

Type 2 diabetes (T2D), cardiovascular disease (CVD), and other obesity-related disorders are conditions associated with insulin resistance (IR), and are major public health epidemics in the United States and worldwide. IR is commonly defined as the relative inability of insulin to mediate disposal of a quantity of glucose, and is nearly universally observed in all T2DM patients. While obesity is a well-established risk factor for IR, it accounts for only about 25% of the variability of IR observed in humans. It has also been observed that apparently healthy individuals have surprisingly wide-ranging capacity for insulin-mediated glucose disposal. While hyperglycemia secondary to hyperinsulinemia does not occur in these compensated individuals, they nonetheless exhibit increased risk for CVD1. Moreover, even obese individuals controlled for the level of adiposity also demonstrate remarkably variant metabolic abnormalities including hypertriglyceridemia, which is also associated with CVD risk2. The identification of new genes and pathways modulating IR risk is needed to provide mechanistic insights into this condition and to identify both biomarkers and therapeutic targets for individuals with T2D, CVD, and other obesity-related disorders.
Human genetic studies have provided important causal insights into IR. The genetic lesions responsible for multiple monogenic forms of IR have been identified and have collectively implicated that IR can be a consequence of either inheritance of protein coding mutations in genes encoding components of insulin receptor signaling (e.g., AKT2 mutations3) or adipose tissue function (e.g., AGPAT2 mutation-associated lipodystrophies4). Studies of these monogenic disorders have provided key insights into IR including the critical role of the adipocyte, but such protein-coding mutations have not been identified in the majority of IR individuals. Recently, genome-wide association studies (GWAS) have provided new genetic insights into T2D risk and IR5, 6. Unlike monogenic studies, GWAS studies have associated common DNA sequence variants (SNPs), largely in non-coding and potentially transcriptional regulatory regions of the genome, with T2D and IR. To date, nearly 60 genomic loci have been associated with IR5, with the top 10 IR-associated loci being replicated in two separate GWAS studies5, 7. Largely due to the non-coding, regulatory nature of the IR-associated SNPs, functional validation of GWAS loci has lagged behind monogenic studies.
Advances in statistical genetics, epigenetics, and genome editing have recently fueled a rapid expansion in the capacity to functionally validate GWAS loci. For example, data from the 1000 Genomes Project, can be used for imputation and genetic fine-mapping to identify all genetically plausible candidate causal/functional variants in each T2D8 or IR locus, such as those modulating the levels of the adipokine adiponectin9. Targeted and consortia efforts to map three-dimensional epigenomes and transcriptomes of disease-relevant tissues and cell types, such as those by RoadMap Epigenomics and Genotype-Tissue Expression (GTEx), have expanded our understanding of specific intergenic genomic regions that have cell type-specific regulatory functions and helped to connect SNPs to their putative effector genes through regulatory element-promoter interactions. In some cases, these studies have clarified potential mechanisms of GWAS loci, especially in loci where a single SNP is associated with a complex disease, but the vast majority of GWAS loci remain functionally unresolved.
In this issue of Circulation Research, Chen et al.10 developed an in vitro knockout (KO) cell screening platform using human preadipocytes and adipocytes to functionally assess candidate gene function(s) in IR-relevant molecular and cellular phenotypes including adipogenesis, lipid metabolism, and insulin signaling (Figure 1). A prevailing mode in the field is that subcutaneous adipocytes function as a buffering system for lipid energy balance and that abnormalities in these functions are associated with IR-associated CVD11. Within ten IR-associated GWAS loci, Chen et al. studied the functions of 16 candidate effector genes that are highly expressed in a human adipocyte differentiation model– the Simpson-Golabi-Behmel syndrome (SGBS) preadipocyte. SGBS preadipocytes resemble subcutaneous preadipocytes and are neither transformed nor immortalized12. Importantly, this cell culture model retains the capacity for adipose differentiation after 50 generations, which makes it amenable to genome editing strategies such as CRISPR/Cas9 to study gene function. The authors utilized Cas9 and triple-gRNAs to create genetic knock-down SGBS preadipocyte (SGBS-KO) models for the following genes: GRB14, FST, PEPD, PDGFC, COBLL1, RSPO3, FAM13A, ANKRD55, MAP3K1, LYPLAL1, ARL15, SYN2, SLC30A10, LOC646736, IRS-1, and PPARG. They extensively characterized genome editing outcomes using amplicon sequencing and knock-down of targeted genes using transcript and protein expression assays.
Figure 1.
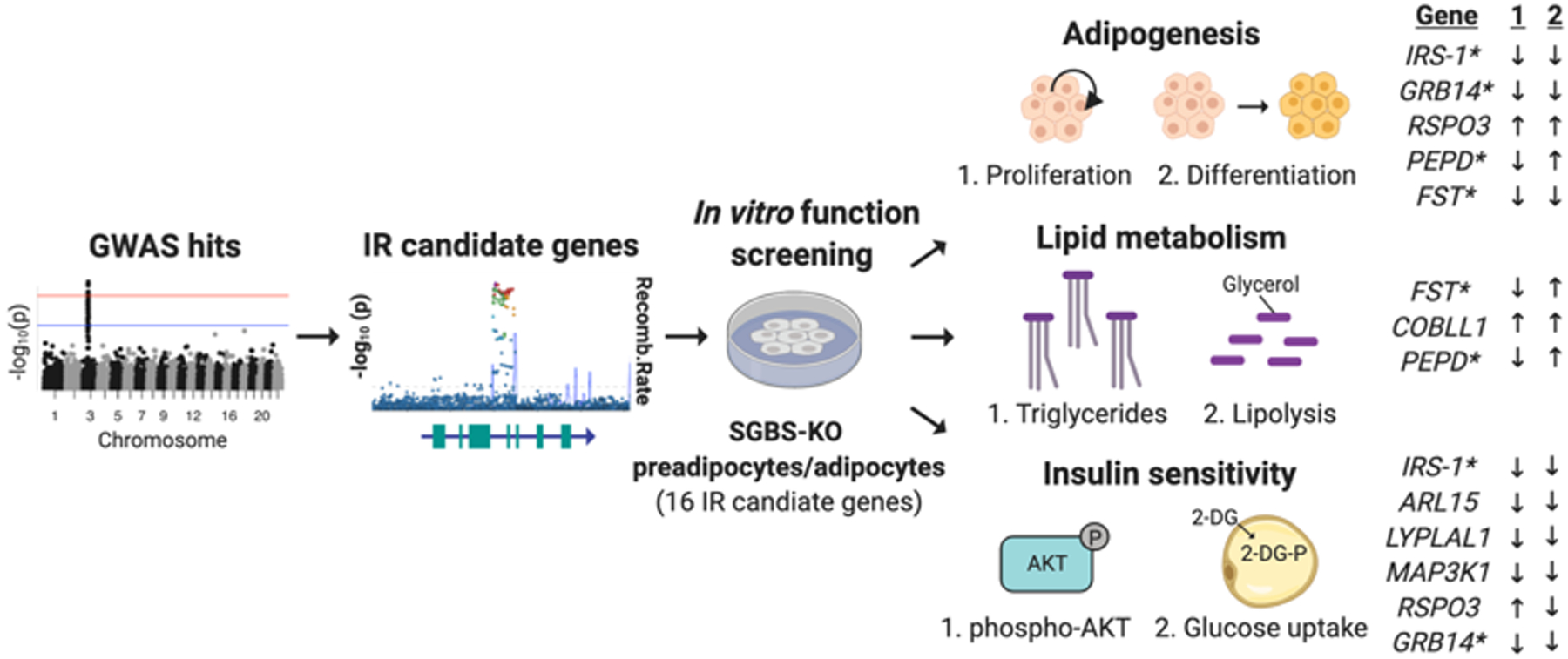
Schematic illustrating the strategy utilized by Chen et al. for the selection of 16 insulin resistance (IR)-associated candidate genes and the panel of assays conducted in a SGBS preadipocyte and adipocyte model. Listed genes demonstrated altered phenotypes in both assays for either adipogenesis, lipid metabolism or insulin sensitivity (* denotes lead SNP is also an eQTL for listed gene). Created with Biorender.
With these SGBS-KO models in hand, Chen et al. then assessed IR-relevant cellular phenotypes. For assessment of adipogenesis, SGBS-KO preadipocytes were studied for proliferation rates and differentiation efficiency. Ten of 16 SGBS-KO lines showed defects in preadipocyte proliferation and/or differentiation, with two knockouts (IRS-1, GRB14) decreasing and one (RSPO3) increasing both parameters. PEPD-KO decreased proliferation and increased differentiation. The heterogenous adipogenesis phenotypes observed in this study are mechanistically compatible with the observation that IR can result from either excess or decreased adipogenesis13. The group also characterized adipocyte lipid metabolism in 16 SGBS-KO adipocyte models. Four (PPARG, GRB14, PEPD and FST) decreased triglyceride accumulation, while one (COBLL1) increased triglyceride accumulation. Eight (PEPD, FST, COBLL1, IRS1, ANKRD55, PDGFC and MAP3K1) increased lipolysis rates. Because both PEPD-KO and FST-KO also decreased triglyceride accumulation and decreased triglyceride accumulation in subcutaneous tissue has been implicated in IR-associated lipodystrophies4, the authors postulated that PEPD and FST may represent new candidate lipodystrophy genes. Finally, SGBS-KO adipocyte insulin sensitivity was assessed through insulin-induced AKT2 phosphorylation and glucose uptake assays. AKT2 serves as a critical signaling regulator for adipose metabolism and plays a role in insulin-stimulated glucose uptake14, and AKT2 mutations are linked to monogenic forms of IR3. Five of the KO lines (IRS-1, ARL15, LYPLAL1, MAP3K1, and GRB14) exhibited both reduced insulin-dependent AKT2 phosphorylation and glucose uptake, suggesting that these genes regulate AKT2-dependent glucose uptake in SGBS adipocytes. RSPO3-KO enhanced insulin-induced AKT2 phosphorylation but decreased glucose uptake, suggesting that RSPO3 may regulate signaling steps between AKT2 phosphorylation and glucose uptake.
In summary, 12 of 16 candidate gene knockouts revealed at least one abnormality in IR-related SGBS cell phenotypes, with several having multiple abnormalities. Chen et al. prioritized seven gene-KOs affecting all three phenotypes (FST, IRS1, GRB14, MAP3K1, PPARG, PDGFC and PEPD), including two (PPARG and IRS1) with previously described adipocyte functions as positive controls, for follow-up analyses. Using the GTEx eQTL Calculator, the authors confirmed that IR-associated SNPs were also linked to altered expression of five of these top candidate IR effector genes (IRS1, GRB14, PDGFC, PEPD, and FST) in subcutaneous adipocytes. The risk allele of the corresponding IR-associated SNP was found to reduce expression for four genes (IRS1, FST, PEPD and PDGFC) but increased expression of one gene (GRB14). The authors also found that of the ten risk alleles studied, five risk alleles were significantly associated with increased CVD and five were nominally associated with CVD.
The study by Chen et al. both expands our knowledge of the genetic regulation of IR-related phenotypes in adipocytes in vitro, and provides important functional insights into potential mechanisms underlying the statistical associations with IR for ten GWAS loci. However, there are several limitations of this study that should be noted. Although Chen et al., linked these putative GWAS effector genes to adipocyte phenotypes, the use of this adipocyte cell model to study candidate IR-associated gene function precludes the opportunity to understood how other cell types within the adipose tissue or in other organs may be affected by these GWAS loci to contribute to IR in humans. In the future, it will be critical to cross-reference lead SNPs with regulatory element usage in other IR-relevant tissues types such as liver and skeletal muscle. For example, Chen et al. reported rs4865796 as an eQTL for FST expression in subcutaneous adipocytes, but it also affects FST expression in skeletal muscle (see GTEx eQTL calculator v8). Moreover, the genes studied here were prioritized in part due to their proximity to the lead IR-associated SNP, but 3D epigenome profiling studies suggest that SNP-containing cis-regulatory elements may skip the nearest gene to regulate the expression of those more distant. Additional studies of chromatin interactions and 3D nuclear organization in human IR-relevant cell types will be critical to provide orthogonal evidence supporting these selected candidates as the true IR GWAS effector genes. Finally, Chen et al. used CRISPR/Cas9 to generate genetic knock-down using multiple gRNAs to create insertions and deletions in protein-coding regions of transcripts that frequently resulted in near complete elimination of target protein expression. This is a suitable method to study gene function, but GWAS SNPs are generally thought to result in more subtle functional changes, such modest changes in transcript levels. Functional studies that employ methods to titrate transcript dosage and better model eQTL-like changes in IR-relevant cell types, or genome engineering approaches to experimentally alter the SNP alleles in an isogenic cell line, will also be important future directions. Nonetheless, Chen et al. eloquently display the utility of genome editing in a human adipocyte model system to study the function of candidate IR GWAS effector genes and their roles in pathophysiologic processes that contribute to IR and IR-associated human disorders. Furthermore, this will work will help propagate new studies to help better understand the pathogenesis of IR, T2D, and CVD.
Sources of Funding:
J.T.H. is supported by funding from the NIH (K08HL125807, R01HL142787, and U01EB028898), F.A.L. is supported by the AHA (PRE35110005). M.L.S. is supported by funding from the ADA (1-18-ACE-15), NIH (R01DK117137), and DOD (W81XWH-18-0401).
Footnotes
Disclosures: None.
References:
- 1.Reaven G. Insulin resistance and coronary heart disease in nondiabetic individuals. Arterioscler Thromb Vasc Biol. 2012;32:1754–9. [DOI] [PubMed] [Google Scholar]
- 2.Stefan N, et al. Metabolically healthy obesity: epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol. 2013;1:152–62. [DOI] [PubMed] [Google Scholar]
- 3.George S, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lightbourne M and Brown RJ. Genetics of Lipodystrophy. Endocrinol Metab Clin North Am. 2017;46:539–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott RA, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44:991–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao W, et al. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat Genet. 2017;49:1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lotta LA, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahajan A, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50:1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu Y, et al. A meta-analysis of genome-wide association studies for adiponectin levels in East Asians identifies a novel locus near WDR11-FGFR2. Hum Mol Genet. 2014;23:1108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Z, et al. Functional Screening of Candidate Causal Genes for Insulin Resistance in Human Preadipocytes and Adipocytes. Circ Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Civelek M, et al. Genetic Regulation of Adipose Gene Expression and Cardio-Metabolic Traits. Am J Hum Genet. 2017;100:428–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer-Posovszky P, et al. Human SGBS cells - a unique tool for studies of human fat cell biology. Obes Facts. 2008;1:184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kahn BB and Flier JS. Obesity and insulin resistance. J Clin Invest. 2000;106:473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer-Posovszky P, et al. Differential function of Akt1 and Akt2 in human adipocytes. Mol Cell Endocrinol. 2012;358:135–43. [DOI] [PubMed] [Google Scholar]