD
- higher-energy collisional dissociation
- TDP
- top-down proteomics
- MDP
- middle-down proteomics
- FTMS
- Fourier transform mass spectrometry
- SNR
- signal-to-noise ratio
- ETD
- electron transfer dissociation
- ECD
- electron capture dissociation
- C-score
- characterization score
- PTMs
- post-translational modifications
- LC-MS
- liquid chromatography-mass spectrometry
- FDR
- false-discovery rate.
REFERENCES
- 1. Cox J., and Mann M. (2011) Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem. 80, 273–299 [DOI] [PubMed] [Google Scholar]
- 2. Gillet L. C., Leitner A., and Aebersold R. (2016) Mass spectrometry applied to bottom-up proteomics: entering the high-throughput era for hypothesis testing. Annu. Rev. Anal. Chem. 9, 449–472 [DOI] [PubMed] [Google Scholar]
- 3. Laskay U. A., Srzentic K., Monod M., and Tsybin Y. O. (2014) Extended bottom-up proteomics with secreted aspartic protease Sap9. J. Proteomics 110, 20–31 [DOI] [PubMed] [Google Scholar]
- 4. Srzentic K., Fornelli L., Laskay U. A., Monod M., Beck A., Ayoub D., and Tsybin Y. O. (2014) Advantages of extended bottom-up proteomics using Sap9 for analysis of monoclonal antibodies. Anal. Chem. 86, 9945–9953 [DOI] [PubMed] [Google Scholar]
- 5. Wu C., Tran J. C., Zamdborg L., Durbin K. R., Li M., Ahlf D. R., Early B. P., Thomas P. M., Sweedler J. V., and Kelleher N. L. (2012) A protease for 'middle-down' proteomics. Nat. Methods 9, 822–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Srzentic K., Zhurov K. O., Lobas A. A., Nikitin G., Fornelli L., Gorshkov M. V., and Tsybin Y. O. (2018) Chemical-Mediated Digestion: An Alternative Realm for Middle-down Proteomics? J. Proteome Res. 17, 2005–2016 [DOI] [PubMed] [Google Scholar]
- 7. Smith L. M., and Kelleher N. L. (2013) Proteoform: a single term describing protein complexity. Nat. Methods 10, 186–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Toby T. K., Fornelli L., and Kelleher N. L. (2016) Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 9, 499–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fornelli L., Toby T. K., Schachner L. F., Doubleday P. F., Srzentic K., DeHart C. J., and Kelleher N. L. (2018) Top-down proteomics: Where we are, where we are going? J. Proteomics 175, 3–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Compton P. D., Kelleher N. L., and Gunawardena J. (2018) Estimating the distribution of protein post-translational modification states by mass spectrometry. J. Proteome Res. 17, 2727–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nesvizhskii A. I., and Aebersold R. (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 [DOI] [PubMed] [Google Scholar]
- 12. Venne A. S., Kollipara L., and Zahedi R. P. (2014) The next level of complexity: crosstalk of posttranslational modifications. Proteomics 14, 513–524 [DOI] [PubMed] [Google Scholar]
- 13. Zheng Y. P., Fornelli L., Compton P. D., Sharma S., Canterbury J., Mullen C., Zabrouskov V., Fellers R. T., Thomas P. M., Licht J. D., Senko M. W., and Kelleher N. L. (2016) Unabridged analysis of human histone H3 by differential top-down mass spectrometry reveals hypermethylated proteoforms from MMSET/NSD2 overexpression. Mol. Cell. Proteomics 15, 776–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ntai I., Fornelli L., DeHart C. J., Hutton J. E., Doubleday P. F., LeDuc R. D., van Nispen A. J., Fellers R. T., Whiteley G., Boja E. S., Rodriguez H., and Kelleher N. L. (2018) Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk. Proc. Natl. Acad. Sci. U.S.A. 115, 4140–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scigelova M., Hornshaw M., Giannakopulos A., and Makarov A. (2011) Fourier transform mass spectrometry. Mol. Cell. Proteomics 10, M111009431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Olsen J. V., Macek B., Lange O., Makarov A., Horning S., and Mann M. (2007) Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 4, 709–712 [DOI] [PubMed] [Google Scholar]
- 17. Durbin K. R., Skinner O. S., Fellers R. T., and Kelleher N. L. (2015) Analyzing internal fragmentation of electrosprayed ubiquitin ions during beam-type collisional dissociation. J. Am. Soc. Mass Spectrom. 26, 782–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhurov K. O., Fornelli L., Wodrich M. D., Laskay U. A., and Tsybin Y. O. (2013) Principles of electron capture and transfer dissociation mass spectrometry applied to peptide and protein structure analysis. Chem. Soc. Rev. 42, 5014–5030 [DOI] [PubMed] [Google Scholar]
- 19. Zubarev R. A., Kelleher N. L., and McLafferty F. W. (1998) Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc. 120, 3265–3266 [DOI] [PubMed] [Google Scholar]
- 20. Syka J. E., Coon J. J., Schroeder M. J., Shabanowitz J., and Hunt D. F. (2004) Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 101, 9528–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fornelli L., Ayoub D., Aizikov K., Liu X. W., Damoc E., Pevzner P. A., Makarov A., Beck A., and Tsybin Y. O. (2017) Top-down analysis of immunoglobulin G isotypes 1 and 2 with electron transfer dissociation on a high-field Orbitrap mass spectrometer. J. Proteomics 159, 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ge Y., Rybakova I. N., Xu Q. G., and Moss R. L. (2009) Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. Proc. Natl. Acad. Sci. U.S.A. 106, 12658–12663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ge Y., Lawhorn B. G., ElNaggar M., Strauss E., Park J. H., Begley T. P., and McLafferty F. W. (2002) Top down characterization of larger proteins (45 kDa) by electron capture dissociation mass spectrometry. J. Am. Chem. Soc. 124, 672–678 [DOI] [PubMed] [Google Scholar]
- 24. Fornelli L., Parra J., Hartmer R., Stoermer C., Lubeck M., and Tsybin Y. O. (2013) Top-down analysis of 30–80 kDa proteins by electron transfer dissociation time-of-flight mass spectrometry. Anal. Bioanal. Chem. 405, 8505–8514 [DOI] [PubMed] [Google Scholar]
- 25. Good D. M., Wirtala M., McAlister G. C., and Coon J. J. (2007) Performance characteristics of electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 6, 1942–1951 [DOI] [PubMed] [Google Scholar]
- 26. Anderson L. C., DeHart C. J., Kaiser N. K., Fellers R. T., Smith D. F., Greer J. B., LeDuc R. D., Blakney G. T., Thomas P. M., Kelleher N. L., and Hendrickson C. L. (2017) Identification and characterization of human proteoforms by top-down LC-21 Tesla FT-ICR mass spectrometry. J. Proteome Res. 16, 1087–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anderson L. C., Karch K. R., Ugrin S. A., Coradin M., English A. M., Sidoli S., Shabanowitz J., Garcia B. A., and Hunt D. F. (2016) Analyses of histone proteoforms using front-end electron transfer dissociation-enabled orbitrap instruments. Mol. Cell. Proteomics 15, 975–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Y., Riley N. M., Sun L., Hebert A. S., Yan X., Westphall M. S., Rush M. J., Zhu G., Champion M. M., Mba Medie F., Champion P. A., Coon J. J., and Dovichi N. J. (2015) Coupling capillary zone electrophoresis with electron transfer dissociation and activated ion electron transfer dissociation for top-down proteomics. Anal. Chem. 87, 5422–5429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Riley N. M., Sikora J. W., Seckler H. S., Greer J. B., Fellers R. T., LeDuc R. D., Westphall M. S., Thomas P. M., Kelleher N. L., and Coon J. J. (2018) The value of activated ion electron transfer dissociation for high-throughput top-down characterization of intact proteins. Anal. Chem. 90, 8553–8560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brodbelt J. S. (2014) Photodissociation mass spectrometry: new tools for characterization of biological molecules. Chem. Soc. Rev. 43, 2757–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Little D. P., Speir J. P., Senko M. W., O'Connor P. B., and McLafferty F. W. (1994) Infrared multiphoton dissociation of large multiply charged ions for biomolecule sequencing. Anal. Chem. 66, 2809–2815 [DOI] [PubMed] [Google Scholar]
- 32. Julian R. R. (2017) The mechanism behind top-down UVPD experiments: making sense of apparent contradictions. J. Am. Soc. Mass Spectrom. 28, 1823–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zabuga A. V., Kamrath M. Z., Boyarkin O. V., and Rizzo T. R. (2014) Fragmentation mechanism of UV-excited peptides in the gas phase. J. Chem. Phys. 141, 154309. [DOI] [PubMed] [Google Scholar]
- 34. Bowers W. D., Delbert S. S., Hunter R. L., and Mciver R. T. (1984) Fragmentation of oligopeptide ions using ultraviolet-laser radiation and fourier-transform mass-spectrometry. J Am Chem. Soc. 106, 7288–7289 [Google Scholar]
- 35. Hunt D. F., Shabanowitz J., and Yates J. R. (1987) Peptide sequence-analysis by laser photodissociation fourier-transform mass-spectrometry. J. Chem. Soc. Chem. Comm. 548–550 [Google Scholar]
- 36. Thompson M. S., Cui W. D., and Reilly J. P. (2004) Fragmentation of singly charged peptide ions by photodissociation at lambda=157 nm. Angew. Chem. Int. Edit. 43, 4791–4794 [DOI] [PubMed] [Google Scholar]
- 37. Fung Y. M. E., Kjeldsen F., Silivra O. A., Chan T. W. D., and Zubarev R. A. (2005) Facile disulfide bond cleavage in gaseous peptide and protein cations by ultraviolet photodissociation at 157 nm. Angew. Chem. Int. Edit. 44, 6399–6403 [DOI] [PubMed] [Google Scholar]
- 38. Madsen J. A., Boutz D. R., and Brodbelt J. S. (2010) Ultrafast ultraviolet photodissociation at 193 nm and its applicability to proteomic workflows. J. Proteome Res. 9, 4205–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shaw J. B., Li W. Z., Holden D. D., Zhang Y., Griep-Raming J., Fellers R. T., Early B. P., Thomas P. M., Kelleher N. L., and Brodbelt J. S. (2013) Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J. Am. Chem. Soc. 135, 12646–12651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tamara S., Dyachenko A., Fort K. L., Makarov A. A., Scheltema R. A., and Heck A. J. (2016) Symmetry of charge partitioning in collisional and UV photon-induced dissociation of protein assemblies. J. Am. Chem. Soc. 138, 10860–10868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morrison L. J., and Brodbelt J. S. (2016) 193 nm ultraviolet photodissociation mass spectrometry of tetrameric protein complexes provides insight into quaternary and secondary protein topology. J. Am. Chem. Soc. 138, 10849–10859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cannon J. R., Cammarata M. B., Robotham S. A., Cotham V. C., Shaw J. B., Fellers R. T., Early B. P., Thomas P. M., Kelleher N. L., and Brodbelt J. S. (2014) Ultraviolet photodissociation for characterization of whole proteins on a chromatographic time scale. Anal. Chem. 86, 2185–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cleland T. P., DeHart C. J., Fellers R. T., VanNispen A. J., Greer J. B., LeDuc R. D., Parker W. R., Thomas P. M., Kelleher N. L., and Brodbelt J. S. (2017) High-throughput analysis of intact human proteins using UVPD and HCD on an orbitrap mass spectrometer. J. Proteome Res. 16, 2072–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dilillo M., de Graaf E. L., Yadav A., Belov M. E., and McDonnell L. A. (2018) Ultraviolet photodissociation of ESI and MALDI generated protein ions on a Q-Exactive mass spectrometer. J. Proteome Res. 18, 557–564 [DOI] [PubMed] [Google Scholar]
- 45. Girod M., Sanader Z., Vojkovic M., Antoine R., MacAleese L., Lemoine J., Bonacic-Koutecky V., and Dugourd P. (2015) UV photodissociation of proline-containing peptide ions: insights from molecular dynamics. J. Am. Soc. Mass Spectrom. 26, 432–443 [DOI] [PubMed] [Google Scholar]
- 46. Halim M. A., Girod M., MacAleese L., Lemoine J., Antoine R., and Dugourd P. (2016) Combined infrared multiphoton dissociation with ultraviolet photodissociation for ubiquitin characterization. J. Am. Soc. Mass Spectrom. 27, 1435–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brodie N. I., Huguet R., Zhang T., Viner R., Zabrouskov V., Pan J. X., Petrotchenko E. V., and Borchers C. H. (2018) Top-down hydrogen-deuterium exchange analysis of protein structures using ultraviolet photodissociation. Anal. Chem. 90, 3079–3082 [DOI] [PubMed] [Google Scholar]
- 48. Fornelli L., Srzentic K., Huguet R., Mullen C., Sharma S., Zabrouskoy V., Fellers R. T., Durbin K. R., Compton P. D., and Kelleher N. L. (2018) Accurate sequence analysis of a monoclonal antibody by top-down and middle-down orbitrap mass spectrometry applying multiple ion activation techniques. Anal. Chem. 90, 8421–8429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McCool E. N., Chen D., Li W., Liu Y., and Sun L. (2019) Capillary zone electrophoresis-tandem mass spectrometry with ultraviolet photodissociation (213 nm) for large-scale top–down proteomics. Anal. Methods 11, 2855–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. LeDuc R. D., Fellers R. T., Early B. P., Greer J. B., Thomas P. M., and Kelleher N. L. (2014) The C-score: a Bayesian framework to sharply improve proteoform scoring in high-throughput top down proteomics. J. Proteome Res. 13, 3231–3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rosa-Garrido M., Chapski D. J., Schmitt A. D., Kimball T. H., Karbassi E., Monte E., Balderas E., Pellegrini M., Shih T. T., Soehalim E., Liem D., Ping P., Galjart N. J., Ren S., Wang Y., Ren B., and Vondriska T. M. (2017) High-Resolution Mapping of Chromatin Conformation in Cardiac Myocytes Reveals Structural Remodeling of the Epigenome in Heart Failure. Circulation 136, 1613–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Durbin K. R., Fornelli L., Fellers R. T., Doubleday P. F., Narita M., and Kelleher N. L. (2016) Quantitation and identification of thousands of human proteoforms below 30 kDa. J. Proteome Res. 15, 976–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tran J. C., and Doucette A. A. (2008) Gel-eluted liquid fraction entrapment electrophoresis: An electrophoretic method for broad molecular weight range proteome separation. Anal. Chem. 80, 1568–1573 [DOI] [PubMed] [Google Scholar]
- 54. Shevchenko A., Wilm M., Vorm O., and Mann M. (1996) Mass spectrometric sequencing of proteins from silver stained polyacrylamide gels. Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 55. Fornelli L., Durbin K. R., Fellers R. T., Early B. P., Greer J. B., LeDuc R. D., Compton P. D., and Kelleher N. L. (2017) Advancing top-down analysis of the human proteome using a benchtop quadrupole-orbitrap mass spectrometer. J. Proteome Res. 16, 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Toby T. K., Fornelli L., Srzentic K., DeHart C. J., Levitsky J., Friedewald J., and Kelleher N. L. (2019) A comprehensive pipeline for translational top-down proteomics from a single blood draw. Nat. Protoc. 14, 119–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Meng F. Y., Cargile B. J., Miller L. M., Forbes A. J., Johnson J. R., and Kelleher N. L. (2001) Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat. Biotechnol. 19, 952–957 [DOI] [PubMed] [Google Scholar]
- 58. Fenyo D., and Beavis R. C. (2003) A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem. 75, 768–774 [DOI] [PubMed] [Google Scholar]
- 59. LeDuc R. D., Fellers R. T., Early B. P., Greer J. B., Shams D. P., Thomas P., and Kelleher N. L. (2019) Accurate estimation of context-dependent false discovery rates in top-down proteomics. Mol. Cell. Proteomics 18, 796–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Higdon R., Haynes W., and Kolker E. (2010) Meta-analysis for Protein identification: a case study on yeast data. Omics 14, 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fellers R. T., Greer J. B., Early B. P., Yu X., LeDuc R. D., Kelleher N. L., and Thomas P. M. (2015) ProSight Lite: graphical software to analyze top-down mass spectrometry data. Proteomics 15, 1235–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. DeHart C. J., Fellers R. T., Fornelli L., Kelleher N. L., and Thomas P. M. (2017) Bioinformatics analysis of top-down mass spectrometry data with ProSight Lite. Methods Mol. Biol. 1558, 381–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Haverland N. A., Skinner O. S., Fellers R. T., Tariq A. A., Early B. P., LeDuc R. D., Fornelli L., Compton P. D., and Kelleher N. L. (2017) Defining gas-phase fragmentation propensities of intact proteins during native top-down mass spectrometry. J. Am. Soc. Mass Spectrom. 28, 1203–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Biemann K. (1990) Nomenclature for Peptide Fragment Ions (Positive-Ions). Method Enzymol. 193, 886–887 [DOI] [PubMed] [Google Scholar]
- 65. Sobott F., Watt S. J., Smith J., Edelmann M. J., Kramer H. B., and Kessler B. M. (2009) Comparison of CID versus ETD based MS/MS fragmentation for the analysis of protein ubiquitination. J. Am. Soc. Mass Spectrom. 20, 1652–1659 [DOI] [PubMed] [Google Scholar]
- 66. Good D. M., Wirtala M., McAlister G. C., and Coon J. J. (2007) Performance characteristics of electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 6, 1942–1951 [DOI] [PubMed] [Google Scholar]
- 67. Mao Y., Valeja S. G., Rouse J. C., Hendrickson C. L., and Marshall A. G. (2013) Top-down structural analysis of an intact monoclonal antibody by electron capture dissociation-Fourier transform ion cyclotron resonance-mass spectrometry. Anal. Chem. 85, 4239–4246 [DOI] [PubMed] [Google Scholar]
- 68. Fornelli L., Damoc E., Thomas P. M., Kelleher N. L., Aizikov K., Denisov E., Makarov A., and Tsybin Y. O. (2012) Analysis of intact monoclonal antibody IgG1 by electron transfer dissociation orbitrap FTMS. Mol. Cell. Proteomics 11, 1758–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Srzentic K., Nagornov K. O., Fornelli L., Lobas A. A., Ayoub D., Kozhinov A. N., Gasilova N., Menin L., Beck A., Gorshkov M. V., Aizikov K., and Tsybin Y. O. (2018) Multiplexed middle-down mass spectrometry as a method for revealing light and heavy chain connectivity in a monoclonal antibody. Anal. Chem. 90, 12527–12535 [DOI] [PubMed] [Google Scholar]
- 70. Riley N. M., Westphall M. S., and Coon J. J. (2015) Activated ion electron transfer dissociation for improved fragmentation of intact proteins. Anal. Chem. 87, 7109–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cotham V. C., and Brodbelt J. S. (2016) Characterization of therapeutic monoclonal antibodies at the subunit-level using middle-down 193 nm ultraviolet photodissociation. Anal. Chem. 88, 4004–4013 [DOI] [PubMed] [Google Scholar]
- 72. Brunner A. M., Lossl P., Liu F., Huguet R., Mullen C., Yamashita M., Zabrouskov V., Makarov A., Altelaar A. F., and Heck A. J. (2015) Benchmarking multiple fragmentation methods on an orbitrap fusion for top-down phospho-proteoform characterization. Anal. Chem. 87, 4152–4158 [DOI] [PubMed] [Google Scholar]
- 73. Meng F., Cargile B. J., Miller L. M., Forbes A. J., Johnson J. R., and Kelleher N. L. (2001) Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat. Biotechnol. 19, 952–957 [DOI] [PubMed] [Google Scholar]
- 74. Senko M. W., Beu S. C., and McLaffertycor F. W. (1995) Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass Spectrom. 6, 229–233 [DOI] [PubMed] [Google Scholar]
- 75. Smith L. M., Thomas P. M., Shortreed M. R., Schaffer L. V., Fellers R. T., LeDuc R. D., Tucholski T., Ge Y., Agar J. N., Anderson L. C., Chamot-Rooke J., Gault J., Loo J. A., Pasa-Tolic L., Robinson C. V., Schluter H., Tsybin Y. O., Vilaseca M., Vizcaino J. A., Danis P. O., and Kelleher N. L. (2019) A five-level classification system for proteoform identifications. Nat. Methods 16, 939–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lyon Y. A., Riggs D., Fornelli L., Compton P. D., and Julian R. R. (2018) The ups and downs of repeated cleavage and internal fragment production in top-down proteomics. J. Am. Soc. Mass Spectrom. 29, 150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dongre A. R., Jones J. L., Somogyi A., and Wysocki V. H. (1996) Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: Evidence for the mobile proton model. J. Am. Chem. Soc. 118, 8365–8374 [Google Scholar]
- 78. Wysocki V. H., Tsaprailis G., Smith L. L., and Breci L. A. (2000) Special feature: Commentary - Mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom. 35, 1399–1406 [DOI] [PubMed] [Google Scholar]
- 79. Bashyal A., Sanders J. D., Holden D. D., and Brodbelt J. S. (2019) Top-down analysis of proteins in low charge states. J. Am. Soc. Mass Spectrom. 30, 704–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Huang Y. Y., Triscari J. M., Tseng G. C., Pasa-Tolic L., Lipton M. S., Smith R. D., and Wysocki V. H. (2005) Statistical characterization of the charge state and residue dependence of low-energy CID peptide dissociation patterns. Anal. Chem. 77, 5800–5813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Huang Y. Y., Tseng G. C., Yuan S. S., Pasa-Tolic L., Lipton M. S., Smith R. D., and Wysocki V. H. (2008) A data-mining scheme for identifying peptide structural motifs responsible for different MS/MS fragmentation intensity patterns. J. Proteome Res. 7, 70–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vaisar T., and Urban J. (1996) Probing the proline effect in CID of protonated peptides. J. Mass Spectrom. 31, 1185–1187 [DOI] [PubMed] [Google Scholar]
- 83. Schaaff T. G., Cargile B. J., Stephenson J. L., and McLuckey S. A. (2000) Ion trap collisional activation of the (M+2H)(2+)-(M+17H)(17+) ions of human hemoglobin beta-chain. Anal. Chem. 72, 899–907 [DOI] [PubMed] [Google Scholar]
- 84. Dong N. P., Zhang L. X., and Liang Y. Z. (2011) A comprehensive investigation of proline fragmentation behavior in low-energy collision-induced dissociation peptide mass spectra. Int. J. Mass Spectrom. 308, 89–97 [Google Scholar]
- 85. Michalski A., Neuhauser N., Cox J., and Mann M. (2012) A systematic investigation into the nature of tryptic HCD spectra. J. Proteome Res. 11, 5479–5491 [DOI] [PubMed] [Google Scholar]
- 86. Talbert L. E., and Julian R. R. (2018) Directed-backbone dissociation following bond-specific carbon-sulfur UVPD at 213 nm. J. Am. Soc. Mass Spectrom. 29, 1760–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li H., Sheng Y., McGee W., Cammarata M., Holden D., and Loo J. A. (2017) Structural characterization of native proteins and protein complexes by electron ionization dissociation-mass spectrometry. Anal. Chem. 89, 2731–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the RAW files referring to LC-MS experiments have been deposited into MassIVE (http://massive.ucsd.edu/) with the identifier MSV000083972.