

Abstract
The pleckstrin homology (PH) domain of general receptor for phosphoionositides 1 (GRP1-PHD) binds specifically to phosphatidylinositol (3,4,5)-triphosphate (PIP3), and acts as a second messenger. Using an extensive array of molecular dynamics (MD) simulations employing highly mobile membrane mimetic (HMMM) model as well as complementary full membrane simulations we capture differentiable binding and dynamics of GRP1-PHD in the presence of membranes containing PC, PS and PIP3 lipids in varying compositions. While GRP1-PHD forms only transient interactions with pure PC membranes, incorporation of anionic lipids resulted in stable membrane-bound configurations. We report the first observation of two distinct PIP3 binding modes on GRP1-PHD, involving PIP3 interactions at a “canonical” and at an “alternate” site, suggesting the possibility of simultaneous binding of multiple anionic lipids. The full membrane simulations confirmed the stability of the membrane bound pose of GRP1-PHD as captured from our HMMM membrane binding simulations. By performing additional steered membrane unbinding simulations and calculating non equilibrium work associated with the process, as well as metadynamics simulations, on the protein bound to full membranes, allowing for more quantitative examination of the binding strength of the GRP1-PHD to the membrane, we demonstrate that along with the bound PIP3, surrounding anionic PS lipids increase the energetic cost of unbinding of GRP1-PHD from the canonical mode, causing them to dissociate more slowly than the alternate mode. Our results demonstrate that concurrent binding of multiple anionic lipids by GRP1-PHD contributes to its membrane affinity, which in turn control its signaling activity.
Introduction
A common step in many cellular signalling pathways is the recruitment of peripheral proteins to the cytoplasmic side of the plasma membrane.1 These signalling processes are predominantly initiated by specific interactions between lipids and peripheral membrane proteins. Although the bulk of the plasma membrane in mammalian cell is composed of neutral phospholipids and cholesterol, monovalent anionic lipids such as phosphatidylserine (PS), which constitute about one-fifth of the plasma membrane, render the cytoplasmic leaflet of the plasma membrane electronegative in nature.2 In general, non-specific electrostatic interactions with the anionic lipids can also drive the localization of the peripheral membrane proteins from the cytosol to the membrane surface.
Lipid-specific interactions between the peripheral proteins and the membrane are pre-dominantly regulated by rare signalling lipids such as multivalent phosphoinositides, which possess high electronegative charges on their headgroup. These lipids are known to regulate various signalling pathways.3 Moreover, depending on the location and degree of phosphorylation of the headgroup, phosphoinositides can exist as multiple isomers of phosphatidylinositol phosphate (PIP) lipids.4,5 Many peripheral membrane proteins recognize and specifically bind to PIP lipids on the membrane surfaces of different organelles in the cell and on the plasma membrane.6–8
PI(4,5)P2 and PI(3,4,5)P3, henceforth referred to as PIP2 and PIP3 respectively, are the two commonly found PIP lipids in the plasma membrane.9 The level of PIP2 in the plasma membrane is around 1–2% and it can be interconverted to PIP3 in response to specific signals. 3,4 For example, PIP2 can be phosphorylated by phosphoinositol-3-kinase (PI3K)10 to produce PIP3, which works as a second messenger in signaling pathways.11 It is known that the signaling lipid PIP3 regulates diverse cellular processes, e.g., cytoskeleton rear-rangements, vesicle trafficking, and apoptosis, by recruiting an array of peripheral signalling proteins to the membrane surface.12–16
Pleckstrin homology (PH) domains are one of the peripheral membrane proteins that are known to selectively bind PIP lipids in the plasma membrane. PH domain of the general receptor for phosphoinositides 1 (GRP1-PHD) binds specifically to PIP3 lipids and excludes PIP2,12 while those belonging to the protein phospholipase C delta 1 (PLCδ1) bind specifically to PIP2 lipids in the plasma membrane.8 In all its homologs, the PH domain has a relatively conserved core with approximately 120 amino acid residues. The core consists of an α-helix and two sandwiched β-sheets, connected by loops that vary in sequence. In a majority of PH domains the loop connecting the β1/β2 strands has the KXn(K/R)XR motif (proposed to constitute the “canonical” PIP3 binding site, henceforth referred to as the C site) which forms binding pockets for the negatively charged headgroup of PIP lipids. However, in other PH domains not possessing this consensus motif, binding of PIP lipids occurs on the face opposite to β1/β2 strand (coined as alternate binding site, henceforth referred to as A site).17,18 Interestingly, a recent crystal structure of the PH domain from ASAP1 suggested that PIP may bind to both C and A sites, thus highlighting the complexity of interactions between the PH domain and anionic lipids in the membrane.19
A number of biophysical techniques including surface plasmon resonance (SPR),22 isothermal titration calorimetry (ITC),23 and förster resonance energy transfer (FRET)12 have been used to gauge the binding affinities of the PH domains for PIP lipids. These studies reported that PH domains selectively bind to PIP2, PIP3 or other PIPs with nanomolar to micromolar affinity.24–26 However, these studies were performed outside the context of a membrane and employed a water soluble analog of PIP, 23 which may not represent PIP lipids in a membrane environment, thus making it difficult to determine how PH domains localize on the PIP-rich membranes.
Molecular dynamics (MD) simulations at various resolutions have been used to investigate protein-lipid interactions in PH,27–30 C2,8 and GLA domains31 and their results optimally complement experimental studies by providing a microscopic view of the lipid-protein interactions determining selectivity and affinity. 32 In the current study we employ MD simulations to investigate the PIP3-specific membrane binding of GRP1-PHD. A high resolution crystal structure of GRP1-PHD bound to I(1,3,4,5)P4 (headgroup of PIP3 lipid) is known (PDB:1FGY)20 (Figure 1), and it has been used in a number of previous computational studies.27,33,34 These studies either employed coarse-grained representation of the system or were performed starting with a membrane embdedded GRP1-PHD, thus they were unable to provide atomistic description of the process of GRP1-PHD association to PIP3-containing anionic membranes.
Figure 1:
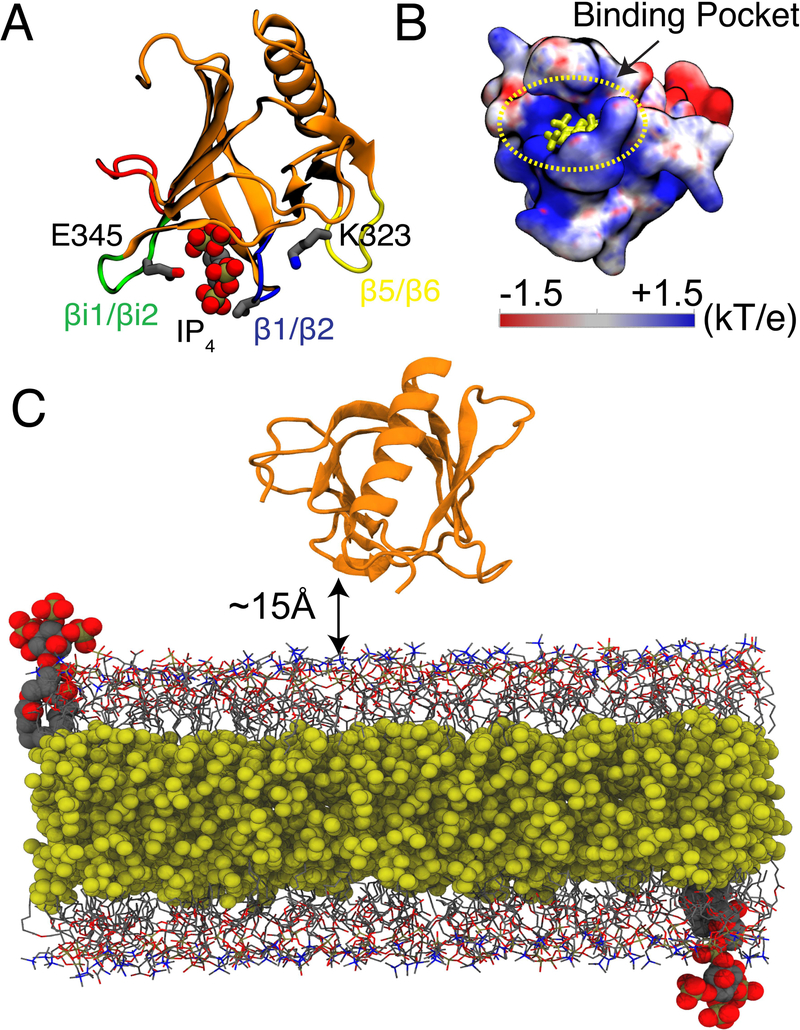
GRP1-PHD structure and simulation system. (A) The crystal structure of GRP1-PHD (PDB:1FGY)20 with co-crystalized IP4 representing the PIP3 lipid headgroup. The membrane interacting loops are as follows: β1/β2 (shown in blue), βi1/βi2 (shown in green), and β5/β6 (shown in yellow). (B) Electrostatic potential map of apo GRP1-PHD, IP4 is shown in yellow. The electrostatic map was generated using APBS software.21 Blue denotes positive potential surface and red denotes negative potential surface. (C) A representative simulation box for the HMMM membrane binding simulations of GRP1-PHD, which is initially placed at least 15 Å away from the membrane surface. Short-tailed PC and PS lipids are shown in grey lines and short-tailed PIP3 lipids (one in each leaflet corresponds to a molar ratio of 1%) are shown in vdW representation. The hydrophobic core of the membrane is filled with an organic solvent (DCLE), shown in yellow vdW representation. Bulk water molecules and ions are not shown for clarity.
Our approach is to use unbiased sampling of the membrane-binding process using atomistic MD simulations to capture lipid-protein interactions responsible for the association and binding of GRP1-PHD to anionic membranes. Through a large set of independent MD simulations of initially unbound GRP1-PHD placed above the lipid-bilayer, we arrive at an unbiased population of PIP3 bound GRP1-PHD. To accelerate the sampling and insertion of GRP1-PHD we used the highly mobile membrane mimetic (HMMM) model,35 which accelerates the lipid diffusion and reorganization in the membrane using an atomistic representation. This approach has been previously used to study binding of other peripheral proteins.36–42 The enhanced sampling of lipid-protein interactions enabled by the use of HMMM combined with multiple independent simulations performed for each condition allowed us to not only observe repeated binding of PIP3 in the canonical (C) mode, but also describe for the first time spontaneous membrane association of GRP1-PHD mediated by an alternate (A) mode43 of of PIP3 binding. The membrane-bound configurations were found stable during the subsequent simulations in which HMMM membranes were converted to full membranes. Through non-equilibrium work calculations and metadynamics simulations performed on the final, bound structure of GRP1-PHD to full membranes, we also evaluate the energetics associated with unbinding of GRP1-PHD from these two binding modes. These findings highlight the role of the membranes specifically PIP3, as an active platform for the localization of GRP1-PHD.
Methods
System setup
GRP1-PHD crystal structure was taken from the Protein Data Bank entry 1FGY20 as a starting structure for the simulations. A C-terminal carboxylate capping group, an N-terminal ammonium capping group, and hydrogen atoms were added using the PSFGEN plugin of VMD (Visual Molecular Dynamics).44 Next, GRP1-PHD was placed in a 63×63×63 Å3 water box using the Solvate plugin of VMD. The solvated GRP1-PHD was then neutralized with Na+ and Cl− ions (150 mM NaCl) using the AUTOIONIZE plugin of VMD, and the system was energy minimized for 1,000 steps and equilibrated for 1ns. The final solution-equilibrated GRP1-PHD was used for all the subsequent membrane binding simulations.
Multiple independent HMMM membranes were constructed using HMMM BUILDER in CHARMM-GUI.45,46 The presence of short tailed lipids and the use of an organic liquid, DCLE, which mimics the bilayer interior, significantly enhance the dynamics of lipids thereby allowing rapid peripheral protein insertion.35,41,47,48 With the aid of this membrane mimetic model, we were able to perform multiple membrane binding simulations of GRP1-PHD in the presence of mixed-lipid membranes containing phosphatidylcholine (PC), phosphatidylserine (PS), and PIP3 in varying compositions (Table 1).
Table 1:
Details of the systems and the simulations performed
| Simulation Type | Membrane Composition | Simulated replicas | Membrane Binding replicas* | PIP3 Bound replicas | Interacting Lipids | Time (ns) per replica |
|---|---|---|---|---|---|---|
| PC | 5 | 2 | NA | PC | 100 | |
| HMMM | PC:PS (80:20) | 9 | 7 | NA | PC, PS | 100 |
| PC:PS:PIP3 (80:19:1) | 9 | 9 | 4 | PC, PS, PIP3 | 100, 150** | |
| Full | PC:PS (80:20) | 1 | 1 | NA | PC, PS | 150 |
| Membrane | PC:PS:PIP3 (80:19:1) | 2*** | 2 | 2 | PC, PS, PIP3 | 150 |
| PC:PS (80:20) | 3 | 1 | NA | PC, PS | 40 | |
| SMD | PC:PS:PIP3 (80:19:1) | 3 | 2 | 2 | PC, PS, PIP3 | 40 |
| WT-MetaD | PC:PS:PIP3 (80:19:1) | 1 | 2 | 2 | PC, PS, PIP3 | 600–900 |
GRP1-PHD interacting with the membrane.
PIP3-bound replicas were extended to 150 ns.
Canonical and Alternate binding modes.
We started by placing GRP1-PHD in the aqueous solution at least 15Å away from the phosphate plane of the cis leaflet of the lipid bilayer. To introduce randomness into the initial orientation of GRP1-PHD with respect to the membrane, we generated different initial configurations of the protein by arbitrarily varying the angle between a vector passing through the core of the β-sandwich, and the membrane normal (θ = 62.2°, 91.7°, 109.4°, 117.8°, 118.3°, 119.1°, 120.8°, 121.8°, and 123.9°). These initial configurations will allow for a better exploration of potential binding poses of the protein to the membrane and sampling of relevant specific lipid-protein interactions. Each replica was simulated for a total of 105 ns (initial equilibration of 5ns followed by 100ns of production). To test the stability of the PIP3-bound GRP1-PHD, the HMMM production run of the representative replicas was further increased to 150 ns. The resulting membrane-bound configurations of GRP1-PHD obtained from these extended HMMM simulations were further equlibrated in full-membrane environments (after conversion of short tailed lipids to full lipids) and used in SMD and metadynamics simulations.
All the simulations were performed using NAMD2,49 CHARMM36 protein and lipid forcefields50,51 and TIP3P water. Long-range electrostatic forces were calculated using the particle mesh Ewald (PME) method.52 Non-bonded forces were calculated with a cutoff of 12 A and a switching distance of 10 Å. A Langevin thermostat using γ = 1ps−1 was used to maintain the system temperature at 310 K. Short-tailed HMMM lipids are best simulated within a fixed area ensemble. The pressure was therefore maintained only along the membrane normal (nPnAT) using a Nosé-Hoover piston.53
Conversion of HMMM model to full membrane
To ensure the membrane-bound GRP1-PHD complex generated using HMMM was stable in full membranes, we converted PIP3-bound GRP1-PHD copies to full membranes using CHARMM-GUI.45,46 The PIP3-bound conformation of GRP1-PHD in full-membrane was initially equlibrated for 10 ns with protein backbone atoms harmonically restrained to their initial positions with a force constant of 1 kcal/mol/Å2. This restraint was released for the following 150 ns production runs.
Analysis
The membrane binding and depth of GRP1-PHD insertion into the lipid bilayer were measured by two metrics: (1) ensemble averaged z position of all Cα atoms of GRP1-PHD with respect to the cis leaflet phosphate plane, and (2) time evolution of the center of mass (COM) of the side-chain heavy atoms of the residues frequently interacting with the membrane and engaging lipids in major ways: R277, V278, and K279. These residues form the tip of the membrane binding β1-β2 loop of the GRP1-PHD. GRP1-PHD was considered to be membrane-bound if the tip of β1/β2 (V278) penetrated below the level of phosphate plane in the membrane.
Furthermore, we have characterized the orientational dynamics of PIP3 headgroup when bound to GRP1-PHD by calculating its tilt angle (θ) with respect to the membrane plane. θ was defined as the angle between the vector passing through P-C4 atoms of the inositol ring and the membrane normal. Similarly, the orientation of membrane-bound GRP1-PHD was monitored by: (1) displaying its dipole moment, and (2) measuring the angle between a vector passing through the Cα atoms of F296 and C292, representing the core of the β-sandwich, and the membrane normal. As these coarse metrics may not provide sufficient details on the nature of GRP1-PHD-lipid interactions, we also quantified specific interactions between lipid headgroups and GRP1-PHD. Based on our previous experience,54 a 3.5 Å heavy-atom cut-off was chosen to define lipid-protein contacts. For the GRP1-PHD-bound PIP3, we have calculated specific contacts between the protein and the PO4 at 3’, 4’ and 5’ positions of the inositol ring.
Non-equilibrium membrane unbinding simulations
To go beyond the qualitative description of lipid-protein interactions, we have performed steered molecular dynamics (SMD)55 simulations with the distance between the COM of the lipid bilayer and the COM of the protein as a reaction coordinate. The full membrane model of membrane-bound GRP1-PHD was used for these simulations. SMD was performed at a rate of 0.25 Å/ns with a harmonic force constant of 7 kcal/mol/Å2. Non-equilibrium work profiles generated from SMD simulations were used to study whether the presence of PIP3 lipid affected the dissociation of GRP1-PHD from the full membranes and to compare the binding strength of C and A sites. To test the reproducibility of the non-equilibrium calculations, 3 independent replicas starting with different initial velocities were simulated for each case. In order to prevent the separation of the two leaflets of the lipid bilayer during the pulling simulations (an artifact which may arise when the COM of the membrane is involved in the reaction coordinate), a z restraint (k = 1 kcal/mol/Å2) was applied to the phosphorous atoms of the lipid bilayer.
Well-tempered metadynamics
We employed well-tempered metadynamics (WT-MetaD)56 calculations in order to further differentiate between C and A binding modes as captured in our membrane binding simulations. In WT-MetaD calculations we used the distance (rather than its projection along a particular axis, e.g., the membrane normal) between the COM of GRP1-PHD and the COM of the PIP3 headgroup (all the heavy atoms of inositol ring), as the reaction coordinate. 57 The WT-MetaD simulations were performed with an initial Gaussian hill-height of 0.25kcal/mol, a bias factor of 10.6, and a Gaussian hill deposition rate of 2ps. Initial position for each WT-MetaD simulation was taken from the last frame of the 150 ns long production run in full-tailed lipid environments. The convergence of the free-energy profile obtained from WT-MetaD, which is also reflected in invariance of free energy minima with the simulation time, was analyzed using time evolution of the added Gaussian height. In WT-MetaD, the height of the added Gaussian is rescaled according to the added bias. At the end of the simulations, we observed the added bias or the Gaussian hill height close to 0, suggesting sampling of the entire energy landscape.
Results and Discussion
The results can be divided primarily into four avenues: (1) characterizing the membrane-bound configuration of GRP1-PHD in terms of binding depth and the orientational dynamics in membranes of varying lipid compositions; (2) enumeration of specific interactions between GRP1-PHD and the anionic lipids and characterization of PIP3-specific binding sites/mode(3) determination of non-equilibrium work profile of GRP1-PHD dissociation from different lipid bilayers; and (4) dissociation free energy of GRP1-PHD bound to PIP3 lipids from C and A binding modes calculated using WT-MetaD.
GRP1-PHD association to PC and PC/PS bilayers
After 100 ns, in 2 of 5 replicas GRP1-PHD was observed to bind to membranes with 100% PC (Figures 2A and S1). In 3 other copies we observed that GRP1-PHD did not associate to the lipid bilayer, rather after making contacts it bounced off the lipid bilayer (Figures 2B,C and S2), suggesting transient and weak membrane binding propensity for the protein under these conditions. This finding is in line with FRET12 and simulation studies27,33 and can be attributed to the lack of strong electrostatic interactions between GRP1-PHD and the zwitterionic lipids.
Figure 2:

Interaction of GRP1-PHD with a pure PC lipid bilayer. Ensemble-averaged Cα profile of the unbound (A) and bound (B) GRP1-PHD, calculated during last the 50 ns of HMMM simulations (see Figure S1 for results of all 5 replicas). The blue bars denote the standard deviation in the residue distances. All the membrane interacting loops are highlighted in yellow. (C) Time evolution of side chain COM of the R277, V278, and K279 in one of the simulations (see Figure S2 for data for all 5 replicas) highlighting how the unbound GRP1-PHD bounces off the lipid bilayer. The configurations of GRP1-PHD are shown in ① and ②, taken from the membrane binding simulations at t=5 ns and t=50 ns, respectively. GRP1-PHD dipole moment is shown by a green arrow to allow for visual inspection of the change in orientation.
In the presence of 20% anionic PS lipids (system simulated with 80:20 PC:PS), 7 out of 9 copies of GRP1-PHD were found to rapidly bind and insert into the membrane and to remain bound for the rest of the simulation time. In all the simulated replicas, the β1/β2 loop makes contact with the lipid bilayer (Figures 3A and S3), highlighting its strong membrane binding propensity. Apart from the β1/β2 loop, in some replicas, the βi1/βi2 and β5/β6 loops also make contact with the membrane, suggesting the presence of multiple membrane binding modes of GRP1-PHD. Analysis of the GRP1-PHD-membrane binding trajectories highlighted that at first the β1/β2 loop comes in contact with the lipid bilayer, working as an initial anchor point, after which it can tumble eitherways to make membrane contacts either via the β5/β6 or the βi1/βi2 loop.
Figure 3:

Interaction of GRP1-PHD with an anionic (PC:PS (80:20)) lipid bilayer. (A) Ensemble-averaged Cα profile of membrane-bound GRP1-PHD calculated during the last 50 ns of HMMM membrane binding simulations shown for a representative simulation out of 9 independent replicas (see Figure S3 for results on all 9 replicas). The Cα profile highlights the interaction of the β1/β2 and β5/β6 loops with the lipid headgroups. All the membrane interacting loops are highlighted in yellow. The blue bars denote the standard deviation in the residue distances. (B) Time evolution of the side-chain COM of R277, V278, and K279 during the initial 100 ns of HMMM (see Figure S4 for results on all 9 replicas) and subsequent 150 ns of full membrane (shown in grey background) simulations. ①, ②, and ③ highlight the membrane recruitment events of GRP1-PHD. Snapshot ④ is the stable conformation of membrane-bound GRP1-PHD after 150 ns of simulation in full membrane following the conversion from HMMM membrane. GRP1-PHD dipole moment is shown by a green arrow.
Furthermore, the time evolution of the positions of the side chain COM of R277, V278, and K279 suggests a shallow binding mode, i.e., the side chains stays near the phosphate plane and do not penetrate deep into the lipid bilayer (Figures 3B, S4, and Table S1). The stability of the resulting membrane-bound GRP1-PHD was further tested by converting the representative membrane bound replicas obtained from HMMM simulations to full membranes. The relative depth of GRP1-PHD insertion remained stable during 150 ns of simulation in full membrane (Figure 3B), highlighting stable binding of GRP1-PHD to PS-containing anionic membranes.
GRP1-PHD association to PC/PS/PIP3 bilayer
To gain insight on how the presence of PIP3 affects membrane binding of GRP1-PHD, we simulated multiple (9) independent replicas of GRP1-PHD in the presence of a PIP3 lipid. In order to reproduce the natural abundance of PIP3 lipids in the plasma membrane which is low,9 starting from our PC:PS membrane, we replaced one PS lipid in each leaflet with a PIP3 lipid, making the bilayer composition PC:PS:PIP3 (80:19:1). All the 9 simulated copies of GRP1-PHD spontaneously and stably bound to the membrane during the HMMM membrane-binding simulations (Figures 4A,B and S5). Interestingly, we observed that our unbiased membrane binding simulations of GRP1-PHD in PIP3-containing bilayers capture similar Cα fingerprints as proposed in a previous EPR study27 (Figure 4A). Similar to the membrane binding simulations of GRP1-PHD in the presence of a PC:PS membrane, the β1/β2 loop makes contacts with the lipid bilayer in all the simulated replicas. Moreover, in some replicas the β5/β6 and βi1/βi2 loops show a clear tendency to bind to the lipid bilayer, suggesting the possibility of multiple membrane binding modes for GRP1-PHD. The binding depth analysis shows that V278 is the deepest membrane binding residue of GRP1-PHD with an average depth of −1.65±2.61Å below the phosphate plane (Figure S6 and Table S1). These results are in close correspondence with a previously published EPR study of GRP1-PHD which predicted V278 formed the closest point of contact with the membrane surface.58
Figure 4:

Interaction of GRP1-PHD with PIP3-containing (PC:PS:PIP3 (80:19:1)) lipid bilayer. Ensemble-averaged Cα profile of GRP1-PHD in representative simulations capturing PIP3 binding to (A) the canonical site (C site) and to (B) the alternate site (A site), calculated over last 100ns of HMMM simulations (see Figure S5 for results on all 9 replicas). In the C site PIP3 is bound to the β1/β2 loop while in the A site it is bound to the β5/β6 loop. Representative snapshots of the canonical (C) and alternate (D) PIP3 binding modes are shown. All the GRP1-PHD residues lying within 3.5 Å of PIP3 are shown. The membrane interacting loops are highlighted in yellow. The blue bars denote the standard deviation in the residue distances.
In 4 of the 9 membrane-bound replicas, GRP1-PHD is found to be bound directly to a PIP3 lipid (Figure 4C,D). In two of the bound replicas PIP3 interacts with the β1/β2 loop (C site), closely corresponding to the position of the co-crystallized PIP3 headgroup (PDB:1FGY) 20(Supp. Video 1). In the other two replicas PIP3 interacts with the β5-β6 loop (A site). To the best of our knowledge this mode of binding has not been captured for GRP1-PHD, but is known to be present in other PH domains. 17,59,60
To test the stability of the C and A binding modes, MD simulations of the PIP3-bound replicas were extended to 150 ns in HMMM membranes and then converted to the full membrane and simulated for another 150 ns. Both the orientation and the relative depth of insertion of GRP1-PHD remained stable in 150 ns of full membrane simulations following the conversion (Figure S7). These full membrane simulations were utilized to characterize the conformational dynamics of GRP1-PHD and PIP3 lipid in the bound complex. Comparison of the orientational dynamics of GRP1-PHD in C and A modes indicates that the PH domain prefers to orient at an angle of 36.9±9.1 and 29±11.9, respectively, with respect to the membrane normal (Figure 5). Using a similar approach, we compared the orientational dynamics of PIP3 in the two binding modes with the free PIP3 (present in the trans leaflet) and observed a narrower orientational distribution of the bound PIP3 lipid, while a much broader orientation was observed for the free PIP3 lipid (Figure 5B).
Figure 5:

Orientational preference of membrane bound GRP1-PHD and protein-bound PIP3 lipid. (A) Orientation angle of GRP1-PHD as captured from the simulations in the full membrane. The orientation angle of GRP1-PHD in the two binding modes was defined as the angle between the vector passing through Cα atoms of C292 and F296 shown in green (long axis of the core-β sandwich) and the membrane normal (z-axis in our simulations). (B) The tilt angle of bound PIP3 lipid in the two binding modes was defined as the angle between the vector passing through the atoms P and C4 of PIP3 and the membrane normal. In the unbound form, PIP3 headgroup was observed to sample all possible orientations, while a narrower orientation distribution was observed in GRP1-PHD-bound PIP3.
The differences in the orientational dynamics of GRP1-PHD and PIP3 lipid in the two binding modes can be attributed to the differential concentration of surrounding anionic PS lipids in the area close to the protein. We analyzed the distribution of PS lipids around the bound GRP1-PHD by calculating their occupancy map in the two binding modes. As compared to A-mode, a larger concentration of anionic PS lipids was observed around GRP1-PHD in C-mode (Figure S8), suggesting drastically different local lipid environment around the bound GRP1-PHD in the two binding modes.
Lipid-specific binding of GRP1-PHD
To provide a microscopic view of PIP3 binding, as well as the location of putative PS-specific hotspots, we analyzed lipid-protein interactions using 150 ns trajectories obtained from full membrane simulations. We started by analyzing the interaction of GRP1-PHD with each of the PS charged groups, namely carboxy (COO−), amino (NH3+), and phosphate (PO4−) groups in both C and A modes. In both binding modes, the β1/β2 loop forms a high number of contacts with the negatively charged PS lipid headgroups (Figure 6A,B), highlighting its importance for the membrane. Interestingly, the βi1/βi2 loop, which forms a significant number of contacts with the PS lipids in A-mode, hardly interacts with GRP1-PHD in C-mode.
Figure 6:

Lipid-protein interactions in PIP3-bound GRP1-PHD. Histograms of the protein residues in contact to the PS charged groups, COO−, PO4− and NH3+ are shown for (A) canonical and for (B) alternate binding modes. The histogram of the protein residues in contact to the phosphate groups of PIP3 (P, P3, P4, and P5) for (C) canonical and for (D) alternate binding modes. The representative snapshots of GRP1-PHD highlighting the residues in contact with PIP3 for the two binding modes are shown in E and F. All the lipidprotein contacts were analyzed using 150- ns full membrane simulations. All the residues are colored according to their interaction with the charged groups.
We further compared the PIP3 binding sites in the two modes by analyzing GRP1-PHD interaction with each of the PIP3 charged groups, namely the terminal phosphates at positions 3 (P3), 4 (P4), and 5 (P5) of the inositol ring, and the phosphodiester group of the headgroup (Figure 6C,D). Overall in both binding modes GRP1-PHD remained associated with the PIP3 lipid during the entire simulation time. K273, R277, K282, R284 and Y298 in C mode, while R283, R322, and K323 in A mode form hydrogen bonds with the PIP3 headgroup (Table S3 and S2). At any given time more than one GRP1-PHD residue were involved in binding with PIP3. The PIP3 canonical binding site (C mode) captured from our unbiased simulations is in close correspondence with the previous NMR, EPR, and the X-ray structure.20,33,58
Differential unbinding of GRP1-PHD from anionic membranes
To probe the dissociation of GRP1-PHD from the anionic membranes, we employed constant velocity SMD simulations on GRP1-PHD bound to full membranes, using the distance between the COM of GRP1-PHD and the COM of the membrane as the reaction coordinate. Figure 7 shows the non-equilibrium work profiles for GRP1-PHD dissociation from the two PIP3 binding modes, as well as from PIP3-free anionic bilayers (PC:PS). The non-equlibrium work profiles show that the amount of work required to dissociate GRP1-PHD from the membrane is higher in the presence of PIP3 lipids, thus corroborating the higher affinity of GRP1-PHD towards PIP3 lipids. 22,61,62 These SMD pulling simulations were repeated three times for each system and consistent behaviors were observed.
Figure 7:

Dissociation of the GRP1-PHD from anionic membranes. (A) Non-equilibrium work profiles for dissociating PHD from the lipid bilayer containing anionic lipids. (B) Starting configuration for pulling simulations which were performed using distance between the COM of the membrane and the COM of PHD as the reaction coordinate. Lipid tails, water and ions are not shown for clarity.
Since non-equilibrium work calculations were not able to fully differentiate between C and A modes, we performed additional free energy calculations.We generated 1D free energy profiles of GRP1-PHD dissociation from C and A modes by using WT-MetaD (Figure 8). A global minimum is observed at GRP1-PHD-PIP3 separation of 20Åwith a well-depth of 8.5 kcal/mol for C-mode, and at a distance of 25 Å with a well depth of 7.1 for A-mode. These profiles suggest favorable binding of GRP1-PHD in both modes, but with a somewhat higher preference for C-mode, which might be attributed to the clustering of PS lipids around the GRP1-PHD in this binding mode (Figure S8). This reveals cooperativity as the key mechanism for membrane localization of GRP1-PHD.
Figure 8:

Free Energy profiles for dissociation of GRP1-PHD from canonical and alternate binding mode calculated with WT-MetaD. Representative snapshots of the PIP3-bound and PIP3-dissociated GRP1-PHD in the canonical mode (left) and the alternate mode (right) are shown at the bottom. The convergence of free energy profiles was analyzed by the time evolution of free energy minima (Figure S9).
Experimentally derived dissociation constants for GRP1-PHD from PIP containing membranes range from 1 μM to 5 nM, depending on the experimental conditions (e.g. presence/absence of background anionic lipids, pH and temperature).22,24,25 which approximately correspond to ΔG values between −8 and −11 kcal/mol. The difference between the free energy values derived in this work and the experimental values can be attributed to factors such as the differences in the overall lipid compositions, particularly the higher PIP concentration in experiments.12,25
Conclusion
We employed a membrane model with enhanced lipid dynamics to capture spontaneous, unbiased binding and insertion of GRP1-PHD to PC, PS, and PIP3-containing anionic membranes at an atomistic level. Membrane binding of GRP1-PHD was found to depend on the presence of anionic lipids, highlighting the importance of electrostatic interactions for the association process. Anionic lipids such as PS and PIP can contribute to the membrane binding affinity of GRP1-PHD, both through nonspecific effects, namely by increasing the overall negative electrostatic potential of the membrane, and through direct, specific binding to the protein. The simulations show that GRP1-PHD can bind to PIP3 and PS lipids in distinct binding modes, suggesting that simultaneous binding of multiple anionic lipids is possible and may contribute towards the specificity of GRP1-PHD for specific lipid compositions. Despite employing different starting configurations for the simulations, we were able to capture a reproducible membrane-bound configuration of GRP1-PHD. The insertion and orientation of membrane-bound GRP1-PHD was found to be preserved in full membranes.
GRP1-PHD was observed to bind PIP3 in two distinct binding modes: a canonical or C mode, and an alternate or A mode (Figure 9). Microscopic examination of lipid-protein contacts and interactions in the two binding sites suggests that in C site, PIP3 interacts mostly with the β1-β2 loop, while in A site, PIP3 interacts primarily with the β5-β6 loop. Furthermore, non-equilibrium work calculations and 1D dissociation free energy profiles demonstrate stronger binding of GRP1-PHD to the C binding mode. The presented results provide a detailed view of lipid dependence of membrane binding GRP1-PHD, specifically distinct roles of anionic lipids in its membrane binding, which in turn control its signalling activity.
Figure 9:

Summary of the observed differential binding modes of GRP1-PHD to anionic membranes. Shown is a schematic membrane consisting of hydrocarbon tails (red and yellow), with the headgroups color-coded according to the simulated systems (golden for neutral PC, green for anionic PS, and yellow for anionic PIP3. The phosphate on the PIP3 are highlighted in red). Also shown is GRP1-PHD used in the simulations (in orange, cartoon representation). Starting with the unbound GRP1-PHD in the aqueous solution three distinct interactions with the membrane were captured: (A) GRP1-PHD interacts preferentially with the anionic PS lipids and never dissociates to interact with the target PIP3 lipids; (B) GRP1-PHD interacts directly with PIP3 in canonical or C binding mode; and (C) GRP1-PHD interacts directly with PIP3 in alternate or A binding mode. Once the GRP1-PHD docks onto the PIP3 headgroup it never dissociates within the simulated time. It is to be emphasized that in C mode, cluster of PS lipids was observed around the PIP3-bound GRP1-PHD, which might contribute towards the higher affinity for this mode of interaction.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Institutes of Health under award numbers P41-GM104601 (to ET), and R01-GM123455 (to ET). We also acknowledge computing resources provided by Blue Waters at National Center for Supercomputing Applications, and Extreme Science and Engineering Discovery Environment (grant MCA06N060 to ET). We would like to acknowledge Beckman Institute Graduate Fellowship for supporting S.P.
References
- (1).Cho W; Stahelin RV Annual Review of Biophysics and Biomolecular Structure 2005, 34, 119–151. [DOI] [PubMed] [Google Scholar]
- (2).Leventis PA; Grinstein S Annual Review of Biophysics 2010, 39, 407–427. [DOI] [PubMed] [Google Scholar]
- (3).Di Paolo G; De Camilli P Nature 2006, 443, 651–657. [DOI] [PubMed] [Google Scholar]
- (4).Kutateladze TG Nature Cell Biology 2010, 6, 507–513. [Google Scholar]
- (5).Stahelin RV; Scott JL; Frick CT Chemistry and Physics of Lipids 2014, 182, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lemmon MA Nature Reviews Molecular Cell Biology 2008, 9, 99–111. [DOI] [PubMed] [Google Scholar]
- (7).Morales KA; Igumenova TI Biochemistry 2012, 51, 3349–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lai CL; Landgraf KE; Voth GA; Falke JJ Journal of Molecular Biology 2010, 402, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Salamon RS; Backer JM Bioessays 2013, 35, 602–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mclaughin S; Murray D Nature 2005, 438, 605–611. [DOI] [PubMed] [Google Scholar]
- (11).Cantley LC Science 2002, 296, 1655–1657. [DOI] [PubMed] [Google Scholar]
- (12).Corbin JA; Dirkx RA; Falke JJ Biochemistry 2004, 43, 16161–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Czech MP Annual Review of Physiology 2003, 65, 791–815. [DOI] [PubMed] [Google Scholar]
- (14).Lemmon MA; Ferguson KM Biochemical Journal 2000, 350, 1–18. [PMC free article] [PubMed] [Google Scholar]
- (15).Vanhaesebroeck B; Leevers SJ; Ahmadi K; Timms J; Katso R; Driscoll PC; Woscholski R; Parker PJ; Waterfield MD Annual Review of Biochemistry 2001, 70, 535–602. [DOI] [PubMed] [Google Scholar]
- (16).Hurley JH Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids 2006, 1761, 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ceccarelli DFJ; Blasutig IM; Goudreault M; Li Z; Ruston J; Pawson T; Sicheri F Journal of Biological Chemistry 2007, 282, 13864–13874. [DOI] [PubMed] [Google Scholar]
- (18).Moravcevic K; Oxley CL; Lemmon MA Structure 2012, 20, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jiang Z; Hess SK; Heinrich F; Lee JC Journal of Physical Chemistry B 2015, 119, 4812–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lietzke SE; Bose S; Cronin T; Klarlund J; Chawla A Molecular Cell 2000, 6, 385–394. [DOI] [PubMed] [Google Scholar]
- (21).Bakerand NA; Sept D; Joseph S; Holst MJ; McCammon JA Proceedings of the National Academy of Sciences, USA 2001, 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Manna D; Albanese A; Park WS; Cho W Journal of Biological Chemistry 2007, 282, 32093–32105. [DOI] [PubMed] [Google Scholar]
- (23).Scott JL; Musselman CA; Adu-Gyamfi E; Kutateladze TG; Stahelin RV Integr. Biol. 2012, 4, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Currie R; Walker K; Gray A; Deak M; Casamayor A; Downes C; Cohen P; Alessi D; Lucocq J Biochemical Journal 1999, 337, 575–583. [PMC free article] [PubMed] [Google Scholar]
- (25).Landgraf KE; Malmberg NJ; Falke JJ Biochemistry 2008, 47, 8301–8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Frech M; Andjelkovic M; Ingley E; Reddy K; Falck J; Hemmings B Journal of Biological Chemistry 1997, 272, 8474–8481. [DOI] [PubMed] [Google Scholar]
- (27).Lai C-L; Srivastava A; Pilling C; Chase AR; Falke JJ; Voth GA Journal of Molecular Biology 2013, 425, 3073–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Naughton FB; Kalli AC; Sansom MS P. Journal of Physical Chemistry Letters 2016, 7, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lumb CN; Je J; Xue Y; Stansfeld PJ; Stahelin RV; Kutateladze TG; Sansom MS P. Structure 2011, 19, 1338–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yamamoto E; Kalli AC; Yasuoka K; Sansom MS P. Structure 2016, 24, 1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ohkubo YZ; Tajkhorshid E Structure 2008, 16, 72–81. [DOI] [PubMed] [Google Scholar]
- (32).Hedger G; Shorthouse D; Koldso H; Sansom MS P. Journal of Physical Chemistry B 2016, 120, 8154–8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lumb CN; He J; Xue Y; Stansfeld PJ; Stahelin RV; Kutateladze TG; Sansom MS P. Structure 2011, 19, 1338–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lumb CN; Sansom MS P. PLoS Computational Biology 2012, 8, e1002607–e1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ohkubo YZ; Pogorelov TV; Arcario MJ; Christensen GA; Tajkhorshid E Biophysical Journal 2012, 102, 2130–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Arcario MJ; Ohkubo YZ; Tajkhorshid E Journal of Physical Chemistry B 2011, 115, 7029–7037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Baylon JL; Lenov IL; Sligar SG; Tajkhorshid E Journal of the American Chemical Society 2013, 135, 8542–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Vermaas JV; Tajkhorshid E Biochimica et Biophysica Acta - Biomembranes 2014, 1838, 3107–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Vermaas JV; Tajkhorshid E Biochemistry 2017, 56, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Muller MP; Wang Y; Morrissey JH; Tajkhorshid E Journal of Thrombosis and Haemostasis 2017, 15, 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vermaas JV; Baylon JL; Arcario MJ; Muller MP; Wu Z; Pogorelov TV; Tajkhorshid E Journal of Membrane Biology 2015, 248, 563–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Muller MP; Jiang T; Sun C; Lihan M; Pant S; Mahinthichaichan P; Trifan A; Tajkhorshid E Chemical Reviews 2019, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Naughton FB; Kalli AC; Sansom MS Journal of Physical Chemistry Letters 2016, 7, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Humphrey W; Dalke A; Schulten K Journal of Molecular Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (45).Jo S; Kim T; Iyer VG; Im W Journal of Computational Chemistry 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- (46).Qi Y; Cheng X; Lee J; Vermaas JV; Pogorelov TV; Tajkhorshid E; Park S; Klauda JB; Im W Biophysical Journal 2015, 109, 2012–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Baylon JL; Vermaas JV; Muller MP; Arcario MJ; Pogorelov TV; Tajkhorshid E Biochimica et Biophysica Acta - Biomembranes 2016, 1858, 1573–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Vermaas JV; Tajkhorshid E Journal of Physical Chemistry B 2014, 118, 1754–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K Journal of Computational Chemistry 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD Journal of Chemical Theory and Computation 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW Journal of Physical Chemistry B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG Journal of Chemical Physics 1995, 103, 8577–8593. [Google Scholar]
- (53).Martyna GJ; Tobias DJ; Klein ML Journal of Chemical Physics 1994, 101, 4177–4189. [Google Scholar]
- (54).Tietjen GT; Baylon JL; Kerr D; Gong Z; Henderson JM; Heffern CT; Meron M; Lin B; Schlossman ML; Adams EJ; Tajkhorshid E; Lee KY C. Biophysical Journal 2017, 113, 1505–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Isralewitz B; Baudry J; Gullingsrud J; Kosztin D; Schulten K Journal of Molecular Graphics and Modeling 2001, 19, 13–25. [DOI] [PubMed] [Google Scholar]
- (56).Barducci A; Bussi G; Parrinello M Physical Review Letters 2008, 100, 020603. [DOI] [PubMed] [Google Scholar]
- (57).Fiorin G; Klein ML; Henin J Molecular Physics 2013, 111, 3345–3362. [Google Scholar]
- (58).Chen H-C; Ziemba BP; Landgraf KE; Corbin JA; Falke JJ PLoS One 2012, 7, e33640–e33650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Hyvnenand M Maciasand MJ Nilgesand M Oschkinatand H Saraste M and, EMBO MW Journal 1995, 14, 4676–4685. [Google Scholar]
- (60).Anand K; Maeda K; Gavin A-C PLoS One 2012, 7, e365216-e365226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kavran JM; Klein DE; Lee A; Falasca M; Isakoff SJ; Skolnik EY; Lemmon MA Journal of Biological Chemistry 1998, 273, 30497–30508. [DOI] [PubMed] [Google Scholar]
- (62).Klarlund JK; Rameh LE; Cantley LC; Buxton JM; Holik JJ; Sakelis C; Patki V; Corvera S; Czech MP Journal of Biological Chemistry 1998, 273, 1859–1862. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.