

Supplemental Digital Content is available in the text
Abstract
Genomic alterations in relapsed B-cell precursor acute lymphoblastic leukemia (BCP-ALL) may provide insight into the role of specific genomic events in relapse development. Along this line, comparisons between the spectrum of alterations in relapses that arise in different upfront treatment protocols may provide valuable information on the association between the tumor genome, protocol components and outcome. Here, we performed a comprehensive characterization of relapsed BCP-ALL cases that developed in the context of 3 completed Dutch upfront studies, ALL8, ALL9, and ALL10. In total, 123 pediatric BCP-ALL relapses and 77 paired samples from primary diagnosis were analyzed for alterations in 22 recurrently affected genes. We found pronounced differences in relapse alterations between the 3 studies. Specifically, CREBBP mutations were observed predominantly in relapses after treatment with ALL8 and ALL10 which, in the latter group, were all detected in medium risk-treated patients. IKZF1 alterations were enriched 2.2-fold (p = 0.01) and 2.9-fold (p < 0.001) in ALL8 and ALL9 relapses compared to diagnosis, respectively, whereas no significant enrichment was found for relapses that were observed after treatment with ALL10. Furthermore, IKZF1 deletions were more frequently preserved from a major clone at diagnosis in relapses after ALL9 compared to relapses after ALL8 and ALL10 (p = 0.03). These data are in line with previous studies showing that the prognostic value of IKZF1 deletions differs between upfront protocols and is particularly strong in the ALL9 regimen. In conclusion, our data reveal a correlation between upfront treatment and the genetic composition of relapsed BCP-ALL.
Introduction
Despite the improved survival rate for B-cell precursor acute lymphoblastic leukemia (BCP-ALL), relapsed ALL remains one of the leading causes of cancer related death in children.1–3 Since ALL relapse involves the outgrowth of therapy resistant leukemic cells, a substantial fraction of genomic alterations identified at relapse can be traced back in samples obtained at diagnosis, either fully clonal or as minor clones.4–12 Furthermore, additional genomic alterations are usually present at relapse, which are acquired during or after therapy and may contribute to relapse development.13–15 Therefore, the origin of genomic alterations identified at relapse, may help identify those lesions that are most strongly correlated with therapy failure.
Multiple studies have been aimed at investigating the genomic landscape of relapsed BCP-ALL.4,11,12,16–20 A significant increase in the median number of copy number alterations (CNAs) and mutations was observed in BCP-ALL relapse samples, when compared to diagnosis.4,11,20 These studies show that recurrently affected genes and pathways at diagnosis, including those involved in B-cell differentiation, cell cycle regulation, and Ras and JAK/STAT signaling, are also observed at relapse, although with different frequencies.4,6,12,20,21 Alterations observed with a higher frequency at relapse, such as those affecting IKZF1, TP53, and genes involved in Ras signaling (KRAS, NRAS, PTPN11, FLT3, and BRAF), may play a role in therapy resistance and could be of prognostic value at the time of diagnosis.6,22–24 By contrast, mutations in drug metabolism (NT5C2, PRPS1) or chromatin modification (CREBBP) regularly appear to arise during initial therapy, and become detectable only at relapse.13–16
Different chemotherapy regimens may have different effects on every leukemic subclone present in a patient, resulting in changes in clonal dynamics and, in some cases, survival of distinct subclones.25 For example, KRAS hotspot mutations were found to be associated with methotrexate resistance and vincristine sensitivity of leukemic cells in vitro and in vivo.20 Furthermore, mutations in NT5C2 and PRPS1 appear to confer selective resistance to 6-mercaptopurine, a fundamental drug used in the maintenance phase of ALL treatment, and leukemic cells with these mutations, even when present at extremely low numbers, have a strong selective advantage during 6-MP treatment, allowing them to grow out into a relapse.14–16,26 In addition, a therapy resistance phenotype has been described for leukemic cells with alterations in IKZF1, a gene in which alterations have a strong prognostic value for relapse development.6,22 However, dependent on the protocol, the predictive value of IKZF1 deletions varies. In the Dutch Childhood Oncology Group (DCOG) trials, where the prognostic effect was much stronger in ALL9 compared to ALL10, while not significant in ALL8.27 These findings suggest that the treatment efficacy of IKZF1-altered BCP-ALL depends on timing and dosage of individual chemotherapeutics used in these protocols. Therefore, the genetic characteristics of relapsed BCP-ALL might be determined by the upfront treatment.
In the period of 1991 to 2012, over 2000 children with newly diagnosed BCP-ALL were enrolled in 3 Dutch ALL treatment trials ALL8, ALL9 and ALL10.3,28,29 Within each trial, patients were stratified in groups based on risk factors to maximize outcome with optimal treatment intensity. However, the parameters used for stratification were very different between the 3 protocols. For example, whereas stratification of patients in ALL8 and ALL9 was solely based on classical clinical and cytogenetic parameters, ALL10 stratification was based on minimal residual disease monitoring.3,28,29 Furthermore, timing and dosage of the various chemotherapeutic drugs differed considerably between protocols and treatment arms and, thus, also between patients with comparable risk factors (Table S1, Supplemental Digital Content). One major protocol-dependent difference in DCOG trials ALL8, ALL9, and ALL10 was the number of glucocorticoids (GCs) in relation to the intensity of additional chemotherapy that was used. In contrast to ALL8 and ALL10, treatment in ALL9 heavily relied on high dose of GCs (dexamethasone) during the induction and maintenance phases, particularly in the ALL9 non-high risk (NHR) treatment arm.29 This variability in upfront treatment protocols creates an interesting opportunity to study the impact of these protocol differences on the genomic architecture of relapsed ALL, which to date has not been systematically investigated.
In this study, we characterized 123 relapsed BCP-ALL samples that were treated upfront by one of 3 DCOG studies ALL8, ALL9 and ALL10. Our data indicate that the composition of genetic alterations in relapsed BCP-ALL is indeed affected by the upfront treatment.
Results
Genomic profiling of relapsed BCP-ALL
To establish the frequency of recurrent genetic alterations in relapsed BCP-ALL samples, we collected 123 BCP-ALL relapse samples from patients that relapsed after upfront treatment according to ALL8 (n = 39), ALL9 (n = 55) or ALL10 (n = 29, Figure S1, Supplemental Digital Content). The included relapses represented approximately half of the total number of relapses that occurred in each of these protocols (Table S2–3, Supplemental Digital Content). To enable quantitative comparisons between these relatively small groups, we focused our study on copy number alterations and mutations in a selected set of 22 genes that were found to be recurrently affected in relapsed BCP-ALL in previous studies.4,6,11–13,16–22 This set of genes includes B-cell differentiation genes (IKZF1, IKZF2, IKZF3, PAX5, and EBF1), genes involved in Ras signaling (KRAS, NRAS, PTPN11, FLT3, and BRAF), JAK-STAT signaling (JAK1, JAK2, JAK3), cell cycle regulation (CDKN2A, CDKN2B, and RB1), and drug metabolism (NT5C2, NR3C1 and BTG1), as well as genes encoding the histone acetyltransferase CREBBP (CBP), the tumor suppressor TP53, and ETS family transcription factor ETV6. Targeted copy number and mutation screening was performed using MLPA and Amplicon-based IonTorrent sequencing, respectively (Table S4–5, Supplemental Digital Content). Using this approach, we detected an average of 1.3 CNAs and 0.6 sequence mutations in the aforementioned genes per relapse sample (range 0–5 and 0–3, respectively).
Genes involved in B-cell differentiation were affected in approximately half of the cases (47%), whereas activating mutations in genes of the Ras pathway were found in 30% of the relapses (Fig. 1A). The most frequent genetic alterations identified in these 123 relapses involved deletions and mutations in IKZF1 (33%), and deletions of the CDKN2A/2B locus (30%; Figs. 1B and S2, Supplemental Digital Content).30 Alterations in ETV6 and PAX5 were found in 25% and 20% of the BCP-ALL relapse samples, and 11% carried mutations in the HAT domain of CREBBP. Ras pathway mutations mostly involved KRAS (15%) or NRAS (8%; Fig. 1B) and BRAF mutations were not identified in our relapse cohort. Four relapses were found to carry NT5C2 mutations, and also BTG1 deletions (7%) and NR3C1 mutations (2%), which may be correlated with GC resistance,4,31 were not common in these relapse samples. IKZF1 alterations (p < 0.0001; Fisher exact test) and mutations in CREBBP and NT5C2 (p < 0.0001 and p < 0.01; Fisher exact test) were more frequently found in relapsed ALL compared to primary diagnosis (Table S6, Supplemental Digital Content), which is in line with previous studies.11,13–15,21
Figure 1.
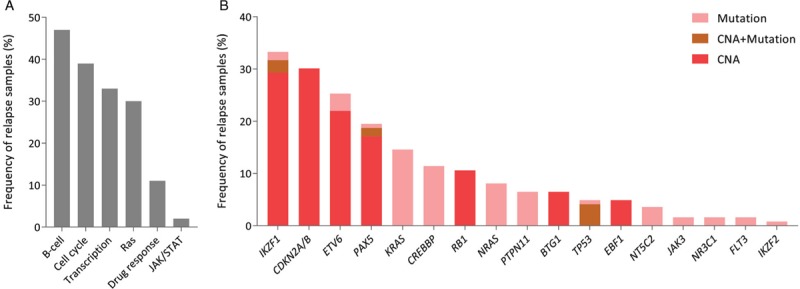
Genomic profiling of relapsed BCP-ALL. (A) Frequency of genetic alterations in pathways studied in relapse samples. B-cell differentiation genes include IKZF1, IKZF2, IKZF3, PAX5, and EBF1. Cell cycle regulation genes include CDKN2A, CDKN2B, TP53, and RB1. Transcription regulators include CREBBP and ETV6. Ras signaling genes include KRAS, NRAS, PTPN11, FLT3, and BRAF. Drug response genes are NT5C2, BTG1, and NR3C1. Genes involved in JAK/STAT signaling are JAK1, JAK2, and JAK3. (B) Frequency of genetic alterations in the genes studied in relapse samples.
Genomic presentation of relapse is influenced by upfront treatment
To address the question whether the genetic makeup of relapse varies between upfront treatment studies, we compared genomic alterations observed in relapses that occurred after treatment according to ALL8, ALL9, and ALL10. First, we compared the prevalence of genetic alterations in ALL relapses between the 3 studies as a whole because frequencies of these alterations at time of diagnosis, so before risk stratification, will be similar. The average number of alterations per relapse sample for the ALL8, ALL9 and ALL10 groups was 1.8, 2.1, and 1.8, respectively, which was not significantly different (Fig. 2A). Compared to diagnosis, we indeed observed differences in the pattern of genetic alterations between relapses observed after these 3 upfront treatment studies. Whereas CREBBP mutations were particularly enriched in relapses from patients treated according to ALL8 and ALL10, mutations in PTPN11 were increased only in ALL9 relapses (Fig. 2B and Table S6, Supplemental Digital Content). IKZF1 alterations seemed to be increased in all 3 relapse groups, although statistical significance was only reached in the ALL8 and ALL9 cohorts, where the increase was 2.2 (p = 0.01, Fisher exact test) and 2.9-fold (p < 0.0001, Fisher exact test), respectively (Fig. 2B and Table S6, Supplemental Digital Content). In a comparison among the 3 relapse groups, only the low number of CREBBP mutations in ALL9 relapses (2%) relative to those observed in ALL8 (21%) and ALL10 (17%) was found to be significant (p = 0.01, Fisher exact test).
Figure 2.
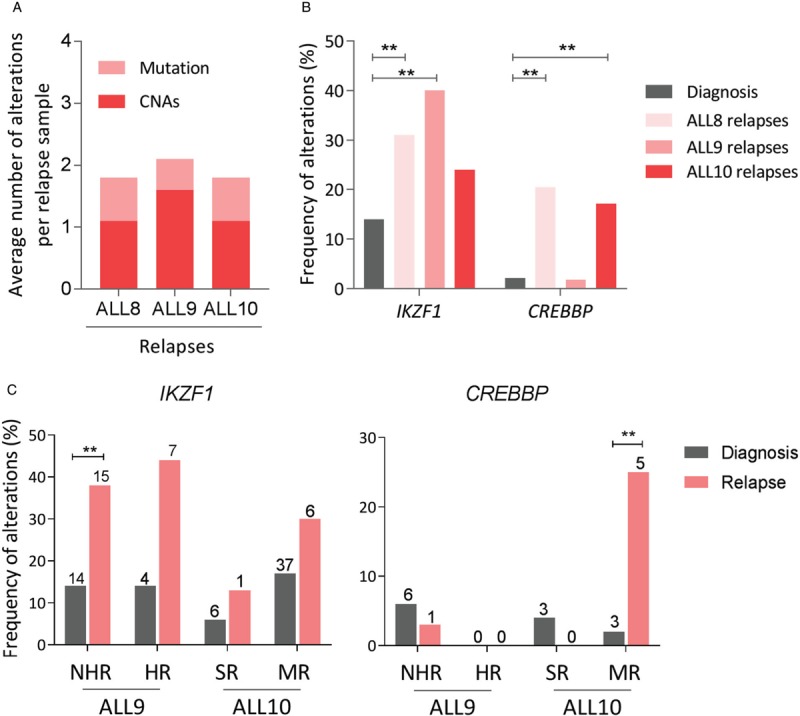
Frequency of genetic alterations in relapse samples classified by upfront treatment protocols. (A) Average number of mutations and CNAs in relapse samples treated by upfront protocols, ALL8, ALL9, and ALL10. (B) Comparison of affected gene frequency between diagnosis and relapse. Only genes showing significant differences between diagnosis and total relapse are depicted. Details of all genes are presented in Table S6, S10. (C) Comparison of affected frequency in IKZF1 and CREBBP between diagnosis and relapse among upfront risk-stratified treatment groups. The number of samples affected is indicated on the top of the bar. Details of the comparison are presented in Table S9, S10. Significant difference (p < 0.01) is indicated by double asterisks.
To further correlate the observed changes to treatment, we analyzed the risk-stratified treatment groups of each protocol separately, limited to more common alterations that also showed unequal distributions between the total cohorts: IKZF1 and CREBBP. We reasoned that differences in genetic alterations between treatment arms at the time of relapse may be caused by both the differential stratification in upfront risk groups at the time of diagnosis, and the impact of treatment itself. To study these effects separately, we first analyzed the distribution of alterations in these genes at the start of treatment, using available MLPA data6,27 and targeted sequence data from diagnosis cohorts. We noticed that patients with IKZF1 deleted leukemia at diagnosis were more frequently stratified in higher risk groups in ALL8 and ALL10, whereas in ALL9 there was an equal distribution of IKZF1 deletions in the NHR and HR groups (Table S7, Supplemental Digital Content). This suggests that in contrast to ALL8 and ALL10, IKZF1 alteration status had no impact on the criteria used for stratification in ALL9. To make a similar analysis for CREBBP mutations observed at diagnosis, we made use of available mutation data in 2 cohorts of ALL9 and ALL10 diagnosis samples. For ALL8, the mutation data at the time of diagnosis were not available. For patients with CREBBP mutations no significant differences in distribution between treatment arms were detected in ALL9 nor in ALL10 (Table S8, Supplemental Digital Content). Thus, the variability of alterations in IKZF1 and CREBBP at time of primary diagnosis is limited.
Next, we compared the deletion and mutation frequencies for these genes in each risk group between diagnosis and relapse. The most pronounced increase in IKZF1 deletions between diagnosis and relapse was observed in ALL9, where both in the NHR and HR group a ∼3-fold increase was observed in relapse compared to diagnosis (Fig. 2C and Table S9, Supplemental Digital Content). The standard risk (SR) and high risk (HR) groups of ALL10 were too small to draw definitive conclusions, but in the larger medium risk (MR) group no significant enrichment of IKZF1 deletions at relapse was observed. With respect to CREBBP, all mutations found at the time of relapse after ALL10 treatment were in the MR-treated arm and were significantly more common than those observed at primary diagnosis (Fig. 2C and Table S9, Supplemental Digital Content). Taken together, these data suggest that, with similar numbers of alterations, the composition of genetic alterations in relapsed BCP-ALL varies between different upfront treatment protocols and risk groups.
Origin of genetic alterations in relapse
An increased frequency of genetic alterations in relapse compared to diagnosis can be the result of therapy resistance of clones that carry this alteration at the time of diagnosis, or by selective outgrowth of sub-clonal or newly acquired alterations. To discriminate between these 2 options, we performed genomic analysis in paired diagnosis-relapse samples. Of 123 relapsed BCP-ALL, paired diagnosis samples were available for 77 patients, of which 62 carried alterations in at least one of the 22 genes observed at relapse. For those 62 cases, all CNAs (n = 103) and sequence mutations (n = 48) identified at relapse were screened in matched samples taken at primary diagnosis using MLPA and Sanger sequencing, respectively (Fig. 3A; Table S10, Supplemental Digital Content). In this way, 47 CNAs (45%) and 16 SNVs (33%) were found to be present in major clones at diagnosis. Again, we first compared alterations between the 3 studies and subsequently between treatment arms separately. The average number of alterations that were absent in the major clone at diagnosis was highest in relapse that occurred after treatment according to ALL8, and lowest in ALL10-associated relapses (Table S11, Supplemental Digital Content).
Figure 3.
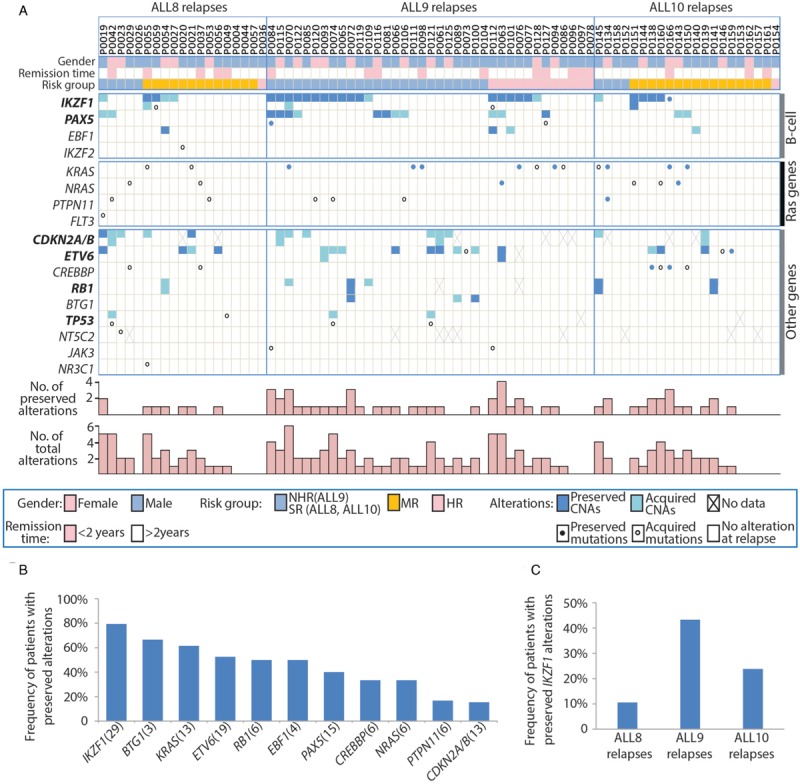
Origin of genetic alterations in relapsed BCP-ALL. (A) Relapse-associated alterations in each patient and their status (preserved or acquired). Patients are displayed in columns sorted by upfront treatment protocols. Genes are shown in rows grouped by their corresponding pathways. For genes indicated in bold, double rows represent homozygous or compound heterozygous alterations. Preserved alterations are shown in dark grey (CNA) or solid dots (mutation), while acquired alterations are depicted in light grey (CNA) or open circles (mutation). Numbers of preserved alterations and total numbers of alterations identified at relapse in each patient are shown at the bottom. Five genes not indicated in this figure (IKZF3, BRAF, FLT3, JAK1, JAK2) were wildtype in all tested samples. Details of all alterations are presented in Table, S10. (B) Frequency of patients that carried preserved alterations in 11 genes studied. The number of patients studied for each gene is displayed in brackets. (C) Frequency of patient with preserved IKZF1 alterations in patients treated with different upfront protocols.
Genetic alterations in B-cell differentiation genes were frequently found to be present already at diagnosis, particularly those in IKZF1. In fact, in 23 of the 29 patients with IKZF1 alterations at relapse (79%), the alteration was preserved from diagnosis (Fig. 3B). The six patients in which the alterations were not observed at diagnosis did not show clustering within a specific treatment arm or study. The number of Ras pathway mutations preserved from the major clone at diagnosis was highly variable between genes, ranging from 8 out of 13 mutations (62%) in KRAS to only 1 out of 6 mutations (17%) in PTPN11 (Fig. 3B). By deep-sequencing of diagnosis samples, which was performed for 21 cases, the majority of Ras mutations (18/22) could be traced backed to the paired diagnosis sample, 7 of which were present as subclones. Deep-sequencing revealed that CREBBP mutations, which were found to be more common in relapses treated after ALL8 and ALL10 compared to ALL9 (Fig. 2C), were predominantly acquired at relapse (4 out of 6; Fig. 3B and Table S10, Supplemental Digital Content). Deletions of the CDKN2A/2B locus, frequently present in a homozygous state, were found to be conserved between diagnosis and relapse in 46% of the cases, but the presence of only 3 MLPA probes in this locus precluded a more detailed comparison. Therefore, we reanalyzed these 13 samples using the ME024 MLPA kit, which contains 23 probes that span a region of 36.6 Mb surrounding the 9p21.3 locus. For only 2 cases (15%), the CDKN2A/2B deletions appeared to be identical, even after comparison with high-density SNP-array-based copy number analysis, whereas in all of the other cases the deletions were different (Fig. S3, Supplemental Digital Content). This suggests that CDKN2A/2B deletions drive leukemogenesis, but are not selected for in upfront therapy.
Next, we analyzed the clonal origin of relapse-associated IKZF1 alterations in the context of upfront treatment intensity within this paired sample cohort. Notably, whereas the frequency of IKZF1 alterations that occurred in ALL8, ALL9, and ALL10 relapses were not significantly different (Fig. 2B and 3A), the fraction of IKZF1 alterations that was preserved from the major clone at diagnosis was found to be only 40% in ALL8 compared to 89% and 83% in ALL9 and ALL10, respectively (Fig. 3A). Therefore, to compare the IKZF1 alterations in the context of upfront treatment, we selectively focused on these preserved IKZF1 alterations. This comparison did result in a striking difference among the 3 studies (p = 0.033, Fisher's exact test; Fig. 3C). A total of 43% (16/37) of relapses treated after ALL9 carried IKZF1 alterations that originated from the major clone at diagnosis, compared to 11% in ALL8 (2/19) and 24% in ALL10 (5/21). The preserved IKZF1 alterations were detected in 44% and 42% of relapses after ALL9-NHR (11/25) and ALL9-HR (5/12), respectively. In the relapses after medium risk regimen of ALL8 and ALL10, 15% (2/13) and 31% (5/16) carried IKZF1 alterations originating from a major clone at diagnosis. No relapses with preserved IKZF1 alterations were observed in patients with upfront standard risk regimen in ALL8 and ALL10. Together, these data suggest that the differences observed at relapse were a direct consequence of the upfront treatment.
Discussion
Several studies have demonstrated that the genomic landscape of relapsed ALL differs from that observed at diagnosis, both in the number of alterations and the genes that are affected.4,5,11,12 These differences likely originate from the combined effect of continuous evolutionary progression of the dividing leukemic cells, and therapy-induced genomic instability and clonal outgrowth. In this study, we have investigated the impact of treatment on the presentation of genetic alterations in relapsed ALL. To enable a good comparison between relapses arising in different upfront treatment protocols, we have focused on a limited set of 22 genes known to be recurrently affected. We collected 123 relapse samples from patients treated according to Dutch Childhood Oncology Group studies ALL8 (1991–1996), ALL9 (1997–2004) and ALL10 (2004–2012). Although more late relapses in ALL8 and younger relapses in ALL10 were included in this study, none of the investigated genes showed significant difference regarding the remission time in ALL8 relapses and diagnosis age in ALL10 relapse (Table S12–13, Supplemental Digital Content). The comparisons revealed that relapses that arise in the context of these upfront treatment protocols show differences in these common genetic alterations, with frequent CREBBP mutations in ALL8 and ALL10 relapses, and IKZF1 alterations being particularly common in relapses from patients treated according to the ALL9 protocols.
CREBBP mutations cluster in the histone acetyl transferase (HAT) domain, which mediates the acetylation of lysine residues in various proteins, including histone 3 lysine 18 and 27 (H3K18 and H3K27).13,32 Mutations affecting CREBBP occur in 18% of relapsed childhood ALL cases, and up to 30% in high hyperdiploid and hypodiploid ALL relapses.13,32–34 These mutations originate from major or minor clones at primary diagnosis, but in a similar number of cases these mutations are newly acquired at the time of relapse.13,34 It has been shown that CREBBP mutations impair H3K18 acetylation in mouse embryonic fibroblasts, ALL cell lines and patient-derived xenografts, and may cause reduced expression of CREBBP targets, including glucocorticoid responsive genes.13,35 Preliminary studies have indicated that CREBBP impairment correlates with a glucocorticoid resistance phenotype but since this could not be demonstrated in in vitro models, this phenomenon has not yet been fully resolved.35 The higher number of CREBBP mutations in ALL8 and ALL10 relapses may be explained by the higher frequency of cases with high hyperdipoid cytogenetics (28% and 25%, respectively) compared to those observed in ALL9 (15%; Table S2, Supplemental Digital Content). Indeed, the majority of CREBBP mutations in our study (62%) were identified in high hyperdiploid cases (Table S3, Supplemental Digital Content). However, since none of the eight high hyperdiploid relapses in ALL9 were found to carry CREBBP mutations, the possibility remains that these mutations are less likely to contribute to relapse in ALL9-based protocols, for example because of the absence of a strong selective pressure for these alterations.
The high frequency of preserved IKZF1 alterations in ALL9 relapses compared to those in ALL8 and ALL10 is remarkable. Many ALL study groups have reported that patients with IKZF1 alterations at primary diagnosis have a higher risk of relapse, but the prognostic value appeared to be different between various protocols.6,27,36–42 This observation already suggests that the contribution of IKZF1 dysfunction on relapse development is dependent on the treatment that patients received at primary diagnosis. We noticed that IKZF1 deletions in children treated according to ALL9 are equally distributed between the risk groups, whereas in ALL10 IKZF1-deleted leukemias are stratified to higher risk groups and, thus, received more intense treatment compared to wildtype leukemias. Since the stratification in ALL10 is mainly based on early therapy response,1–3 this observation indirectly suggests that IKZF1-alterations influence the response during the first courses of ALL10 treatment. We previously demonstrated that IKZF1 alterations substantially impair the GC-response of both normal and leukemic cells,43 suggesting that the contribution of IKZF1 alterations on therapy resistance and relapse may be dependent on the intensity of the GCs used in the upfront treatment. The current study is consistent with this notion, showing a particularly high percentage of preserved IKZF1 alterations in patients that relapsed after ALL9-based treatment, a treatment regimen which heavily relies on high doses the GC dexamethasone (Table S1, Supplemental Digital Content).29 In fact, the median risk arm of ALL10, which uses similar doses of dexamethasone as the ALL9 arms, has relatively lower number of preserved IKZF1 alterations, suggesting that the other administered components in ALL10-MR are able to overcome the IKZF1-induced GC resistance in a subset of cases. The observations on preserved IKZF1 alterations also illustrate the importance of including genomic data at the time of diagnosis to determine the contribution of specific genetic events to relapse development.44–46
The differences in common genetic alterations of relapses observed in these 3 upfront treatment studies provide insights that will aid further improvements of childhood ALL therapy by adapting risk group stratification or study design. We have recently shown that the combined loss of IKZF1 and BTG1 results in even worse survival rates compared to loss of IKZF1 alone.47 Furthermore, several studies have demonstrated that IKZF1 deletions in the presence of specific other gene deletions are associated with poorer survival, which provides a tool for refined risk stratification that will be implemented in future ALL treatment studies.44–46 Alterations affecting IKZF1 result in impaired lymphoid maturation of precursor B-cells, and are highly frequent in BCR-ABL1-positive leukemia.48–50 A study by Churchman et al revealed that IKZF1 alterations induce a stem-cell like phenotype and act synergistically with Arf alterations to drive BCR-ABL1-positive ALL in mice.51 Strikingly, the poor response to tyrosine kinase inhibitors (TKI) observed in IKZF1-deleted BCR-ABL1-positive ALL, which is a tailored therapy for individuals with this type of leukemia,52,53 could be reversed with retinoids, thus providing an important treatment opportunity that deserves further exploration, both in BCR-ABL1-positive and negative ALL.51,54 Taken together, these studies provide promising new concepts for targeting the pathways that are affected by IKZF1 deletions.
Glucocorticoids (GCs) are key components in ALL treatment.55 Our results suggest that upfront protocols like ALL9, which rely for a significant part on high GCs and lack, for instance, intensive asparaginase courses, are less effective in eradicating leukemias with IKZF1 alterations. This would imply that such protocols are less favorable for the future treatment of patients with IKZF1 deletions.
In conclusion, we have shown that genetic characterization of relapsed ALL in the context of upfront treatment reveals striking differences in the composition of common genetic alterations of relapse while providing important insight into mechanisms of therapy response. Our data indicate that therapy that relies on high doses of GC results in a higher percentage of IKZF1-altered relapses. These studies may guide the design of new protocols, particularly when extended to larger sample series and more detailed drug comparisons.
Materials and methods
Patient material and DNA isolation
Relapse samples from a total of 123 BCP-ALL patients were included in this study, all of which were obtained from the DCOG biobank with informed consent. These patients were treated according to one of 3 DCOG treatment studies ALL8 (n = 39), ALL9 (n = 55) or ALL10 (n = 29). The patients included were a good representation of the total cohort of relapsed patients from these trials with respect to gender, risk group, remission time, and ploidy status at diagnosis, with the exception of remission time in ALL8 relapses (Table S2, Supplemental Digital Content). No infant ALL patients were included in this cohort. Patient characteristics are listed in Table S3 (Supplemental Digital Content). To investigate whether relapse-associated genetic alterations were also present at diagnosis, 77 available paired diagnosis samples were analyzed. Mononuclear cells were isolated by Ficoll gradient separation and stored in liquid nitrogen. Genomic DNA was extracted from mononuclear cells and purified using a QIAamp purification kit (Qiagen, Venlo, The Netherlands).
Multiplex ligation-dependent probe amplification
A targeted screen for copy number alterations (CNAs) in BTG1, CDKN2A/2B, EBF1, ETV6, IKZF1, PAX5, RB1, and TP53 (Table S4) in diagnosis and relapse samples was performed by multiplex ligation-dependent probe amplification (MLPA)56 using SALSA MLPA kits P335, ME024 and P056 (MRC-Holland, Amsterdam, The Netherlands) according to manufacturer's instructions.
Targeted sequencing and data validation
Sequence mutations in 21 genes (Table S4) were determined in the relapse cohort using Ion AmpliSeq-based sequencing (Life Technologies, CA). A total of 10 ng genomic DNA was used in each AmpliSeq PCR reaction. After barcoding and adaptor ligation, products were purified with Agencourt AMPure XP beads (Beckman Coulter Genomics, High Wycombe, UK). Emulsion PCR was performed using OneTouch 200 Template kit (Life Technologies, CA). Polymerase and sequencing primers were added to final enriched spheres before loading onto the chip. Sequence reads were mapped and analyzed using SeqNext software V4.1.1 (JSI, Ettenheim, Germany). Validations were performed by Sanger sequencing. Hotpot regions for NT5C2 (exons 11 and 15) were sequenced in 104 relapses using Sanger sequencing. All primers are listed in Table S5. Available complete remission samples were used to confirm the somatic status of mutations.
Statistical analysis
Statistical analyses were carried out using R version 3.3.2, Pearson Chi-square and Fisher exact tests were used to compare categorical variables. A two-sided p value below 0.05 was considered statistically significant.
Supplementary Material
Footnotes
Citation: Yu J, Waanders E, van Reijmersdal SV, Antić Ž, van Bosbeek CM, Sonneveld E, de Groot H, Fiocco M, Kessel AG, van Leeuwen FN, Pieters R, Hoogerbrugge PM, Kuiper RP. Upfront treatment influences the composition of genetic alterations in relapsed pediatric B-cell precursor acute lymphoblastic leukemia. HemaSphere, 2020;00:00. http://dx.doi.org/10.1097/HS9.0000000000000318
This work was supported by grants from the Dutch Cancer Society (KUN2012-5366 to EW), KiKa (KiKa 150 to RPK, FNvL, and PMH), and the China Scholarship Council (CSC 201304910347 to JY).
The authors report no conflicts of interest.
References
- 1.Pui C-H, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. [DOI] [PubMed] [Google Scholar]
- 2.Kamps WA, van der Pal-de Bruin KM, Veerman AJP, et al. Long-term results of Dutch Childhood Oncology Group studies for children with acute lymphoblastic leukemia from 1984 to 2004. Leukemia. 2009;24:309–319. [DOI] [PubMed] [Google Scholar]
- 3.Pieters R, De Groot-Kruseman H, Van der Velden VH, et al. Successful therapy reduction and intensification for childhood acute lymphoblastic leukemia based on minimal residual disease monitoring: study ALL10 from the Dutch Childhood Oncology Group. J Clin Oncol. 2016;34 22:2591–2601. [DOI] [PubMed] [Google Scholar]
- 4.Mullighan CG, Phillips LA, Su X, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang JJ, Bhojwani D, Yang W, et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008;112:4178–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuiper RP, Waanders E, van der Velden VHJ, van Reijmersdal SV, Venkatachalam R, Scheijen B, et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia. 2010;24 7:1258–1264. [DOI] [PubMed] [Google Scholar]
- 7.Davidsson J, Paulsson K, Lindgren D, et al. Relapsed childhood high hyperdiploid acute lymphoblastic leukemia: presence of preleukemic ancestral clones and the secondary nature of microdeletions and RTK-RAS mutations. Leukemia. 2010;24:924–931. [DOI] [PubMed] [Google Scholar]
- 8.Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–361. [DOI] [PubMed] [Google Scholar]
- 9.Kuster L, Grausenburger R, Fuka G, et al. ETV6/RUNX1-positive relapses evolve from an ancestral clone and frequently acquire deletions of genes implicated in glucocorticoid signaling. Blood. 2011;117:2658–2667. [DOI] [PubMed] [Google Scholar]
- 10.van Delft FW, Horsley S, Colman S, et al. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood. 2011;117:6247–6254. [DOI] [PubMed] [Google Scholar]
- 11.Ma X, Edmonson M, Yergeau D, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun. 2015;6:6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oshima K, Khiabanian H, da Silva-Almeida AC, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2016;113:11306–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mullighan CG, Zhang J, Kasper LH, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer JA, Wang J, Hogan LE, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet. 2013;45:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tzoneva G, Perez-Garcia A, Carpenter Z, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19:368–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li B, Li H, Bai Y, et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med. 2015;21:563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindqvist CM, Lundmark A, Nordlund J, et al. Deep targeted sequencing in pediatric acute lymphoblastic leukemia unveils distinct mutational patterns between genetic subtypes and novel relapse-associated genes. Oncotarget. 2016;7:64071–64088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding L-W, Sun Q-Y, Tan K-T, et al. Mutational landscape of pediatric acute lymphoblastic leukemia. Cancer Res. 2017;77:390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spinella J-F, Richer C, Cassart P, et al. Mutational dynamics of early and late relapsed childhood ALL: rapid clonal expansion and long-term dormancy. Blood Adv. 2018;2:177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartzman O, Savino AM, Gombert M, et al. Suppressors and activators of JAK-STAT signaling at diagnosis and relapse of acute lymphoblastic leukemia in Down syndrome. Proc Natl Acad Sci U S A. 2017;114:E4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Mullighan CG, Harvey RC, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood. 2011;118:3080–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute Lymphoblastic Leukemia. N Engl J Med. 2009;360:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hof J, Krentz S, van Schewick C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29:3185–3193. [DOI] [PubMed] [Google Scholar]
- 24.Irving J, Matheson E, Minto L, et al. Ras pathway mutations are highly prevalent in relapsed childhood acute lymphoblastic leukaemia, may act as relapse-drivers and confer sensitivity to MEK inhibition. Blood. 2014;124:3420–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tzoneva G, Dieck CL, Oshima K, et al. Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature. 2018;553:511–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van der Veer A, Waanders E, Pieters R, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood. 2013;122:2622–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamps WA, Bökkerink JPM, Hakvoort-Cammel F, et al. BFM-oriented treatment for children with acute lymphoblastic leukemia without cranial irradiation and treatment reduction for standard risk patients: results of DCLSG protocol ALL-8 (1991-1996). Leukemia. 2002;16:1099–1111. [DOI] [PubMed] [Google Scholar]
- 29.Veerman AJ, Kamps WA, van den Berg H, et al. Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol. 2009;10:957–966. [DOI] [PubMed] [Google Scholar]
- 30.Kuiper RP, Schoenmakers EFPM, van Reijmersdal SV, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21:1258–1266. [DOI] [PubMed] [Google Scholar]
- 31.van Galen JC, Kuiper RP, van Emst L, et al. BTG1 regulates glucocorticoid receptor autoinduction in acute lymphoblastic leukemia. Blood. 2010;115:4810–4819. [DOI] [PubMed] [Google Scholar]
- 32.Inthal A, Zeitlhofer P, Zeginigg M, et al. CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia. 2012;26:1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nature genetics. 2013;45:242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malinowska-Ozdowy K, Frech C, Schönegger A, et al. KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. 2015;29:1656–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dixon ZA, Nicholson L, Zeppetzauer M, et al. CREBBP knockdown enhances RAS/RAF/MEK/ERK signaling in Ras pathway mutated acute lymphoblastic leukemia but does not modulate chemotherapeutic response. Haematologica. 2017;102:736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asai D, Imamura T, Suenobu SI, et al. IKZF1 deletion is associated with a poor outcome in pediatric B-cell precursor acute lymphoblastic leukemia in Japan. Cancer Med. 2013;2:412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olsson L, Castor A, Behrendtz M, et al. Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011. Leukemia. 2014;28:302–310. [DOI] [PubMed] [Google Scholar]
- 38.Palmi C, Valsecchi MG, Longinotti G, et al. What is the relevance of Ikaros gene deletions as a prognostic marker in pediatric Philadelphia-negative B-cell precursor acute lymphoblastic leukemia? Haematologica. 2013;98:1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dörge P, Meissner B, Zimmermann M, et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica. 2013;98:428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moorman AV, Enshaei A, Schwab C, et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. 2014;124:1434–1444. [DOI] [PubMed] [Google Scholar]
- 41.Clappier E, Grardel N, Bakkus M, et al. IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children's Leukemia Group study 58951. Leukemia. 2015;29:2154–2161. [DOI] [PubMed] [Google Scholar]
- 42.Olsson L, Ivanov Öfverholm I, Norén-Nyström U, et al. The clinical impact of IKZF1 deletions in paediatric B-cell precursor acute lymphoblastic leukaemia is independent of minimal residual disease stratification in Nordic Society for Paediatric Haematology and Oncology treatment protocols used between 1992 and 2013. Br J Haematol. 2015;170:847–858. [DOI] [PubMed] [Google Scholar]
- 43.Marke R, Havinga J, Cloos J, et al. Tumor suppressor IKZF1 mediates glucocorticoid resistance in B-cell precursor acute lymphoblastic leukemia. Leukemia. 2016;30:1599–1603. [DOI] [PubMed] [Google Scholar]
- 44.Irving JA, Enshaei A, Parker CA, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2016;128:911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stanulla M, Dagdan E, Zaliova M, et al. IKZF1plus defines a new minimal residual disease–dependent very-poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36:1240–1249. [DOI] [PubMed] [Google Scholar]
- 46.Hamadeh L, Enshaei A, Schwab C, et al. Validation of the United Kingdom copy-number alteration classifier in 3239 children with B-cell precursor ALL. Blood Adv. 2019;3:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scheijen B, Boer JM, Marke R, et al. Tumor suppressors BTG1 and IKZF1 cooperate during mouse leukemia development and increase relapse risk in B-cell precursor acute lymphoblastic leukemia patients. Haematologica. 2017;102:541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Joshi I, Yoshida T, Jena N, et al. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat Immunol. 2014;15:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwickert TA, Tagoh H, Gultekin S, et al. Stage-specific control of early B cell development by the transcription factor Ikaros. Nat Immunol. 2014;15:283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–114. [DOI] [PubMed] [Google Scholar]
- 51.Churchman Michelle L, Low J, Qu C, et al. Efficacy of retinoids in IKZF1-mutated BCR-ABL1 acute lymphoblastic leukemia. Cancer Cell. 2015;28:343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinelli G, Iacobucci I, Storlazzi CT, et al. IKZF1 (Ikaros) deletions in BCR-ABL1–Positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: A GIMEMA AL WP Report. J Clin Oncol. 2009;27:5202–5207. [DOI] [PubMed] [Google Scholar]
- 53.Van der Veer A, Zaliova M, Mottadelli F, et al. IKZF1 status as a prognostic feature in BCR-ABL1–positive childhood ALL. Blood. 2014;123:1691–1698. [DOI] [PubMed] [Google Scholar]
- 54.Churchman ML, Mullighan CG. Ikaros: Exploiting and targeting the hematopoietic stem cell niche in B-progenitor acute lymphoblastic leukemia. Exp Hematol. 2017;46:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pui C-H, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–178. [DOI] [PubMed] [Google Scholar]
- 56.Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucl Acids Res. 2002;30:e57–e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.