

Summary
Acute myocardial infarction (AMI), is associated with activation of various cells, including platelets that form monocyte-platelet complexes (MPC). Here, we analyzed MPC in vivo and in vitro and investigated the abilities of different monocyte subclasses to form MPC, characteristics of the cells involved in MPC formation, and MPC changes in AMI. We identified MPC by co-staining for platelet antigen CD41a and monocyte antigens CD14 and CD16. Platelet activation was evaluated from expression of phosphatidylserine (PS) as revealed by Annexin V. Monocytes of different classes disproportionately formed MPC: although classical monocytes (CD14++CD16-) constituted the majority, MPC were preferentially formed by intermediate monocytes (CD14++CD16+). CD41-positive events in MPC exposed more PS than the circulating ones. AMI was associated with a 50% increase in circulating monocytes and with a threefold increase in MPC, in particular in those formed by classical monocytes. In AMI patients, MPC formed by intermediate monocytes contained more CD41a-positive events than MPC with other monocyte subsets whereas in controls, MPC formed by classical monocytes carried more platelets than other MPC. The sizes of some of the CD41a+ events in MPC were smaller than those of regular platelets and seem to represent platelet-derived EV. Binding of EVs to monocytes was confirmed in in vitro experiments when monocytes were co-incubated platelet-generated EV. There was an association between the increase in MPC and their composition, and in-patient complications of AMI. MPC in AMI patients may play an important role in this pathology and can be used as an AMI correlate.
Keywords: Monocyte subsets, monocyte-platelet complexes, extracellular vesicles, acute myocardial infarction, flow cytometry
Introduction
Acute cardiovascular syndrome (ACS) is associated with a general activation of the immune system that includes activation of many cells, in particular monocytes and platelets, leading to destabilization and rupture of coronary atherosclerotic plaques and to acute myocardial infarction (AMI) (1–2). One of the manifestations of the activation of monocytes and platelets is the formation of monocyte-platelet complexes (MPC). These complexes are predominantly formed via specific interaction of P-selectin on activated platelets with P-selectin glycoprotein ligand-1, which is expressed by monocytes and further stabilized by additional integrin adhesion between these cells (3). Increase in MPC numbers has been reported for various pathologies associated with inflammation and platelet activation, in particular for cardiovascular diseases (4), inflammatory bowel disease (5), diabetes mellitus (6), asthma (7) and HIV disease (8,9). Platelet aggregation with monocytes is not merely a marker of platelet activation (10); it may also contribute to pathogenesis by upregulating monocyte surface adhesion molecules (11) and enhancing cytokine production (12).
Previously, attempts were made to link MPC with the clinical parameters of AMI patients. General MPC counts in the context of ACS were proposed as an early marker of AMI (13). However, MPC are diverse in the status of the involved platelets and monocytes. Tapp et al. observed a prominent increase in CD14++CD16+ monocyte counts, which correlated with peak troponin and plasma interleukin (IL)-6 and IL-10 levels (14 ). Zhou et al. showed that persistent CD14++CD16+ monocytosis as well as increased levels of CD14++CD16+ MPC had predictive values for adverse cardiovascular events in a two-year follow-up (15).
In spite of extensive studies, the physiological role of MPC remains unclear. In particular, the diversity of MPC has not been fully investigated. Here, we aimed to investigate what types of monocytes and platelets are involved in MPC in healthy donors and AMI patients. We confirmed that monocytes of different subpopulations differentially aggregate with platelets, and the numbers and composition of MPC are changed in patients with AMI. We found that the patterns of platelet aggregation with different monocytes in control and AMI patients were different. Moreover, our data are in agreement that some of these aggregates involve not only platelets but also platelet-generated extracellular vesicles (EV) (16). Aggregation of monocytes with platelet EVs was further studied in experiments in vitro, where we found they aggregated with monocytes more efficiently than those from healthy controls. Understanding the mechanisms of the formation of these aggregates and the role of MPC in pathological processes requires an analysis of different types of MPC in health and disease. Identification of these mechanisms and their changes in patients with AMI may provide an additional diagnostic tool for this disease.
Methods
Patients.
We consecutively enrolled AMI patients with and without persistent ST segment elevation (STEMI and NSTEMI) diagnosed according to the Third Universal Definition of Myocardial Infarction (17) at the moment of admission to the I.V. Davydovsky Moscow City Hospital. The control group included healthy volunteers with no signs of possible coronary artery disease and negative results on the treadmill test. All AMI patients enrolled in our study received loading doses 250–500 mg of aspirin and 300–600 mg of clopidogrel on average 40 min before hospital admission and blood collection for MPC analysis (18). Also, seven patients were anticoagulated with unfractionated heparin, and one patient temporarily refused to take any emergency therapy. Exclusion criteria comprised factors that could affect monocyte count (infectious disease, active cancer, hemodynamically significant valvular heart disease, cardiogenic shock, renal failure, and pregnancy). Additionally, no STEMI patients had a history of left ventricle (LV) dysfunction. All study patients received standard treatment according to current guidelines (18); 93.5% of AMI patients underwent primary percutaneous intervention. The study was performed in accordance with the Helsinki declaration and was approved by the MSMSU Ethics Committee. All participants provided written informed consent. Leukocyte and platelet counts were determined with the clinical automated blood analyzer Drew3-PAC (Drew Scientific, Miami Lakes, FL, USA). We calculated the monocyte and MPC concentrations on the basis of the leukocyte count and percentage in each population, estimated with flow-cytometric analysis.
Sample preparation for MPC detection.
Peripheral venous blood was sampled by means of direct venipuncture into a 3.2% sodium citrate-containing tube (Sarstedt, Numbrecht, Germany). To avoid in vitro cell aggregation, we discarded the first 3 ml of blood and collected the rest into tubes that contained 3.2% Na citrate and fixed blood within 1 min after collection, together with an equal volume of 0.5% formaldehyde (Thermo Scientific, Rockford, IL, USA) in PBS, for 15 min at room temperature. To evaluate whether, in spite of all these measures, significant numbers of MPC are nevertheless formed in vitro, we measured the kinetics of their formation in unfixed blood (Figure S1) (19). In our experiment, MPC percentage increased at a linear rate of 0.42±0.26% MPC per minute within the first hour without fixation. For flow analysis, a 30-μL sample of fixed blood was immunolabeled for 20 min in the dark with 20 μL of fluorescent mouse anti-human monoclonal antibody mix: 4 μL of anti-CD45-eFluor450 (eBioscience, San Diego, CA, USA), 3 μL of anti-CD14-PerCP Cy5.5 (Biolegend, San Diego, CA, USA), 3 μL of anti-CD16-AlexaFluor488 (Biolegend,), and 10 μL of anti-CD41a-APC (BD Biosciences, San Jose, CA, USA), containing mouse IgG at 2% (Life Technologies, Eugene, OR, USA) to prevent non-specific binding. Mouse IgG1κ isotype controls were IgG1κ-AlexaFluor488 and IgG1κ-eFluor450 from eBioscience; mouse IgG1κ-PerCP Cy5.5 were from Biolegend; and mouse IgG1κ-APC were from BD Biosciences. The samples were diluted with 2 ml of 0.5% formaldehyde and subjected to flow cytometry. Annexin V (Ann-V) staining was as described in Supplementary Methods.
MPC quantification with flow cytometry.
We characterized both monocytes and platelets using fluorescent antibodies against specific markers of these cells. Monocytes were recognized from the expression of CD14 and CD16, whereas platelets from expression of CD41a. MPC were defined as double-positive events carrying both platelets and monocyte markers (CD41a/CD14). The first gate defined for each patient was the “leukocytes” gate on a CD45/SSC dot-plot (Figure S2A). We identified monocytes in the leukocyte gate using a CD45/CD14 dot-plot (Figure S2B). Monocytes were further divided into subclasses according to their expression of CD14 and CD16 (Figure S2D) (20). Next, the involvement of these monocyte subsets in MPC was evaluated (Figure S2D1–D3).
Formation of MPC in vitro by isolated monocytes and platelets/EV.
Monocytes were isolated from blood drawn into a tube with EDTA. Red blood cells were lysed with EL Buffer (Qiagen, Hilden, Germany) within 15 min at RT. Leukocytes were washed twice with PBS at 400 g, 10 min, at 4°C. The MACS Pan Monocyte Isolation Kit (Miltenyi Biotec, CA, USA) was used to isolate monocytes from other leukocytes. To remove contaminating platelets and existing MPC we added biotin-labeled antibody against CD41a (250 ng per 25 million PBMCs; Abcam, MA, USA) to the antibody cocktail provided with the kit, together with a twofold amount of anti-biotin magnetic beads. The resulting monocytes contained less than 10% residual MPC. Monocytes were cultured in RPMI supplemented with 10% FBS. Washed platelets were prepared according to Cazenave et al. with some modifications (21) (see Supplementary Methods). Platelet-derived EV were isolated according to Dinkla et al. (22) with some modifications as described in Supplementary Methods. The number of platelets was determined with a hematological analyzer, and the number of monocytes was evaluated in the counting chamber. The platelets were added to monocytes in a ratio of 200:1. Co-incubation was performed at 37°C in a horizontal shaker (ES-20; Biosan, Riga, Latvia). We performed three experiments in doublets for each data point, assessing MPC after 5 and 30 min of co-cultivation. Then, 30 μL of sample was fixed with 0.5% formaldehyde, and in 15 min it was further stained for 20 min in the dark with a fluorescent mouse anti-human monoclonal antibody mix of 3 μL of anti-CD14-PE-Cy7 (Biolegend), 3 μL of anti-CD16-FITC (eBioscience,), and 2 μL of anti-CD42a-BV421 (BD Biosciences); 2% of the mix was mouse IgG (Life Technologies, Eugene, OR, USA). The sample was diluted with 200 ml of 0.5% formaldehyde and subjected to flow cytometry. The gating strategy for evaluation of MPC is illustrated in Figure S3.
Statistical analysis is described in Supplementary Methods.
Results
Patient characteristics
Thirty-one patients with AMI (27 STEMI and 4 NSTEMI) and twenty-eight healthy volunteers were recruited for this study (Table S1). Mean age, body mass index (BMI), arterial hypertension, diabetes mellitus, smoking, dyslipidemia, and several serum/plasma parameters (creatinine and lipid profiles except for HDL and triglycerides) were comparable between AMI patients and controls. Patients with AMI had higher levels of high-sensitivity C-reactive protein and higher white blood cell and monocyte counts, consistent with activation of inflammation and clonal hematopoiesis during atherosclerosis progression (23). There was male gender prevalence in the AMI group, but the above-mentioned parameters were not significantly different for males and females in both the AMI and the control groups.
The summary of emergency prehospital antiplatelet therapy received by the enrolled AMI patients is presented in Methods and in Table S2. There were no significant differences in MPC fractions between patients who received lower (300 mg) or higher (600 mg) doses of clopidogrel or who received clopidogrel with heparin (7.43±3.37% vs. 12.84±11.14% vs. 11.51±8.72%, respectively; p=0.682). The results showed that prehospital emergency antiplatelet therapy did not affect the formation of MPC in AMI patients
The time from the last onset of angina to admission and blood drawing did not exceed 24 h (1.4–24 h; mean=7.88±8.06 h). We observed significant differences (Table S3) in MPC percentages between early admission (< 4 h) and later admission (> 4 h), in agreement with Furman et. al. 2001 (13). The total duration of coronary instability preceding the acute myocardial infarction was 1.4–168 h. Neither longer nor shorter periods of angina nor the number of onsets of angina during these periods had a significant effect on the fraction of MPC.
Circulating monocytes and platelets in blood of healthy volunteers
Monocytes.
The average total monocyte count in this group was 478.12±52.14 cells/μL of blood (Table 1). Blood monocytes were identified in a leukocyte gate among other CD45-positive cells and were further divided into subtypes by expression of CD14 and CD16 (20). We distinguished three subsets of monocytes: classical (CD14++CD16-), intermediate (CD14++CD16+), and non-classical (CD14+CD16++) (Figure S2A–D). The prevalent subpopulation was of classical monocytes, constituting about 90% of the entire monocyte population. The remaining 10% were shared almost equally between non-classical and intermediate monocytes (Table 1).
Table 1.
Circulating and MPC-associated monocytes in blood of healthy individuals and patients with AMI.
| Total monocytes | Classical monocytes |
Intermediate monocytes |
Non-classical monocytes |
|
|---|---|---|---|---|
| Healthy patients | ||||
| Circulating monocytes | 478.12±52.14 | 437.62±34.79 (90.34±0.56%) |
30.59±3.89 (6.14 ±0.52%) |
15.12±2.08 (3.01±0.29%) |
| MPC | 20.62±1.86 (4.35±0.23%) |
14.48±1.60 (3.23±0.16%) |
3.06±0.68 (10.29±1.37%) |
0.44±0.27 (3.23±0.32%) |
| AMI patients | ||||
| Circulating monocytes | 630.65 ± 49.83 | 580.24 ±33.17 (90.44 ±0.89%) |
41.24± 4,85 (6.50 ±0.65%) |
15.28±2.06 (2.59±0.36%) |
| MPC | 63.89±7.74 (11.13±1.61%) |
40.74±5.47 (7.90±1.32%) |
7.31± 1.50 (18.41 ±1.99%) |
1.09±0.26 (6.76±0.84%) |
Data are presented as mean concentrations (counts/μL) ± SEM of monocytes circulated as single cells or as a part of MPC. For monocytes, data in parentheses denote percentages of circulating monocyte subsets among the total monocytes in peripheral blood. For MPC, data in parentheses denote the percentage of monocytes among total monocytes or individual monocyte subsets.
Platelets.
The average platelet count in the group of healthy individuals was 252.13±15.74 cells/μL (Table S1). With flow cytometry, we identified platelets from the expression of integrin CD41a. The CD41a fluorescence profile revealed two distinct populations differing by lower and higher CD41a expression (MFI 222 [203; 254] vs. 1,489 [1,258; 1,759] (Figure S4A; Table 2). Also, these two populations corresponded to two different patterns of FSC (Figure S4B). We estimated the sizes of the particles corresponding to these events using fluorescent MegaMix-FSC polystyrene beads (24). Events with low fluorescence had sizes of less than 0.9 μm and thus seem to represent predominantly extracellular vesicles rather than integral platelets. When the platelet population was treated with Ca2+ ionophore A23187, which stimulates the release of platelets EV (25), the number of events in the low-fluorescence peak increased (Figure S4C).
Table 2.
Median Fluorescence Intensity (MFI) of CD41a-positive events in blood of healthy volunteers and in patients with AMI.
| Total CD41a+ events (platelet with EV) |
Total MPC | MPC with classical monocytes |
MPC with intermediate monocytes |
MPC with non-classical monocytes |
||
|---|---|---|---|---|---|---|
|
Healthy controls |
MFI | 1,406 [1,204;685] | 1,371 [1,125;1,664] | 1,368 [970;1,853] | 1,049 [655;1649] | 1,223 [450;2,104] |
| MFI ratios | 1 | 0.975 | 0.973 | 0.746 | 0.870 | |
|
AMI patients |
MFI | 1,332 [1,145;1,649] | 1,174 [689;1,386] | 1,016 [350;1,616] | 1,467 [816;4,066] | 966 [451;1,764] |
| MFI ratios | 1 | 0.881 | 0.763 | 1.101 | 0.725 | |
| p | 0.7170 | 0.0694 | 0.0044 | 0.0123 | 0.2394 |
Data are presented as medians [25th–75th percentile]. Between groups, comparison was performed with the Mann-Whitney test.
To characterize platelet populations further, we assessed their activation from phosphatidylserine (PS) exposure as measured by Annexin V (Ann-V) binding (26). As shown in Figure 1, in plasma of healthy volunteers Ann-V-positive events were revealed predominantly in the area of the dot plot that presumably corresponded to EVs, where they constituted 30.4% [15.4; 34.1], while in the area corresponding to integral platelets, Ann-V positivity was revealed on average in 1% [0.8; 2] of events (n=14) (Table 3).
Figure 1: Phosphatidylserine exposure on CD41a-positive events.
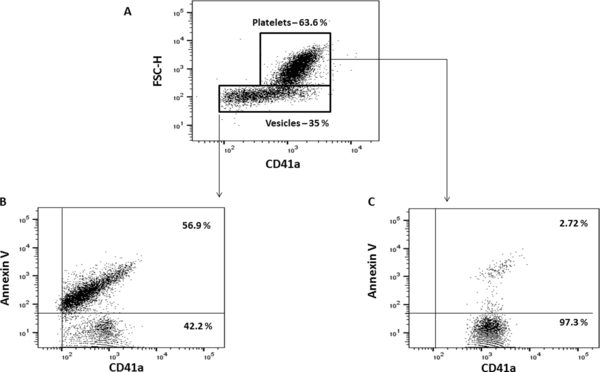
Plasma was stained for CD41a, and PS was detected with Ann V-FITC. A. CD41a-APC/FSC dot plot to identify CD41a+ populations. B, C. Staining of CD41a+ events with Ann V-FITC. A representative experiment out of eleven with samples from AMI patients is shown.
Table 3.
Fractions of Ann V/CD41a-positive events in plasma and in blood of healthy volunteers and patients with AMI.
| Control | AMI | p | ||
|---|---|---|---|---|
| Plasma | Annexin V+/CD41a+ in the EV gate, % | 30.4 [15.4;34.1] | 41.2 [25.6;59.2] | 0.0138 |
| Annexin V+/CD41a+ in the platelet gate, % | 1 [0.8;2] | 1.3 [0.8;2.3] | 0.2284 | |
| Annexin V+/CD41a+, total events, % | 17.2 [11.9;25.3] | 28.1 [18;52.4] | 0.0350 | |
| Blood | Annexin V+/CD41a+, % of CD41a positive events | 2.3 [1.5;3] | 3.2 [2;4.4] | 0.512 |
| Annexin V+/CD41a+, % of the total MPC | 39.3 [30.4;49.5] | 41.2 [31.9;54.1] | 0.716 |
Data are presented as medians [25th–75th percentile] of percentages. Comparison between groups was performed with the Mann-Whitney test.
Monocyte-platelet complexes in blood of healthy volunteers
MPC were defined by positivity for the platelet marker CD41a in the monocyte population. On average, 4.35±0.23% (n=28) of all monocytes were associated with platelets, resulting in a concentration of MPC of 20.62±1.86/μL (Table 1).
Monocytes in MPC.
The contributions of different classes of monocytes to the MPC were not proportional to the presence of these monocytes in blood. Out of monocytes of the intermediate subset, 10.29±1.37% were associated with platelets. Monocytes of classical and non-classical phenotypes contributed respectively 3.23±0.16% and 3.23±0.32% of their populations to MPC (Table 1). As the result of disproportional involvement of different monocytes in MPC, the distribution of free monocytes was significantly different from the distributions of monocytes of different types in MPC (p=0.0004 for classical, p=0.0001 for intermediate, and p=0.020 for non-classical). Since the classical monocytes prevailed in blood and far outnumbered the intermediate ones, MPC that involved the classical monocytes constituted the majority of MPC, on average 78.5±2.1%, while MPC with non-classical and intermediate monocytes constituted on average 4.3±1.7% and 13.1±1.6% of all MPC, respectively (Table 4).
Table 4.
Distribution of monocyte subsets in MPC
| Control | AMI | p | |
|---|---|---|---|
| Classical | 78.5 ± 2.1 | 84.3 ± 1.8 | 0.0201 |
| Intermediate | 13.1 ± 1.6 | 10.8 ± 1.4 | 0.1384 |
| Non-classical | 4.3 ± 1.7 | 2.1 ± 0.4 | 0.1626 |
Monocyte subsets in MPC were determined with flow cytometry in the MPC gate on CD14/CD16 dot-plot. Data are presented as means ± SEM of percentages of monocytes in MPC; p was estimated between AMI and healthy control with the Mann-Whitney test.
Platelets in MPC.
We evaluated MFI for CD41a staining in MPC. The MFI of complexes varied depending on the type of monocytes involved in them (Table 2). MFI in MPC with intermediate monocytes was significantly lower (p=0.0078) than in MPC with monocytes of two other types. PS exposure in MPC (Table 3) was significantly higher than in the circulating CD41a+ population (39.3% [30.4; 49.5] vs 2.3% [1.5; 3]; n=13, p=0.0010).
Circulating monocytes, platelets, and MPC in blood of AMI patients
We compared the involvement of monocytes and platelets in MPC of patients with AMI with that of the healthy volunteers. In AMI patients the numbers of both free monocyte and MPC were increased. No statistical differences in the numbers of monocytes and MPC were found between patients with STEMI and NSTEMI AMI (Table S4). These groups were further analyzed together.
Monocytes.
The number of total monocytes significantly increased: from 478.12±52.14 cells/μL in control to 630.65±49.83 cells/μL in AMI patients (n=31, p=0.0017). However, their distribution across different subclasses was not different from that in healthy individuals. The concentration of MPC in AMI patients increased from 20.62±1.86 per μL in healthy volunteers to 63.89±7.74 per μL in AMI patients (Table 1). This increase was predominantly the result of a significant increase of monocyte involvement in MPC (from 4.35±0.23% of the total monocytes in healthy control to 11.13±1.61% in AMI patients; p=0.00004).
All types of monocytes contributed to the increase in MPC counts, albeit not equally (Figure 2). Predominantly, this increase was due to the contribution of classical monocytes (~3-fold increase), while two other classes increased ~1.8-fold. As the result, there was a statistically significant difference in the distribution of different subtypes of monocytes in MPC between AMI patients and healthy controls, with a predominant increase in AMI patients of MPC formed by classical monocytes (p=0.0201) (Table 4).
Figure 2: Recruitment of monocytes of different subsets into MPC in patients with AMI and in healthy controls.
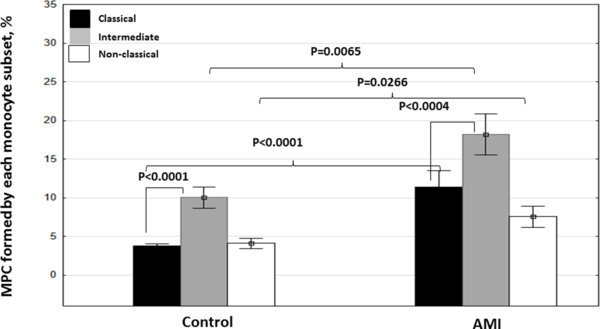
The fractions of MPC within monocyte subsets as defined by CD41a+/CD14+ in the gates of classical (CD14++CD16-), intermediate (CD14++CD16+), and non-classical (CD14+CD16++) monocytes. Data are presented as means ± SEM (n=28 for controls and n=31 for AMI). Indicated are p values as estimated with the Mann-Whitney U-test for two independent groups (control and AMI) or with the Wilcoxon Matched Pairs Test for comparisons within one group.
Platelets.
The numbers of circulating platelets in AMI patients remained as in control: on average 228.71±11.13 cells/μL vs. 252.13 ± 15.74 cells/μL; n=31, p=0.1610. Also, the distribution of CD41a+ events between high and low fluorescence peaks in AMI patients was not different from control (p=0.0848), although the fraction of low-intensity CD41a increased (from 21.96 ±2.62% to 41.9±8.86%; n= 12).
We found a significant increase (p=0.0350) in PS exposure in the number of CD41a+ events in plasma, as measured from Ann-V binding, in patients with AMI compared with healthy controls (Figure 3 and Table 3). This difference was predominantly caused by the increase in the PS positive events of smaller size that corresponded to EV (30.4% [15.4; 34.1] in the control group (n=14) vs. 41.2% [25.6; 59.2] in AMI patients (n=11), p=0.0138).
Figure 3. Phosphatidylserine exposure on platelets/EV of healthy donors and patients with AMI.
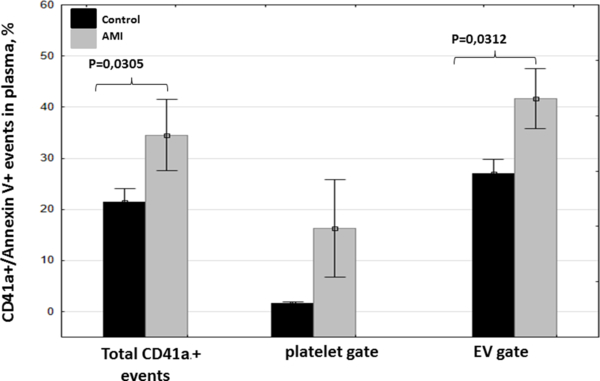
We estimated PS exposure on platelets and on platelet-generated EV in the total CD41a+ population and in two subpopulations by staining with Ann V-FITC. Data are presented as means ± SEM (n=13 for controls and n=11 for AMI).
In MPC of the AMI patients we found an increase in MFI for CD41a compared with healthy controls, especially in MPC that involve intermediate monocytes (p=0.0123) (Figure 4, Table 2). As in controls, PS exposure in MPC was increased compared with circulating platelets/EV (41.2 [31.9; 54.1] vs. 3.2 [2; 4.4]; p=0.0015) (Table 3).
Figure 4. MFI of CD41a in MPC with different monocyte subsets in healthy controls and in patients with AMI.
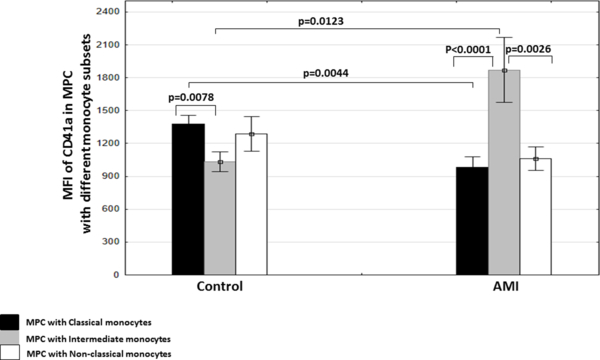
MPC with different monocyte subsets were identified with flow cytometry as CD41a+/CD14+ events. Data are presented as means ± SEM (n=28 for controls and n=31 for AMI). Indicated are p values as estimated with the Mann-Whitney U-test for two independent groups (control and AMI) or with the Wilcoxon Matched Pairs Test for comparisons inside one group.
In summary, in blood of AMI patients there was an increase of free circulating monocytes as well as of MPC. CD41a MFI of MPC with intermediate monocytes in AMI patients was the highest compared with MFI of MPC with classical and non-classical monocytes and was significantly higher than that in the control group.
Formation of MPC in vitro
To further investigate MPC formation, we performed in vitro experiments with isolated monocytes and platelets from blood of control and AMI patients. These experiments allowed us to study aggregation of monocytes with platelets and EVs separately.
Monocytes were exposed to either platelets (blocked with PGE-1 or activated with ADP) or platelet-derived EV. Both for platelets, activated or not, and for EV after 30 min of co-incubation we observed an increase in MPC (Figure 5). Cells from AMI patients form 1.58 ± 0.40 more MPC than cells from controls. Also, EVs generated by platelets from AMI patients form 1.66 ± 0.29 more MPC than in controls. Thus, platelets as well as platelet-derived EV were able to form complexes with monocytes. We used CD42a fluorescence as a measure of platelet/platelet EV aggregation with monocytes. MFI of aggregates stained with anti-CD42a antibodies increased with the time of co-incubation. We stained the in vitro -formed MPC with Ann-V. In experiments on monocyte incubation with activated platelets the fraction of Ann-V-positive MPC was approximately three-fold larger than that when monocytes were incubated with non-activated platelets (from 20 ± 5.2 % to 70.7 ± 14.9).
Figure 5. MPC formed in vitro by isolated monocytes with isolated platelet-derived EV or isolated platelets.

A. Fraction of MPC formed 5 (black) and 30 (grey) min in the course of incubation of monocytes with EV, with platelets blocked with PGE-1 or activated with ADP; B. CD42a MFI in all the events (grey bars) and specifically in MPC (black bars); C. Fractions of MPC formed between platelets or platelet-derived EV with each separate monocyte subset: classical (black), intermediate (grey), and non-classical (hatched). Data are represented as means ± SEM. Statistical differences were calculated as per Student’s t-test and were considered significant if the p value was below 0.05. The p value for all significant differences is indicated in the figure next to the corresponding columns.
Finally, we evaluated the ability of different monocyte subsets to form MPC in vitro. Intermediate monocytes were more extensively involved in MPC formation during the first minutes of co-cultivation, in the cases both of control and of AMI. Also, this was true for platelet-derived EV (Figure 5C). Within 30 min of co-cultivation, in both cases, non-classical monocytes and finally classical monocytes form more MPC, although in case of AMI, the number of such MPC was larger.
Relationship of MPC with in-hospital complications
We compared the fractions of different MPC in patients with and without AMI complications, in particular with LV aneurysm and massive intracoronary thrombosis, observed during coronary angiography, and with acute heart failure Killip class 1–3, separately or in total as a composite end-point registered within 7 days post-admission. In our observational study, among 31 consecutively enrolled AMI patients, during 7 days post-AMI we did not register any deaths or early post-infarction angina. Cardiogenic shock at admission was an exclusion criterion for this study. Both total MPC and the fractions of MPC with each monocyte subset were significantly higher with AMI patients who suffered AMI complications (composite end-point) (Table 5). Patients with LV aneurysm registered on admission compared with patients without LV aneurysm had a significantly higher fraction of total MPC (p=0.0165) as well as a significantly higher fraction of MPC with classical monocytes (p=0.0283). It is of note that intermediate monocyte concentrations were significantly lower with patients who suffered from LV aneurysm and intracoronary thrombosis registered on Day 7. Based on logistic regression analysis, MPC> = 6% (b = 0.5643, p <0.05, regression accuracy 94%) is an independent predictor of in-patient complications (composite endpoint) of AMI.
Table 5.
Comparison of monocyte profiles and MPC content in AMI patients with or without in-hospital complications.
| Parameters | Complications | No complications | p |
|---|---|---|---|
| Composite end-point | No composite end-point | ||
| % MPC | 14.28±2.60 | 7.31±1.07 | 0.0053* |
| MPC concentration | 78.33±9.03 | 46.36±6.39 | 0.0290* |
| % MPC with classical monocytes | 10.68±1.61 | 4.48±0.41 | 0.0002* |
| % MPC with intermediate monocytes | 21.87±1.94 | 14.15±2.72 | 0.0358* |
| % MPC with non-classical monocytes | 8.61±0.97 | 4.47±0.54 | 0.0151* |
| MPC concentration with classical monocytes | 52.02±6.58 | 26.85±2.71 | 0.0025* |
| MPC concentration with intermediate monocytes | 6.80±1.21 | 7.93±2.73 | 0.9301 |
| MPC concentration with non-classical monocytes | 1.36±0.33 | 0.74±0.18 | 0.2344 |
| % AnnexinV+ total events in plasma from all CD41a+ events | 43.80±5.72 | 24.00±4.27 | 0.3168 |
| % AnnexinV+ platelets in plasma from all CD41a+ platelets | 37.42±8.13 | 1.00±0.18 | 0.0276* |
| % AnnexinV+ vesicles in plasma from all CD41a+ vesicles | 57.92±3.70 | 31.41±4.23 | 0.0446* |
| LV aneurism on day 1 | No LV aneurism on day 1 | ||
| % MPC with classical monocytes | 9.00±1.26 | 7.01±2.05 | 0.0283* |
| % AnnexinV+ platelets in plasma from all CD41a+ platelets | 37.42±8.13 | 1.00±0.18 | 0.0276* |
| % AnnexinV+ vesicles in plasma from all CD41a+ vesicles | 57.92±3.70 | 31.41±4.23 | 0.0446* |
| LV aneurism on day 7 | No LV aneurism on day 7 | ||
| % AnnexinV+ vesicles in plasma from all CD41a+ vesicles | 63.93±6.49 | 32.26±3.31 | 0.0278* |
| Concentration of intermediate monocytes | 27.59±5.18 | 48.82±5.50 | 0.0433* |
| Intracoronary thrombosis | No intracoronary thrombosis | ||
| Concentration of intermediate monocytes | 20.64±6.85 | 45.43±5.34 | 0.0433* |
Data are presented as means ± SEM.
p<0.05 according to Mann-Witney U-test.
We also analyzed the amounts of circulating platelet-derived EV and activated platelets in AMI patients measured in the Ann-V binding assay and compared their numbers with the occurrence of AMI complications within 7 days post-admission (Table 5). Both platelet-derived EV (p=0.0446) and activated platelets (p=0.0276) were significantly higher in patients with a composite endpoint of AMI complications. Development of left ventricular aneurism positively correlated only with vesicular expression of Ann-=V (p=0.0433).
We performed multivariate linear regression analysis to estimate potential influence of co-morbidities and standard cardiovascular risk factors (age, sex, smoking, obesity, general cholesterol, LPLD, hyperlipidemia, prior CVD, diabetes mellitus, and arterial hypertension) as well as background medications on the results to establish predictive value for MPC for in-hospital complications. We calculated the regression coefficients, standard errors and p-values for all the standard cardiovascular risk factors as well as for MPC percentage, concentration of different monocyte subpopulations, Annexin V percentage, and MFI level for CD41a staining in MPC and observed that cardiovascular risk factor didn’t contribute to in-patient complications (Table S5A). MPC percentage didn’t reach statistical significance (p=0.1399) to influence on in-hospital complications. But, percentage of intermediate monocytes as well as their absolute amount in complexes with platelets did associate with in-hospital complications (p=0.0223 and p=0.0055 respectively) (Table 6). Then we adjusted all parameters for age and sex and re-analyzed data. Only absolute amount of intermediate MPC percentage was positively and independently associated with the development of in-patient complications of AMI, and this association persisted after adjustment (p=0.0079, Table 6, Table S5B–C).
Table 6.
Multivariate regression analysis of the predictive value of monocyte parameters for composite endpoint of in-patient complications of AMI. Significant factors are marked with asterisk.
| B ± SE | P | |
|---|---|---|
| Multivariate analysis | ||
| Intermediate monocytes (CD14++CD16+), % | 0,183751356 | 0,02228189* |
| MPC with intermediate monocytes, count in 1 uL | 0,274223664 | 0,00548509* |
| Adjusted for age | ||
| MPC with intermediate monocytes, count in 1 uL | 0,3020323 | 0,00797015* |
| Adjusted for sex | ||
| Intermediate monocytes (CD14++CD16+), % | 0,1614142 | 0,02581077* |
| MPC with intermediate monocytes, count in 1 uL | 0,3083062 | 0,00614551* |
B, regression coefficient; SE, standard error; MPC, monocyte-platelet complexes.
Discussion
Immune activation is associated with many human diseases and is now considered to be an important causative factor for various pathologies (27–29). Recently, it was found immune activation is associated with the release by activated cells of nanosized EV that mediate cell-cell communication and play an important role in immune activation (30–31). In cardiovascular diseases, EV are released predominantly by activated platelets (14–16, 32–33). Activation of platelets as well as of monocytes results in an increase in their adhesion to various surfaces as well as aggregation with other cells, in particular with monocytes, forming MPC in blood (34). The numbers of MPC are increased in the course of certain pathologies (35, 36), in particular in AMI, where these complexes are considered to be a marker of this disease (13). Earlier, Tapp et. al. (14) thoroughly described the changes in monocytes of different subsets following STEMI.
Monocytes in MPC may play pro-inflammatory and pro-atherogenic functions, creating an atherogenic milieu at the vascular wall and thus supporting plaque formation (37–38). On the other hand, monocytes could use activated platelets as vehicles to be delivered to the infarcted heart to resolve inflammation and repair tissue (39). Such apparently opposite effects may be due to the fact that different MPC with monocytes and platelets in different physiological states play different roles in human physiology (40). However, the diversity of MPC has not been fully investigated. Therefore, here we analyzed the involvement of different monocytes and platelets in MPC in healthy volunteers and investigated the changes that occur in patients with AMI.
Since platelets are readily activated in vitro, it was important that the MPCs that we analyzed were formed in vivo rather than after blood collection. In our study, we took measures to prevent in vitro aggregation. In particular: (i) we discarded the first 3 ml of blood, which may contain platelets activated because of the puncture; (ii) we used 3.2% Na citrate, which unlike other anticoagulants has been shown neither to increase in vitro platelet activation nor to dissolve already formed complexes; (iii) we excluded harsh vortexing, centrifugation, RBC lysis, Ficoll gradient, and exposure to cold, as suggested by Harding et al. (19); (iv) we fixed blood within 1 min after blood collection. Finally, to evaluate whether, in spite of all these measures, significant numbers of MPC are nevertheless formed in vitro, we measured the kinetics of their formation after the blood was collected but not fixed. In our experiment, MPC fractions increased with a linear rate of ~0.42% MPC per min within the first hour without fixation. Therefore, the majority of MPC analyzed in the present study have been formed in vivo.
Circulating monocytes are classified in three subgroups according to their phenotypes: classical CD14++CD16-, non-classical CD14+CD16++, and intermediate CD14++CD16+ (20). Using flow cytometry, we evaluated the involvement of each of these types of monocytes in MPC. The distribution of circulating monocytes according to these classes in healthy controls was similar to that reported earlier: about 90% were monocytes of the classical phenotype (Table 1). However, their involvement in MPC was not proportional to their presence in circulation. Indeed, although non-classical and intermediate monocytes constituted the minority, these monocytes entered MPC at disproportionally higher numbers. Nevertheless, since the overwhelming majority of circulating monocytes were classical, the majority of MPC contained monocytes of this type. Why monocytes of a particular phenotype are predominantly involved in MPC, or whether they change their phenotype upon binding to platelets, remains to be understood. The preferential ability of intermediate monocytes to aggregate with platelets is retained in vitro: in our experiments with isolated monocytes and platelets, intermediate monocytes seem to be more extensively involved in MPC formation. The same is true for platelet-derived EV (Figure 5C).
Temporal balance between monocyte subsets may play a significant role in inflammation (41): Several studies have reported on possible involvement of intermediate monocytes in general or in MPC in particular in the inflammatory process (14–15, 42). However, the causative role of intermediate monocytes in inflammation remains unclear.
In our study, we observed changes in AMI patients in the ability to form MPC for all types of monocytes. The majority of AMI patients were STEMI. We added four NSTEMI patients to this group since we did not find statistically significant differences in monocyte and MPC between these two types of patients. However, larger cohorts are needed to study whether NSTEMI and STEMI patients differ in the pattern of MPC formation.
AMI was associated with significant changes in the pattern of MPC formation. First, the numbers both of circulating monocytes and of MPC increased. This increase was not the result of a mere increase in circulating monocytes, as their number increased by ~50% whereas the number of MPC more than tripled. Second, the increase in MPC number was predominantly due to classical monocytes, which in the AMI patients seem to be more prone to aggregate with platelets. Nevertheless, since the increase in MPC involved all types of monocytes, in AMI patients all monocytes and/or platelets seem to be more prone to form aggregates. The increased number of MPC with cells from AMI patients was observed also in vitro.
Since AMI is associated with platelet activation (43), we investigated whether MPC were formed predominantly by activated platelets. Since CD41a used in the current work to identify platelets is expressed constitutively and thus did not reflect their activation status (44), we used PS exposure as evaluated from Ann-V binding, as a marker of platelet activation (45). When we compared the exposure of PS on platelets that are circulating as free cells with that in MPC, we found that platelets in the latter were more activated both for healthy individuals and AMI patients. The increased ability of activated platelets to aggregate with monocytes was confirmed in experiments in vitro when platelet activation more than tripled the fraction of Ann-V-positive MPC. The increase of the ability of activated platelets to form aggregates with monocytes may be related to their expression of P-selectin (3). Non-activated platelets may also form complexes with monocytes but not via P-selectin (46).
We can readily distinguish platelets from EV among circulating particles, as in flow cytometry these events have different pattern of FSC and CD41a fluorescence. Also, events corresponding to EV were smaller than the typical platelets as evaluated with sizing beads. Finally, in confirmation that these events represented EV, their amount was significantly increased upon treating platelets with Ca2+ ionophore A23187, which stimulates the release of EVs by platelets (47). These EV exposed more PS on their surface than did the general platelet population, since EV were released predominantly by activated platelets characterized by high PS exposure, in particular at the sites of microvesicle shedding (48).
Although we can distinguish circulating EV from circulating platelets, it was difficult to distinguish them in complexes with monocytes. MPC CD41a positivity may reflect binding not of integral platelets but also platelet EV. Therefore, our analysis of in vivo samples provided only indirect evidence of the formation of monocyte-EV aggregates. The existence of monocyte-EV aggregates was recently observed directly (16).
To study such aggregates, we performed experiments in vitro, which generally confirmed the in vivo data regarding aggregation of monocytes with platelets and platelet-derived EVs. Intermediate monocytes in vitro retained their increased ability to form aggregates with platelets. In vitro experiments allowed us to focus on aggregation of monocytes with EV: Isolated EVs readily aggregated with monocytes predominantly of intermediate phenotype. Finally, as with the in vivo, in vitro the number of MPC was higher in the case of cells from AMI patients.
Several parameters including MPC counts and their compositions at patient admission can be used in the prognosis of the in-hospital complications of AMI. These results are in agreement with the results of a recent cross-sectional study (15, 49). Also, in future, dynamic changes of MPC may be useful to monitor the efficiency and to estimate the duration of antiplatelet therapy. However, translation of our results into clinical practice requires larger cohorts of patients and longer follow-up periods.
In summary, our results confirm published data and provide new information regarding the patterns of MPC in AMI patients. In particular, we found that the patterns of platelet aggregation with monocytes were different in AMI patients and controls: (i) in AMI patients MPC formed by intermediate monocytes carry more platelets whereas in healthy controls more platelets aggregated with classical monocytes; (ii) the numbers of MPC in AMI patients, being already higher than in controls, were further increased if these patients suffered various in-hospital complications; (iii) On the basis of the CD41a fluorescence of the antibody-stained MPC, some of the aggregates seem to consist of monocytes and platelet-derived EVs ; (iii) aggregation of monocytes with platelet EV occurs in in vitro experiments; (iv) these experiments demonstrated that monocytes from AMI patients aggregate with both platelets and platelet EVs more efficiently than monocytes from controls.
The role of MPC in human pathology, in particular in cardiovascular disease, cannot be understood without first dissecting the diversity of these complexes and characterizing them in normal and pathologic settings. This was the aim of the current work. The dramatic quantitative and qualitative changes in MPC in AMI patients indicate their important physiological role and may be used as correlates for AMI.
Supplementary Material
Scientific category Atherosclerosis and Ischaemic Disease
Acknowledgements
We thank Barry Alpher for editing and improving the English style.
Financial Support
The work of M.L., N.P., M.V., A.S., and E.V was supported by grant #14.B25.31.0016 of the Russian Federation Government and by grant # 18-75-10123 of the Russian Scientific Fund. The work of L.M. and A.A. was supported by the Intramural Program of the National Institute of Child Health and Human Development.
Footnotes
Disclosure of Conflicts of Interest
The authors declare no competing financial interests.
References
- 1.Shantsila E, Lip GYH. Monocytes in Acute Coronary Syndromes. Arterioscler Thromb Vasc Biol 2009; 29: 1433–1438. [DOI] [PubMed] [Google Scholar]
- 2.Fang L, Moore X-L, Dart AM, et al. Systemic inflammatory response following acute myocardial infarction. J Geriatr Cardiol 2015; 12: 305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cerletti C, de Gaetano G, Lorenzet R. Platelet-leukocyte interactions: Multiple links between inflammation, blood coagulation and vascular risk. Mediterr J Hematol Infect Dis 2010; 2: e2010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furman MI, Benoit SE, Barnard MR, et al. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J Am Coll Cardiol 1998; 31: 352–358. [DOI] [PubMed] [Google Scholar]
- 5.Irving PM, Macey MG, Shah U, et al. Formation of platelet-leukocyte aggregates in inflammatory bowel disease. Inflamm Bowel Dis 2004; 10: 361–72. [DOI] [PubMed] [Google Scholar]
- 6.Ferroni P, Basili S, Falco A, et al. Platelet activation in type 2 diabetes mellitus. J Thromb Haemost 2004; 2: 1282–91. [DOI] [PubMed] [Google Scholar]
- 7.Johansson MW, Han ST, Gunderson KA, et al. Platelet activation, P-selectin, and eosinophil b1-integrin activation in asthma. Am. J. Respir. Crit. Care Med. 2012; 185: 498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh MV, Davidson DC, Jackson JW, et al. Characterization of Platelet − Monocyte Complexes in HIV-1 − Infected Individuals: Possible Role in HIV-Associated Neuroinflammation. J Immunol 2014; 192: 4674–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayne E, Funderburg NT, Sieg SF, et al. Increased Platelet and Microparticle Activation in HIV Infection: Upregulation of P-Selectin and Tissue Factor Expression. J Acquir Immune Defic Syndr 2012; 59: 340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michelson AD, Barnard MR, Krueger LA, et al. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 2001; 104: 1533–1537. [DOI] [PubMed] [Google Scholar]
- 11.da Costa Martins PA, van Gils JM, Mol A, et al. Platelet binding to monocytes increases the adhesive properties of monocytes by up-regulating the expression and functionality of b1 and b2 integrins. J Leukoc Biol 2006; 79: 499–507. [DOI] [PubMed] [Google Scholar]
- 12.Li N, Hu H, Lindqvist M, et al. Platelet-leukocyte cross talk in whole blood. Arterioscler Thromb Vasc Biol 2000; 20: 2702–8. [DOI] [PubMed] [Google Scholar]
- 13.Furman MI, Barnard MR, Krueger LA, et al. Circulating Monocyte-Platelet Aggregates Are an Early Marker of Acute Myocardial Infarction. J Am Coll Cardiol 2001; 38: 1002–1006. [DOI] [PubMed] [Google Scholar]
- 14.Tapp LD, Shantsila E, Wrigley BJ, et al. The CD14++CD16+ monocyte subset and monocyte-platelet interactions in patients with ST-elevation myocardial infarction. J Thromb Haemost 2012; 10: 1231–1241. [DOI] [PubMed] [Google Scholar]
- 15.Zhou X, Liu XL, Ji WJ, et al. The kinetics of circulating monocyte subsets and monocyte-platelet aggregates in the acute phase of ST-elevation myocardial infarction associations with 2-year cardiovascular events. Medicine (Baltimore) 2016; 95: e3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss R, Gröger M, Rauscher S, et al. Differential Interaction of Platelet-Derived Extracellular Vesicles with Leukocyte Subsets in Human Whole Blood. Scientific Reports 2018; 8:6598; 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J 2012; 33: 2551–2567. [DOI] [PubMed] [Google Scholar]
- 18.Ibanez B, James S, Agewall S, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society. Eur Heart J 2018; 39: 119–177. [DOI] [PubMed] [Google Scholar]
- 19.Harding SA, Din JN, Sarma J, et al. Flow cytometric analysis of circulating platelet-monocyte aggregates in whole blood: Methodological considerations. Thromb Haemost 2007; 98: 451–456. [PubMed] [Google Scholar]
- 20.Weber C, Eduard S, Hristov M, et al. Role and analysis of monocyte subsets in cardiovascular disease. Joint consensus document of the European Society of Cardiology (ESC) Working Groups ‘Atherosclerosis & Vascular Biology’ and ‘Thrombosis’. Thromb Haemost 2016; 116: 626–637. [DOI] [PubMed] [Google Scholar]
- 21.Cazenave J, Ohlmann P, Cassel D, et al. Preparation of washed platelet suspensions from human and rodent blood. Methods Mol Biol 2004; 272: 13–28. [DOI] [PubMed] [Google Scholar]
- 22.Dinkla S, Brock R, Joosten I, et al. Gateway to understanding microparticles: standardized isolation and identification of plasma membrane-derived vesicles. Nanomedicine 2013; 8: 1657–1668. [DOI] [PubMed] [Google Scholar]
- 23.Murphy AJ, Tall AR. Disordered haematopoiesis and athero-thrombosis. Eur Heart J 2016; 37: 1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poncelet P, Robert S, Bouriche T, et al. Standardized Counting of Circulating Platelet Microparticles Using Currently Available Flow Cytometers and Scatter-Based Triggering: Forward or Side Scatter? Cytom Part A 2016; 89A: 148–158. [DOI] [PubMed] [Google Scholar]
- 25.Ayers L, Kohler M, Harrison P, et al. Measurement of circulating cell-derived microparticles by flow cytometry: Sources of variability within the assay. Thromb Res 2011; 127: 370–377. [DOI] [PubMed] [Google Scholar]
- 26.Panteleev MA, Ananyeva NM, Greco NJ., et al. Two subpopulations of thrombin-activated platelets differ in their binding of the components of the intrinsic factor X-activating complex. J Thromb Haemost 2005; 3: 2545–2553. [DOI] [PubMed] [Google Scholar]
- 27.Margolis LB. Immunoactivation at the crossroads of human diseases. Am J Med 2015; 128: 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Funderburg NT, Mayne E, Sieg SF, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood 2010; 115: 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grivel J-C, Ivanova O, Pinegina N, et al. Activation of T lymphocytes in atherosclerotic plaques. Arterioscler Thromb Vasc Biol 2011; 31: 2929–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robbins PD, Morelli AE. Regulation of Immune Responses by Extracellular Vesicles. Nat Rev Immunol 2014; 14: 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gutiérrez-Vázquez C, Villarroya-Beltri C, Mittelbrunn M, et al. Transfer of extracellular vesicles during immune cell-cell interactions. Immunol Rev 2013; 251: 125–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Badimon L, Suades R, Fuentes E, et al. Role of Platelet-Derived Microvesicles As Crosstalk Mediators in Atherothrombosis and Future Pharmacology Targets: A Link between Inflammation, Atherosclerosis, and Thrombosis. Front Pharmacol 2016; 7: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vagida M, Arakelyan A, Lebedeva A, et al. Flow analysis of individual blood extracellular vesicles in acute coronary syndrome. Platelets 2017; 28: 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Totani L, Evangelista V. Platelet–Leukocyte Interactions in Cardiovascular Disease and Beyond. Arter Thromb Vasc Biol 2010; 30: 2357–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarma J, Laan CA, Alam S, et al. Increased Platelet Binding to Circulating Monocytes in Acute Coronary Syndromes. Circulation 2002; 105: 2166–2171. [DOI] [PubMed] [Google Scholar]
- 36.Wrigley BJ, Shantsila E, Tapp LD, et al. Increased formation of monocyte-platelet aggregates in ischemic heart failure. Circ Heart Fail 2013; 6: 127–135. [DOI] [PubMed] [Google Scholar]
- 37.Lindemann S, Kramer B, Daub K, et al. Molecular pathways used by platelets to initiate and accelerate atherogenesis. Curr Opin Lipidol 2007; 18: 566–573. [DOI] [PubMed] [Google Scholar]
- 38.Shantsila E, Lip GYH. The role of monocytes in thrombotic disorders. Insights from tissue factor, monocyte-platelet aggregates and novel mechanisms. Thromb Haemost 2009; 102: 916–924. [DOI] [PubMed] [Google Scholar]
- 39.Ed Rainger G, Chimen M, Harrison MJ, et al. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets 2015; 26: 507–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: Protagonists of infarct inflammation and repair after myocardial infarction. Circulation 2010; 121: 2437–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghattas A, Griffiths HR, Devitt A, et al. Monocytes in Coronary Artery Disease and Atherosclerosis. Where Are We Now? J Am Coll Cardiol 2013; 62: 1541–1551. [DOI] [PubMed] [Google Scholar]
- 42.Rogacev KS, Cremers B, Zawada AM, et al. CD14++CD16+ Monocytes Independently Predict Cardiovascular Events. J Am Coll Cardiol 2012; 60: 1512–1520. [DOI] [PubMed] [Google Scholar]
- 43.Langford E, Wainwright R, Martin J. Platelet Activation in Acute Myocardial Infarction and Unstable Angina Is Inhibited by Nitric Oxide Donors. Arter Thromb Vasc Biol 1996; 16: 51–55. [DOI] [PubMed] [Google Scholar]
- 44.Michelson AD. Evaluation Of Platelet Function By Flow Cytometry. J Pathophysiol Haemost Thromb 2006; 35: 67–82. [DOI] [PubMed] [Google Scholar]
- 45.Dale GL. Coated-platelets: an emerging component of the procoagulant response. J Thromb Haemost 2005; 3: 2185–2192. [DOI] [PubMed] [Google Scholar]
- 46.Rinder HM, Bonan JL, Rinder CS, et al. Activated and Unactivated Platelet Adhesion to Monocytes and Neutrophils. Blood 1991; 78: 1760–1769. [PubMed] [Google Scholar]
- 47.Żmigrodzka M, Guzera M, Miśkiewicz A, et al. The biology of extracellular vesicles with focus on platelet microparticles and their role in cancer development and progression. Tumor Biol 2016; 37: 14391–14401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muralidharan-Chari V, Clancy JW, Sedgwick A, et al. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci 2010; 123: 1603–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiva-blanch G, Laake K, Myhre P, et al. Platelet-, monocyte-derived and tissue factor-carrying circulating microparticles are related to acute myocardial infarction severity. PLoS One 2017; 12: e0172558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.