

ABSTRACT
Progeroid syndromes induced by mutations in lamin A or in its interactors – named progeroid laminopathies – are model systems for the dissection of the molecular pathways causing physiological and premature aging. A large amount of data, based mainly on the Hutchinson Gilford Progeria syndrome (HGPS), one of the best characterized progeroid laminopathy, has highlighted the role of lamins in multiple DNA activities, including replication, repair, chromatin organization and telomere function. On the other hand, the phenotypes generated by mutations affecting genes directly acting on DNA function, as mutations in the helicases WRN and BLM or in the polymerase polδ, share many of the traits of progeroid laminopathies. These evidences support the hypothesis of a concerted implication of DNA function and lamins in aging. We focus here on these aspects to contribute to the comprehension of the driving forces acting in progeroid syndromes and premature aging.
KEYWORDS: Lamin, DNA replication, progeria, nuclear lamina, aging, DNA damage
Progeroid syndromes are rare genetic diseases characterized by reduced lifespan and by premature appearance of symptoms related to physiological aging. For a majority of progeroid syndromes the causative mutation has been identified pointing to the role of DNA function and of lamins in these diseases. We report here on these syndromes, on their genetics and on the molecular mechanisms involved in generating the related phenotypes, focusing on the interrelationships between lamins and DNA function.
Mutations affecting DNA function cause progeroid syndromes
Given the direct connection between DNA function and cell senescence and the implication of senescence in aging[1], it is not surprising that many progeroid syndromes are caused by mutations in genes encoding for DNA repair and DNA maintenance enzymes (Tables 1 and 2). These include the Werner Syndrome caused by mutations in the DNA helicase WRN[2], the Bloom Syndrome caused by mutation in the helicase BLM [3], the Rothmund-Thomson syndrome caused by mutations in the gene RECQL4 [4], another DNA helicase, the Cockayne syndrome involving either the ERCC8 or ERCC6 genes, that are implicated in DNA repair [5], the Xeroderma pigmentosum also caused by mutations in the DNA repair machinery elements [6], the Dyskeratosis congenita involving genes implicated in telomere function, including TERT, TERC and DKC1 [7], and the mosaic variegated aneuploidy syndrome caused by genes implicated in cell division[8].
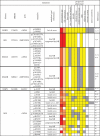
Table 1.
Progeroid syndromes caused by lamin and DNA function linked genes. The table highlights common characteristics of genetically different progeroid syndromes. The colored map indicates that progeroid traits are prevalently modulated in the same direction in progerias generated by either lamin (LMNA, ZMPSTE24) or DNA function (POLD1, SPRTN, AKTIP) mutations. hm: homozygous; hz: heterozygous; AD: autosomic dominant; AR: autosomic recessive; de novo: sporadic mutation; RJALS: Ruijs-Alfs syndrome; SCC: squamous cells carcinoma; HCC: hepatocellular carcinoma; H: hypomorphic mutation; kof: knock out first; * model organism mouse.
 |
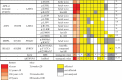 |
Table 2.
Dissection of DNA functions impaired in progeroid syndromes of different origins. Alterations in one or more of the DNA functions, caused by mutations in the genes indicated, result in a premature aging phenotype. DC: Dyskeratosis congenita; WS: Werner syndrome; BS: Bloom syndrome; R-TS: Rothmund-Thomson syndrome; CS: Cockayne syndrome; XP: Xeroderma pigmentosum. Nd: not determined.
| DNA functions altered in progeroid syndromes |
|||||||
|---|---|---|---|---|---|---|---|
| Disease | Altered gene | Telomere metabolism | Chromatin remodeling | DNA transcription | DNA replication | DNA repair | References |
| HGPS, MADA, APS, AWS | LMNA | altered | altered | altered | altered | altered | [15,23,29,50,104] |
| DC | TERT/TERC | altered | nd | nd | nd | nd | [7] |
| DC | DKC1 | altered | nd | nd | nd | nd | [7] |
| - | AKTIP | altered | altered | nd | altered | nd | [63,65] |
| WS | WRN | altered | altered | altered | altered | altered | [105–109] |
| BS | BLM | altered | altered | altered | altered | altered | [110–113] |
| R-TS | RECQL4 | altered | nd | nd | altered | altered | [109] |
| CS | ERCC8/ERCC6 | nd | nd | nd | nd | altered | [5] |
| RJALS | SPRTN | nd | nd | nd | altered | altered | [62,114] |
| MDPL | POLD1 | nd | nd | nd | altered | altered | [70] |
| XP | XPA | nd | nd | nd | nd | altered | [31] |
Mutations of nuclear envelope components cause progeroid syndromes
Along with DNA function genes, a major genetic cause of progeroid syndromes are mutations of nuclear envelope factors or of their interactors [9], which defines the group of progeroid laminopathies. Differently from other premature aging syndromes, progeroid laminopathies have early onset, are usually pediatric diseases, and display more severe symptoms of aging. Another feature that distinguishes progeroid laminopathies from the other premature aging syndromes, is the absence of increased cancer susceptibility (Table 1).
The most characterized progeroid laminopathy is Hutchinson Gilford Progeria syndrome (HGPS), which is caused by a mutation in the LMNA gene generating a mutant lamin A indicated as progerin [10,11]. HGPS is an extremely rare, dominant genetic disorder affecting 1 in 8 million live newborns, with a prevalence of 1 in 20 million living individuals (The Progeria Research Foundation, 2018; https://www.progeriaresearch.org/prf-by-the-numbers/). Children affected by HGPS show severe growth retardation, alopecia, loss of subcutaneous fat, prominent eyes and scalp veins, thin skin, joint stiffness and reduced bone density. They suffer from osteoporosis, atherosclerosis and cardiovascular diseases, that finally cause their premature death at an average age of 14 years [12,13]. Patients with HGPS do not develop childhood tumors, and progerin exerts a tumor-protective function [9]. Along with this, cells from HGPS show different aberrant phenotypes including misshapen nuclei and a disorganized nuclear lamina and envelope [14], altered gene expression [15], cellular senescence [16], genomic instability, revealed by the persistent DNA damage response activation and defects in the repair pathways [17,18], impaired chromatin organization [19–21] and abnormal telomere metabolism [22,23]. This wide variety of aberrant phenotypic features likely reflects the high number of different, and essential, cellular processes in which lamin A is involved.
Restrictive Dermopathy (RD) is another example of lamin related progeroid syndrome. Its causative mutations are found in the gene ZMPSTE24, whose product is implicated in the maturation of lamin A. RD clinical phenotype differs from HGPS on that it is a perinatal lethal disease characterized by a severe intrauterine growth delay. After birth, affected children display a characteristic thin, translucent, tight skin, and a typical facial dysmorphism characterized by a small pinched nose, micrognathia and mouth in a fixed ‘o’ shape. RD patients suffer from joint contractures, respiratory insufficiency in which the respiratory failure most often leads to neonatal death within several weeks of birth [24].
Along with HGPS and RD, nuclear envelope related progerias include the Atypical Werner syndrome (AWS), the Atypical Progeria syndrome (APS), the MandibuloAcral Dysplasia type A (MADA), which have been all related to mutations in LMNA, and MandibuloAcral Dysplasia type B (MADB) which has been related to mutations in ZMPSTE24. MADA is a less severe progeroid laminopathy as compared to HGPS and RD. The clinical features appear at 4 years of age and become more severe with increasing age. This disorder is characterized by growth retardation, postnatal onset of craniofacial anomalies with mandibular hypoplasia, skeletal abnormalities with progressive distal phalanges and clavicular osteolysis. Some patients show progeroid features such as thin nose, sparse, brittle hair, and sclerodermatous skin. Among other typical features there are lipodystrophy and metabolic complications due to insulin resistance [25].
Another case of progeroid laminopathy, deserving particular attention in relation with DNA function, is the Nestor Guillermo Progeria syndrome (NGPS). NGPS is defined as chronic progeria because it has a slow clinical course and long survival despite its early onset. Patients display growth retardation, although less pronounced than HGPS patients, micrognatia, lipoatrophy and major skeletal deformation with severe osteoporosis and osteolysis [12,26]. NGPS is caused by mutations in BANF1 gene encoding for the barrier-to-autointegration factor 1 (BAF1) protein. BAF1 binds the nuclear lamina related LEM proteins LAP2α, Emerin, and MAN1, and acts on DNA function by binding chromatin and histones [18,26,27]. NGPS is thus suggestive of a strong connection between nuclear lamina and DNA function in generating the progeroid phenotype.
isorder characterized by growth retardation, postnatal onset of craniofacial anomalies with mandibular hypo-plasia, skeletal abnorm alities with progressive distal phalanges, and clavicular osteolysis, in addition to skin changes like mottled pigmentati on and atrophy [Young et al., 1971; Freidenberg et al., 1992; Tudisco et al., 2000; Simha and Garg, 2002]. Some patients show progeroid (premature aging) features such as thin nose, sparse, brittle hair, and sclerodermatous (stiff and parched) skin. Among other typical features we can find lipodystrophy and metabolic complications due to insulin resistance and diabete
Genomic instability is a common trait of progeroid syndromes
Genomic instability is a trait of progeroid syndromes deriving from both mutations in lamin and in factors controlling DNA function, and is emerging as driver of aging. In fact both physiological and premature aging can be read as the consequence of cell senescence, which, in turn is generated by genomic damage.
Progerin expressing samples accumulate DNA damage, which is highlighted by the presence of intranuclear γH2AX positive foci that are induced by DNA double strand breaks (DSBs). Foci increase in number with continued passage and, at the same time, cell growth rate slows down till reaching premature senescence [28,29]. Along with this, HGPS samples exhibit persistent DNA damage checkpoint activation, including the phosphorylation of the DNA damage signaling element ATM, the activation of the checkpoint regulators Chk1 and Chk2 and that of the guardian of the genome p53 [28,29]. Moreover, DNA repair is impaired in laminopathic cells. Fibroblasts from HGPS patients and mouse embryonic fibroblasts (MEFs) derived from progeroid models, such as Zmpste24 knock out mice, are extremely sensitive to genotoxic agents as camptothecin and etoposide, which are DSBs inducers, but also to UV irradiation that typically activates the Nucleotide Excision Repair (NER) pathway [30]. This increased sensitivity is mechanistically explained by the observation that laminopathic cells display impaired recruitment of DNA repair proteins at DSBs, which then remain unrepaired. Indeed, an aberrant repair scheme has been observed in HGPS cells, characterized by delayed or inefficient recruitment of the DNA damage response factor 53BP1, of the Non Homologous End Joining (NHEJ) repair protein Ku70 and of the Homology directed repair (HDR) elements Rad50, Rad51, and by aberrant sequestration of the NER component XPA [31] at DSBs. These alterations block the correct DSB repair sequence and cause general impairment of NER [32].
Altered protein-protein interaction bridges DNA function to nuclear envelope in progeroid syndromes
HGPS patients present de novo mutations in exon 11 of LMNA gene leading to the activation of a cryptic splice site that brings to the synthesis of progerin. Progerin does not contain the lamin A cleavage site for Zmpste24, for this reason the protein does not undergo the final step of processing, giving rise to a permanently farnesylated mutant protein. In a subset of progeroid laminopathies, in MADB and RD for example, the disease is caused by compound heterozygous and homozygous mutations in FACE1 gene that encodes for Zmpste24. The mutations give rise to defective protein activity, total loss of function in RD, and lead to the accumulation of a prelamin A form that is not properly processed and retains the farnesylated C-terminus.
These results have suggested the idea that different progerias share a common functional cause in the accumulation of mutant forms of permanently farnesylated lamin A, that could be toxic for the cell. Based on this assumption, therapeutic approaches for progerias have been focused on drugs inhibiting the farnesylation of progerin and prelamin A, including lonafarnib, pravastatin and zoledronic acid [33]. However, the recent identification of progeroid clinical phenotypes not related to the accumulation of farnesylated lamin A have highlighted that this is not the full explanation for the cellular defects observed in these diseases. Indeed, for example, the majority of APS and AWS cases are caused by different mutations in exons of LMNA gene, that do not give rise to splice variants of the protein generating the accumulation of farnesylated products, but are nonetheless characterized by a progeroid phenotype. There are exceptions in which APS and AWS are linked to mutations causing both amino acid changes and the formation of truncated versions of lamin A, that theoretically could remain farnesylated [34,35]. However, its not clear if they contribute to phenotype and amino acid changes seem more important, at least in one case [36], for the pathological phenotype. Along with this, the identification of the causative mutation of NGPS in BANF1 has further underlined that progeroid laminopathies do not necessarily arise from the accumulation of toxic farnesylated mutant lamin A [18]. Accordingly, cell treatments with farnesyltransferase inhibitors, used to treat HGPS patients, revert only some phenotypic aberrant cellular traits, such as nuclear misshapen, but not all of them. For example, they do not avoid senescence or DNA damage activation in HGPS cells [28]. The same result was obtained by in vivo studies. The treatment with farnesyltransferase inhibitors of two progeria mouse models, ZMPSTE24-deficient mice and LMNAHG/+ knock in mice, ameliorates the disease phenotype but not completely, and all treated mice eventually develop severe phenotypes and prematurely die [37–39].
Further evidence supporting the idea that permanent farnesylation is not the only driver in progeria, is the recent finding that a mutant progerin, which is not farnesylated as in HGPS, when expressed in normal cells fully mimics HGPS progerin in its ability to promote senescence [40]. Similar results were obtained also in vivo, in a mouse model expressing a non-farnesylated version of progerin, LMNAnHG/+, in which the cysteine of the CaaX motif was replaced with a serine. These animals develop the disease phenotype, although in a milder form if compared to mice expressing farnesylated progerin [38]. The same group reported a second mouse model expressing a different progerin, LMNAcsmHG/+, that cannot be farnesylated as well, in which a total disease recovery was observed [41], indicating that, actually, which is the importance of permanent farnesylated products in generating pathology is not yet fully clarified.
Altered protein-protein interaction and protein mislocalization or sequestration are now proposed as the origin for the multiplicity of phenotypic traits of progeroid laminopathies. The hypothesis is that an organized lamin A network is necessary for coordinating cellular processes through the formation of macrocomplexes including specific partners. Mutant lamin A networks create abnormal interactions of proteins that act in DNA function, which, in turn, impinges on their correct activity [12,40,42,43]. This hypothesis rests on the discovery that lamin A mutants and, in particular, progerin, differentially interact with a subset of lamin A interacting proteins, and this could account for the deregulation of important cellular processes coordinated by lamins [43–46]. For example, progerin shows an altered interaction with transcription factors including Prx1 and Ing1. It induces their mislocalization and this contributes to the HGPS phenotype [15,46,47]. Progerin was also proven to be unable to interact with members of NURD chromatin remodeling complex, which was shown to contribute to chromatin defects observed in HGPS [21]. Differential interactions of progeroid cells were also observed for DNA repair proteins. Among these, DNA-PKcs, Ku80 and Ku70[48], which interact with lamin A, lose their interaction with progerin. This result indicates that lamin A is functional to the maintenance of a nucleoplasmic pool of DNA repair proteins, which is expected to facilitate their rapid recruitment to sites of DNA damage. The inability of progerin to bind them causes DNA repair impairment, leading to genomic instability [48]. Recently it was demonstrated that progerin interacts preferentially with a pivotal element of the DNA replication machinery, the proliferating cell nuclear antigen (PCNA), which clamps DNA at the forks and favors the processivity of DNA replication [49]. Differently from wild type lamin A, progerin sequesters PCNA far away from replicative forks, causing fork stalling and consequent DNA damage [40,50]. PCNA mislocalization, altering proper DNA replication, accounts for multiple cellular defects of progeroid cells including persistent DNA repair activation and genomic instability [40,50].
The accumulation of different forms, both non farnesylated and farnesylated, of prelamin A, a part from progerin, to toxic level is associated to MADA, MADB and RD progeroid laminopathies and has been also reported in association with cellular stress and senescence [51]. Recent studies analyzing the molecular basis of prelamin A-related chromatin organization changes observed in patients cells, further support the idea that aberrant lamin A could create abnormal interactions of proteins that could affect genome stability. Indeed it was demonstrated that BAF1 interacts preferentially with prelamin A rather than with mature lamin A and this interaction affects BAF1 cellular localization inducing its nuclear retention when prelamin A is abnormally accumulated. This, in turn, induces mislocalization of other chromatin remodeling factors altering the chromatin status [52]. Moreover prelamin A-BAF1 interaction is compromised by the BANF1 gene mutation occurring in NGPS and this could account, at least in part, to the pathological cellular phenotype [53]. Prelamin A is also able to interact with PCNA better than mature lamin A. Indeed, it was demonstrated that the presence of prelamin A interferes with DNA replication fork stability by sequestering PCNA away from its canonical interaction with lamin A [54].
These studies taken together indicate that progeroid defects are not only linked to farnesylation, but also to the alteration of macrocomplex formation caused by dysfunctional lamin A.
DNA replication is altered in progeroid syndromes
Different lines of evidence support the hypothesis that a prominent source of DNA damage in progeroid samples is DNA replication impairment. It has been observed that in patients’ HGPS cells and in ectopically expressing progerin cells the S-phase is prolonged, and DNA damage occurs predominantly during this phase [40]. Moreover, γH2AX foci were identified as co-localizing with phospho-RPA32 (Ser33), which is a marker of DNA replication stalled forks [50]. It was also assessed that DNA damage foci are positive for MCM7, which is a component of the DNA replication complex, remaining on site when forks are stalled or collapsed. Finally, as anticipated above, DNA replication impairment in HGPS has been linked to PCNA activity. More specifically, it has been demonstrated that progerin sequesters PCNA, creating PCNA positive intracellular aggregates which localize away from replicating DNA, this causes processivity defects, indicated by the decreased rate of replication fork progression observed in HGPS cells, along with replication stress and consequent DSBs formation [40,50].
Several groups have focused on the molecular dissection of the link between lamins and DNA replication. A first elementary aspect underlining this connection is that lamins are present at replication sites early in S-phase. Another direct relation is that lamins interact with DNA polδ and ϵ [55]. Moreover, A-type lamins are required for the elongation stage of replication [56,57]. In fact, the disruption of the lamina, obtained by using a dominant negative lamin A mutant, induces not only the mislocalization of PCNA but also that of the Replication Factor C (RFC) complex, which as PCNA, is essential for the elongation phase of DNA replication [56,57]. A-type lamins are also required for the resolution of stalled replication forks [58]. Lamin A/C depleted cells are indeed unable to restart forks after a replication stress resulting in shorter track length and in the formation of chromosomal aberrations [58]. Biochemical evidences further support the link between lamins and the replication machinery. In fact, a direct interaction between lamins and PCNA occurs through their highly conserved Ig-fold domain and this interaction is important for PCNA positioning on chromatin [59].
The relation between DNA replication and progerias is supported also from studies of progeroid syndromes caused by mutations in DNA replication genes. For example, mutations in SPRTN, encoding the PCNA interacting protein Spartan, with a role in the error-prone translesional DNA synthesis (TLS) [60], were identified in three AWS patients [61]. Spartan dysfunction leads to sustained DNA replication stress characterized by decreased DNA replication fork progression speed, incomplete DNA replication and impaired lesion bypass. These replication defects cause DNA damage that, in turn, induces aging or cancer, due to a leakage of G2/M checkpoint observed in cells from these patients [61,62]. Mice bearing hypomorphic alleles of SPRTN are growth retarded, show lipodistrophy, develop cataracts, lordokyphosis and cachexia at a young age [62]. This phenotype recalls those of other progeria models.
We observed progeroid traits in a mouse model defective in a gene implicated in DNA function named AKTIP (in humans and Ft1 in mouse) [63–65]. We described that AKTIP dysfunction causes DNA replication and telomere defects, which in turn generate DNA damage activation and cell senescence [63]. Consistently with the connection of AKTIP both with lamins and DNA function, hypomorphic mice (Ft1kof/kof, Ft1 Knock out first) displayed premature aging defects, including reduced body size, lipodistrophy, altered bone density and kyphosis [64]. We demonstrated that the phenotype is partly rescued by co-depletion of p53, pointing to the interplay between DNA damage and progeria. We also defined that AKTIP binds to the replication factors PCNA and RPA70, altogether falling into the triangular connection described here among lamins, progerias and DNA function [65].
A further link between DNA function and lamins comes from studies on mutation of POLD1. Heterozygous mutations in POLD1 have been found in patients with Mandibular hypoplasia displaying Deafness, Progeroid features and Lipodystrophy (MDPL) [66–69]. POLD1 encodes for the catalytic subunit, called p125 subunit or A unit, of polδ, the polymerase responsible for DNA lagging synthesis also involved in DNA repair [70]. Polδ has been shown to interact with lamin A/C in early S-phase [55]. Currently it is not known which is the impact of POLD1 mutations at molecular level, but it was suggested that heterozygous mutant POLD1 leads to increased numbers of stalled replication forks, which would then trigger cellular senescence [68].
Concluding remarks
Despite the rarity of progeroid syndromes, researches aimed at the understanding of the molecular pathways involved in these diseases are important, not only to find new therapeutic approaches, but also to understand the mechanisms leading to physiological aging. Among progeroid laminopathies the most widely studied is HGPS, caused by mutations leading to accumulation of aberrant lamin A that induces a wide range of pathological phenotypes, both at organismal and cellular level. In HGPS and other laminopathies the large array of aberrant phenotypes reflects the fact that lamins coordinate a variety of fundamental molecular mechanisms, ranging from chromatin organization and regulation, to nuclear structural organization and functioning. The pathogenic molecular mechanism of HGPS was initially attributed to the presence of the farnesyl group in progerin. For this reason therapeutic approaches have aimed at blocking progerin farnesylation. Recently, however, a further important interpretation has emerged from studies on progerias. The hypothesis is that mutated lamins interact differently with their partners creating an aberrant intranuclear macrocomplex scenario. Central processes are affected by this intranuclear misorganization including, importantly, DNA replication and repair. Many players are involved in this aberrant picture, including the DNA processivity factor PCNA.
The identification of DNA replication and genomic instability as drivers of the premature aging phenotype in HGPS and other progeroid diseases will help identifying new therapeutic approaches for these devastating pathologies. In addition, considering that progeroid syndromes recapitulate aspects of physiological aging, the dissection of the mediators of the disease phenotype will also give hints into the coveted comprehension of the complex, multifaceted process of aging in humans.
Funding Statement
This work was supported by the Fondazione Telethon [GEP15033]; MIUR Ateneo; MIUR Ateneo; Progeria Research Foundation [PRF 2016-67]; EMBO short-term fellowship 2017 to MC.
Acknowledgments
This work has been supported by MIUR Ateneo grants to IS and FV, by Telethon grant GEP15033 and Progeria Research foundation grant 2016-67 to IS.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Lopez-Otin C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013;153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bachrati CZ, Hickson ID.. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003;374:577–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bachrati CZ, Hickson ID. RecQ helicases: guardian angels of the DNA replication fork. Chromosoma. 2008;117:219–233. [DOI] [PubMed] [Google Scholar]
- [4].Vennos EM, James WD. Rothmund-Thomson syndrome. Dermatol Clin. 1995;13:143–150. [PubMed] [Google Scholar]
- [5].Iyama T, Wilson DM 3rd.. Elements that regulate the DNA damage response of proteins defective in cockayne syndrome. J Mol Biol. 2016;428:62–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dokal I. Dyskeratosis congenita. Hematology/The Educ Program Am Soc Hematol Am Soc Hematol Educ Program. 2011;2011:480–486. [DOI] [PubMed] [Google Scholar]
- [8].Suijkerbuijk SJ, Van Osch MH, Bos FL, et al. Molecular causes for BUBR1 dysfunction in the human cancer predisposition syndrome mosaic variegated aneuploidy. Cancer Res. 2010;70:4891–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kubben N, Misteli T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat Rev Mol Cell Biol. 2017;18:595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].De Sandre-Giovannoli A, Bernard R, Cau P, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. [DOI] [PubMed] [Google Scholar]
- [11].Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cau P, Navarro C, Harhouri K, et al. Nuclear matrix, nuclear envelope and premature aging syndromes in a translational research perspective. Semin Cell Dev Biol. 2014;29:125–147. [DOI] [PubMed] [Google Scholar]
- [13].Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140:2603–2624. [DOI] [PubMed] [Google Scholar]
- [14].Goldman RD, Shumaker DK, Erdos MR, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Prokocimer M, Barkan R, Gruenbaum Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell. 2013;12:533–543. [DOI] [PubMed] [Google Scholar]
- [16].Varela I, Cadinanos J, Pendas AM, et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–568. [DOI] [PubMed] [Google Scholar]
- [17].Gonzalez-Suarez I, Gonzalo S. Nurturing the genome: A-type lamins preserve genomic stability. Nucleus. 2010;1:129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hutchison CJ. The role of DNA damage in laminopathy progeroid syndromes. Biochem Soc Trans. 2011;39:1715–1718. [DOI] [PubMed] [Google Scholar]
- [19].Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shumaker DK, Dechat T, Kohlmaier A, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103:8703–8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pegoraro G, Kubben N, Wickert U, et al. Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol. 2009;11:1261–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gonzalez-Suarez I, Redwood A, Perkins S, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009;28:2414–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Benson E, Lee S, Aaronson S. Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J Cell Sci. 2010;123:2605–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McKenna T, Sola Carvajal A, Eriksson M. Skin disease in laminopathy-associated premature aging. J Invest Dermatol. 2015;135:2577–2583. [DOI] [PubMed] [Google Scholar]
- [25].Garavelli L, D’Apice MR, Rivieri F, et al. Mandibuloacral dysplasia type A in childhood. Am J Med Genet A. 2009;149A:2258–2264. [DOI] [PubMed] [Google Scholar]
- [26].Cabanillas R, Cadinanos J, Villameytide JA, et al. Nestor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet A. 2011;155A:2617–2625. [DOI] [PubMed] [Google Scholar]
- [27].Puente XS, Quesada V, Osorio FG, et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet. 2011;88:650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu Y, Rusinol A, Sinensky M, et al. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J Cell Sci. 2006;119:4644–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Musich PR, Zou Y. Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging. 2009;1:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu B, Wang J, Chan K, et al. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11:780–785. [DOI] [PubMed] [Google Scholar]
- [31].Sugitani N, Sivley RM, Perry KE, et al. XPA: A key scaffold for human nucleotide excision repair. DNA Repair. 2016;44:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu Y, Wang Y, Rusinol AE, et al. Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. FASEB J. 2008;22:603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gordon LB, Kleinman ME, Massaro J, et al. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation. 2016;134:114–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fukuchi K, Katsuya T, Sugimoto K, et al. LMNA mutation in a 45 year old Japanese subject with Hutchinson-Gilford progeria syndrome. J Med Genet. 2004;41:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Doh YJ, Kim HK, Jung ED, et al. Novel LMNA gene mutation in a patient with atypical Werner’s syndrome. Korean J Intern Med. 2009;24:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen L, Lee L, Kudlow BA, et al. LMNA mutations in atypical Werner’s syndrome. Lancet. 2003;362:440–445. [DOI] [PubMed] [Google Scholar]
- [37].Yang SH, Meta M, Qiao X, et al. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest. 2006;116:2115–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yang SH, Andres DA, Spielmann HP, et al. Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J Clin Invest. 2008;118:3291–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fong LG, Frost D, Meta M, et al. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–1623. [DOI] [PubMed] [Google Scholar]
- [40].Wheaton K, Campuzano D, Ma W, et al. Progerin-induced replication stress facilitates premature senescence in Hutchinson-Gilford progeria syndrome. Mol Cell Biol. 2017;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yang SH, Chang SY, Ren S, et al. Absence of progeria-like disease phenotypes in knock-in mice expressing a non-farnesylated version of progerin. Hum Mol Genet. 2011;20:436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Burla R, La Torre M, Saggio I. Mammalian telomeres and their partnership with lamins. Nucleus. 2016;7:187–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Serebryannyy L, Misteli T. Protein sequestration at the nuclear periphery as a potential regulatory mechanism in premature aging. J Cell Biol. 2017;217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dobrzynska A, Gonzalo S, Shanahan C, et al. The nuclear lamina in health and disease. Nucleus. 2016;7:233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dittmer TA, Sahni N, Kubben N, et al. Systematic identification of pathological lamin A interactors. Mol Biol Cell. 2014;25:1493–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kubben N, Voncken JW, Demmers J, et al. Identification of differential protein interactors of lamin A and progerin. Nucleus. 2010;1:513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Han X, Feng X, Rattner JB, et al. Tethering by lamin A stabilizes and targets the ING1 tumour suppressor. Nat Cell Biol. 2008;10:1333–1340. [DOI] [PubMed] [Google Scholar]
- [48].Kinoshita D, Nagasawa A, Shimizu I, et al. Progerin impairs vascular smooth muscle cell growth via the DNA damage response pathway. Oncotarget. 2017;8:34045–34056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. [DOI] [PubMed] [Google Scholar]
- [50].Hilton BA, Liu J, Cartwright BM, et al. Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes. FASEB J. 2017;31:3882–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cenni V, D’Apice MR, Garagnani P, et al. Mandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing. Ageing Res Rev. 2018;42:1–13. [DOI] [PubMed] [Google Scholar]
- [52].Capanni C, Squarzoni S, Cenni V, et al. Familial partial lipodystrophy, mandibuloacral dysplasia and restrictive dermopathy feature barrier-to-autointegration factor (BAF) nuclear redistribution. Cell Cycle. 2012;11:3568–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Loi M, Cenni V, Duchi S, et al. Barrier-to-autointegration factor (BAF) involvement in prelamin A-related chromatin organization changes. Oncotarget. 2016;7:15662–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cobb AM, Murray TV, Warren DT, et al. Disruption of PCNA-lamins A/C interactions by prelamin A induces DNA replication fork stalling. Nucleus. 2016;7:498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Vaara M, Itkonen H, Hillukkala T, et al. Segregation of replicative DNA polymerases during S phase: DNA polymerase epsilon, but not DNA polymerases alpha/delta, are associated with lamins throughout S phase in human cells. J Biol Chem. 2012;287:33327–33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Goldman RD, Gruenbaum Y, Moir RD, et al. Nuclear lamins: building blocks of nuclear architecture. Genes Dev. 2002;16:533–547. [DOI] [PubMed] [Google Scholar]
- [57].Spann TP, Moir RD, Goldman AE, et al. Disruption of nuclear lamin organization alters the distribution of replication factors and inhibits DNA synthesis. J Cell Biol. 1997;136:1201–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Singh M, Hunt CR, Pandita RK, et al. Lamin A/C depletion enhances DNA damage-induced stalled replication fork arrest. Mol Cell Biol. 2013;33:1210–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Shumaker DK, Solimando L, Sengupta K, et al. The highly conserved nuclear lamin Ig-fold binds to PCNA: its role in DNA replication. J Cell Biol. 2008;181:269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kim MS, Machida Y, Vashisht AA, et al. Regulation of error-prone translesion synthesis by Spartan/C1orf124. Nucleic Acids Research. 2013;41:1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lessel D, Vaz B, Halder S, et al. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet. 2014;46:1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Maskey RS, Kim MS, Baker DJ, et al. Spartan deficiency causes genomic instability and progeroid phenotypes. Nat Commun. 2014;5:5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Burla R, Carcuro M, Raffa GD, et al. AKTIP/Ft1, a new shelterin-interacting factor required for telomere maintenance. PLoS Genetics. 2015;11:e1005167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].La Torre M, Merigliano C, Burla R, et al. Mice with reduced expression of the telomere-associated protein Ft1 develop p53-sensitive progeroid traits. Aging Cell. 2018 Apr 10:e12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Burla R, Carcuro M, Torre ML, et al. The telomeric protein AKTIP interacts with A- and B-type lamins and is involved in regulation of cellular senescence. Open Biology. 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lessel D, Hisama FM, Szakszon K, et al. POLD1 germline mutations in patients initially diagnosed with Werner syndrome. Hum Mutat. 2015;36:1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pelosini C, Martinelli S, Ceccarini G, et al. Identification of a novel mutation in the polymerase delta 1 (POLD1) gene in a lipodystrophic patient affected by mandibular hypoplasia, deafness, progeroid features (MDPL) syndrome. Metabolism. 2014;63:1385–1389. [DOI] [PubMed] [Google Scholar]
- [68].Weedon MN, Ellard S, Prindle MJ, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45:947–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Shastry S, Simha V, Godbole K, et al. A novel syndrome of mandibular hypoplasia, deafness, and progeroid features associated with lipodystrophy, undescended testes, and male hypogonadism. J Clin Endocrinol Metab. 2010;95:E192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nicolas E, Golemis EA, Arora S. POLD1: central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene. 2016;590:128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Pereira S, Bourgeois P, Navarro C, et al. HGPS and related premature aging disorders: from genomic identification to the first therapeutic approaches. Mech Ageing Dev. 2008;129:449–459. [DOI] [PubMed] [Google Scholar]
- [72].Navarro CL, De Sandre-Giovannoli A, Bernard R, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13:2493–2503. [DOI] [PubMed] [Google Scholar]
- [73].Ahmad Z, Phadke SR, Arch E, et al. Homozygous null mutations in ZMPSTE24 in restrictive dermopathy: evidence of genetic heterogeneity. Clin Genet. 2012;81:158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Filesi I, Gullotta F, Lattanzi G, et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics. 2005;23:150–158. [DOI] [PubMed] [Google Scholar]
- [75].Lombardi F, Gullotta F, Columbaro M, et al. Compound heterozygosity for mutations in LMNA in a patient with a myopathic and lipodystrophic mandibuloacral dysplasia type A phenotype. J Clin Endocrinol Metab. 2007;92:4467–4471. [DOI] [PubMed] [Google Scholar]
- [76].Cao H, Hegele RA. LMNA is mutated in Hutchinson-Gilford progeria (MIM 176670) but not in Wiedemann-Rautenstrauch progeroid syndrome (MIM 264090). J Hum Genet. 2003;48:271–274. [DOI] [PubMed] [Google Scholar]
- [77].Yassaee VR, Khojaste A, Hashemi-Gorji F, et al. A novel homozygous LMNA mutation (p.Met540Ile) causes mandibuloacral dysplasia type a. Gene. 2016;577:8–13. [DOI] [PubMed] [Google Scholar]
- [78].Luo DQ, Wang XZ, Meng Y, et al. Mandibuloacral dysplasia type A-associated progeria caused by homozygous LMNA mutation in a family from Southern China. BMC Pediatr. 2014;14:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kosho T, Takahashi J, Momose T, et al. Mandibuloacral dysplasia and a novel LMNA mutation in a woman with severe progressive skeletal changes. Am J Med Genet A. 2007;143A:2598–2603. [DOI] [PubMed] [Google Scholar]
- [80].Garg A, Cogulu O, Ozkinay F, et al. A novel homozygous Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia. J Clin Endocrinol Metab. 2005;90:5259–5264. [DOI] [PubMed] [Google Scholar]
- [81].Ozer L, Unsal E, Aktuna S, et al. Mandibuloacral dysplasia and LMNA A529V mutation in Turkish patients with severe skeletal changes and absent breast development. Clin Dysmorphol. 2016;25:91–97. [DOI] [PubMed] [Google Scholar]
- [82].Ahmad Z, Zackai E, Medne L, et al. Early onset mandibuloacral dysplasia due to compound heterozygous mutations in ZMPSTE24. Am J Med Genet A. 2010;152A:2703–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Akinci B, Sankella S, Gilpin C, et al. Progeroid syndrome patients with ZMPSTE24 deficiency could benefit when treated with rapamycin and dimethylsulfoxide. Cold Spring Harb Mol Case Stud. 2017;3:a001339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Agarwal AK, Fryns JP, Auchus RJ, et al. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. [DOI] [PubMed] [Google Scholar]
- [85].Agarwal AK, Zhou XJ, Hall RK, et al. Focal segmental glomerulosclerosis in patients with mandibuloacral dysplasia owing to ZMPSTE24 deficiency. J Investig Med. 2006;54:208–213. [DOI] [PubMed] [Google Scholar]
- [86].Garg A, Subramanyam L, Agarwal AK, et al. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab. 2009;94:4971–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Mory PB, Crispim F, Kasamatsu T, et al. Atypical generalized lipoatrophy and severe insulin resistance due to a heterozygous LMNA. p.T10I Mutation Arq Bras Endocrinol Metabol. 2008;52:1252–1256. [DOI] [PubMed] [Google Scholar]
- [88].Csoka AB, Cao H, Sammak PJ, et al. Novel lamin A/C gene (LMNA) mutations in atypical progeroid syndromes. J Med Genet. 2004;41:304–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lee S, Park SM, Kim HJ, et al. Genomic diagnosis by whole genome sequencing in a Korean family with atypical progeroid syndrome. J Dermatol. 2015;42:1149–1152. [DOI] [PubMed] [Google Scholar]
- [90].Doubaj Y, De Sandre-Giovannoli A, Vera EV, et al. An inherited LMNA gene mutation in atypical Progeria syndrome. Am J Med Genet A. 2012;158A:2881–2887. [DOI] [PubMed] [Google Scholar]
- [91].Gonzalez-Quereda L, Delgadillo V, Juan-Mateu J, et al. LMNA mutation in progeroid syndrome in association with strokes. Eur J Med Genet. 2011;54:e576–9. [DOI] [PubMed] [Google Scholar]
- [92].Kirschner J, Brune T, Wehnert M, et al. p.S143F mutation in lamin A/C: a new phenotype combining myopathy and progeria. Ann Neurol. 2005;57:148–151. [DOI] [PubMed] [Google Scholar]
- [93].Zirn B, Kress W, Grimm T, et al. Association of homozygous LMNA mutation R471C with new phenotype: mandibuloacral dysplasia, progeria, and rigid spine muscular dystrophy. Am J Med Genet A. 2008;146A:1049–1054. [DOI] [PubMed] [Google Scholar]
- [94].Liang L, Zhang H, Gu X. Homozygous LMNA mutation R527C in atypical Hutchinson-Gilford progeria syndrome: evidence for autosomal recessive inheritance. Acta Paediatr. 2009;98:1365–1368. [DOI] [PubMed] [Google Scholar]
- [95].Verstraeten VL, Broers JL, Van Steensel MA, et al. Compound heterozygosity for mutations in LMNA causes a progeria syndrome without prelamin A accumulation. Hum Mol Genet. 2006;15:2509–2522. [DOI] [PubMed] [Google Scholar]
- [96].Van Esch H, Agarwal AK, Debeer P, et al. A homozygous mutation in the lamin A/C gene associated with a novel syndrome of arthropathy, tendinous calcinosis, and progeroid features. J Clin Endocrinol Metab. 2006;91:517–521. [DOI] [PubMed] [Google Scholar]
- [97].Soria-Valles C, Carrero D, Gabau E, et al. Novel LMNA mutations cause an aggressive atypical neonatal progeria without progerin accumulation. J Med Genet. 2016. [DOI] [PubMed] [Google Scholar]
- [98].Madej-Pilarczyk A, Rosinska-Borkowska D, Rekawek J, et al. Progeroid syndrome with scleroderma-like skin changes associated with homozygous R435C LMNA mutation. Am J Med Genet A. 2009;149A:2387–2392. [DOI] [PubMed] [Google Scholar]
- [99].Nguyen D, Leistritz DF, Turner L, et al. Collagen expression in fibroblasts with a novel LMNA mutation. Biochem Biophys Res Commun. 2007;352:603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Guo X, Ling C, Liu Y, et al. Case of novel lamin A/C mutation manifesting as atypical progeroid syndrome and cardiomyopathy. Can J Cardiol. 2016;32:1166 e29–31. [DOI] [PubMed] [Google Scholar]
- [101].Renard D, Fourcade G, Milhaud D, et al. Novel LMNA mutation in atypical Werner syndrome presenting with ischemic disease. Stroke. 2009;40:e11–4. [DOI] [PubMed] [Google Scholar]
- [102].Kane MS, Lindsay ME, Judge DP, et al. LMNA-associated cardiocutaneous progeria: an inherited autosomal dominant premature aging syndrome with late onset. Am J Med Genet A. 2013;161A:1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Ruijs MW, Van Andel RN, Oshima J, et al. Atypical progeroid syndrome: an unknown helicase gene defect? Am J Med Genet A. 2003;116A:295–299. [DOI] [PubMed] [Google Scholar]
- [104].Decker ML, Chavez E, Vulto I, et al. Telomere length in Hutchinson-Gilford progeria syndrome. Mech Ageing Dev. 2009;130:377–383. [DOI] [PubMed] [Google Scholar]
- [105].Crabbe L, Jauch A, Naeger CM, et al. Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc Natl Acad Sci U S A. 2007;104:2205–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Zhang W, Li J, Suzuki K, et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015;348:1160–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Shin S, Lee J, Yoo S, et al. Active control of repetitive structural transitions between replication forks and holliday junctions by Werner syndrome helicase. Structure. 2016;24:1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Tang W, Robles AI, Beyer RP, et al. The Werner syndrome RECQ helicase targets G4 DNA in human cells to modulate transcription. Hum Mol Genet. 2016;25:2060–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Singh DK, Ghosh AK, Croteau DL, et al. RecQ helicases in DNA double strand break repair and telomere maintenance. Mutat Res. 2012;736:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Nguyen GH, Tang W, Robles AI, et al. Regulation of gene expression by the BLM helicase correlates with the presence of G-quadruplex DNA motifs. Proc Natl Acad Sci U S A. 2014;111:9905–9910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Zimmermann M, Kibe T, Kabir S, et al. TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes Dev. 2014;28:2477–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Machwe A, Xiao L, Groden J, et al. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry. 2006;45:13939–13946. [DOI] [PubMed] [Google Scholar]
- [113].Chan KL, North PS, Hickson ID. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007;26:3397–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Lopez-Mosqueda J, Maddi K, Prgomet S, et al. SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]