

Abstract
Glucose dehydrogenases are important auxiliary enzymes in biocatalysis, employed in the regeneration of reduced nicotinamide cofactors for oxidoreductase catalysed reactions. Here we report the identification and characterization of a novel glucose-1-dehydrogenase (GDH) from Paenibacillus pini that prefers NAD+ as cofactor over NADP+. The purified recombinant P. pini GDH displayed a specific activity of 247.5 U/mg. The enzyme was stable in the pH range 4–8.5 and exhibited excellent thermostability till 50 °C for 24 h, even in the absence of NaCl or glycerol. Paenibacillus pini GDH was also tolerant to organic solvents, demonstrating its potential for recycling cofactors for biotransformation. The potential application of the enzyme was evaluated by coupling with a NAD+-dependent alcohol dehydrogenase for the reduction of acetophenone and ethyl-4-chloro-3-oxo-butanoate. Conversions higher than 95% were achieved within 2 h with low enzyme loading using lyophilized cell lysate, suggesting that P. pini GDH could be highly effective for recycling NADH in redox biocatalysis.
Electronic supplementary material
The online version of this article (10.1007/s12088-019-00834-w) contains supplementary material, which is available to authorized users.
Keywords: Cofactor regeneration, Oxidoreductase, Dehydrogenase, Biocatalysis, Paenibacillus pini
Introduction
Oxidoreductase enzymes such as alcohol dehydrogenases or ketoreductases have immense potential for application in industrial biocatalysis for the enantiopure synthesis of pharmaceutical precursors [1]. However, their need for stoichiometric amounts of reduced nicotinamide (NAD(P)H) cofactors makes the process prohibitively expensive. An efficient system to recycle these cofactors in situ during the course of the reaction is therefore a critical component in determining the efficiency and commercial viability of redox biocatalytic processes. A number of biological cofactor recycle systems [2] have been developed based on (1) addition of co-substrates such as isopropanol (2) auxiliary enzymes (glucose dehydrogenase, formate dehydrogenase, phosphite dehydrogenase, NADH oxidase) and (3) photosynthetic regeneration of NADPH [3]. Glucose-1-dehydrogenase (GDH) catalyzes the oxidation of glucose to glucono-δ-lactone, simultaneously generating the reduced cofactor NAD(P)H from NAD(P)+. GDHs and formate dehydrogenases (FDH), which regenerate NADH during the conversion of formate to CO2, are widely applied as auxiliary enzymes in redox biocatalysis for the regeneration of nicotinamide cofactors [4]. GDH enzyme has been characterized from different organisms [5–10] including Bacillus subtilis, Bacillus megaterium, Sulfolobus solfataricus, Gluconobacter oxydans, Bacillus amyloliquefaciens, Lysinibacillus sphaericus, Bacillus thuringenesis and Thermoplasma acidophilum. GDHs from different sources can vary widely in their preference for cofactors NAD+ or NADP+. The GDH from Bacillus subtilis is often preferred for biocatalytic processes [11, 12], owing to its dual cofactor preference and high expression levels in E. coli host.
Alcohol dehydrogenases (ADH) catalyze the enantioselective reduction of ketones to chiral alcohols, and are employed in the synthesis of intermediates for cholesterol-lowering drugs [13]. In recent years, NADH-specific alcohol dehydrogenases have been reported by our group (from Acetobacter aceti, [13]) and others [14], which exhibit significant activity on ketone substrates like β-ketoesters and acetophenone derivatives. NAD+ is more preferable for cell-free biotransformation in industrial applications due to its lower cost and stability of over a wide range of pH and temperature conditions as compared to NADP+ [15]. While FDH can regenerate NADH efficiently and offer certain process advantages, their expression levels and specific activities (4–10 U/mg) are significantly less than those of GDH (200–500 U/mg) [16]. Hence, a GDH with high activity using NAD+ as cofactor would be essential to enable faster biocatalysis with NADH-specific oxidoreductases.
In this study, we have mined a new GDH from P. pini based on sequence similarity with known GDH genes. Paenibacillus pini GDH was cloned and expressed as a recombinant protein in E. coli; it exhibited high catalytic efficiency on glucose and significant preference for NAD+ as the cofactor. We have further characterized the stability and tolerance of the enzyme in organic solvents in order to evaluate the suitability of the P. pini GDH in industrial biocatalysis.
Materials and Methods
Chemicals and Media
Luria-Bertani medium (LB), yeast extract and carbenicillin used for cultivation were procured from HiMedia, India. Nicotinamide cofactors (NAD+ and NADP+) were obtained from Oriental Yeast Co. Ltd. (Japan). Enzymes, kits and reagents for genetic manipulation were purchased from New England Biolabs (USA). Primers were synthesized by Eurofins Genomics (Bangalore, India). All other chemicals used were of analytical grade and obtained from Sigma Aldrich USA.
Construction of Recombinant Plasmids and Expression of GDH
Sequences coding for GDHs were identified from blastp hits using Bacillus subtilis GDH (GenBank Accession KIU10883.1) as a query. Three identified gene sequences coding for GDHs (Table 1) were codon-optimized for expression in E. coli and synthesized from Invitrogen, Thermo Fisher Scientific (USA). The genes were first PCR-amplified using a high-fidelity Q5 polymerase employing specific primers (Fig. S1). PCR was performed in a gradient thermocycler using conditions: 98 °C/2 min, 30 cycles of [98 °C/20 s, 72 °C/30 s, 72 °C/45 s] and final extension 72 °C/5 min. Restriction digestion of the amplified PCR product and pET21a vector was performed using NdeI and XhoI fast digest (Fermentas, USA). Following gel extraction of cut DNA fragments, ligation was performed using T4 DNA ligase at 24 °C for 2 h. The recombinant plasmids were transformed into E. coli TOP10 cells, and following sequence verification further introduced into E. coli BL21 (DE3) cells for GDH expression.
Table 1.
Details of GDH sequences mined in the study. GDH activity was measured using 20 mM glucose and 0.1 mM NAD at 25 °C
| Source organism | Accession number | Protein expression | GDH activity (U/ml lysate) |
|---|---|---|---|
| Paenibacillus pini | WP_036650715.1 | Soluble | 40 (NAD) |
| Bacillus firmus | WP_0353305881 | Insoluble | 0.9 (NAD/NADP) |
| Oscillatoria sp. PCC10802 | WP_017717521.1 | Insoluble | 0.6 (NAD/NADP) |
Escherichia coli BL21_pET21a_GDH cells were cultivated using a shake flask fed-batch protocol according to Sohoni et al. [17]. The overnight grown seed culture was inoculated (3% v/v) into 500 ml flask containing 100 ml LB medium with 150 µg/ml carbenicillin and was cultivated at 32 °C and 180 rpm. Nutrient shots of glycerol (30 mM final concentration) and yeast extract (1% w/v final concentration) were added at 6 h and 9 h after inoculation. Protein expression was induced by the addition of 1 mM IPTG at 8 h after inoculation, combined with lowering of temperature to 25 °C post-induction. Cells were harvested 16 h after induction and GDH expression was checked on SDS-PAGE and by estimating GDH activity in cell-free lysate.
Purification of P. pini GDH
Recombinant P. pini GDH expressed in E. coli BL21 (DE3) was purified via ammonium sulphate precipitation and anion exchange chromatography. All purification steps were carried out at 4 °C. Cells from 200 ml culture were harvested by centrifugation at 8000 rpm for 10 min, washed with 50 mM phosphate buffer pH 6.4 and lysed by sonication. Powdered (NH4)2SO4 was added slowly to the cell-free lysate while stirring. The protein precipitate recovered from 30–60% saturation of (NH4)2SO4 was resuspended in 20 mM potassium phosphate buffer pH 6.4 and dialyzed against the same buffer. The dialysate was further loaded on a 5 ml Q-Sepharose (GE Healthcare) column pre-equilibrated in 20 mM phosphate buffer pH 6.4. Following washing with the same buffer to remove unbound proteins, elution was performed with a step gradient of NaCl from 100–500 mM. Eluted fractions displaying GDH activity were pooled and concentrated to 3 mg/ml using AmiconUltra15 (Millipore, USA) 30 kDa centrifugal filter. This sample was loaded on a size exclusion column (HiLoad 16/600 Superdex 200, GE Healthcare) and eluted at 1 ml/min with 20 mM phosphate buffer pH 6.4 containing 100 mM NaCl. The final purified enzyme was evaluated using SDS-PAGE and activity assay. Protein concentration was estimated by Bradford method [18] using Bovine serum albumin as a standard.
Denaturing SDS-PAGE was performed using a 5% stacking gel and 12% separating gel [19], and gels were stained with Coomassie Brilliant Blue R-250. GDH subunit molecular weight was estimated by comparing its electrophoretic mobility with marker proteins. Native molecular weight was determined using a size exclusion column (HiLoad 16/600 Superdex 200, GE Healthcare), with marker proteins Ribonuclease (13KDa), Carbonic anhydrase (29KDa), Conalbumin (75KDa) and Goat IgG (150KDa).
GDH Activity Assay
GDH activity was assayed by measuring the change in absorbance at 340 nm in 100 mM phosphate buffer pH 6.4 at 37 °C with 200 mM d-glucose and 2 mM NAD+ using a UV–Vis spectrophotometer (UV-1800, Shimadzu, Singapore). A340 was monitored over 5 min and concentration of NADH was calculated using the extinction coefficient 6220 M−1 cm−1. One unit of enzyme activity was defined as the amount of enzyme that catalyzed the formation of 1 μmol NADH per minute.
Effect of pH and Temperature on Activity and Stability of P. pini GDH
Optimum pH for P. pini GDH was determined by measuring the enzyme activity in different buffers ranging from pH 4.0–9.0 (100 mM). Optimum temperature for the enzyme was evaluated by measuring activity at different temperatures ranging (20–75 °C) at pH 6.4. Glucose and NAD+ concentrations were used as described in Sect. 2.4. The pH stability profile of P. pini GDH was determined by incubating the enzyme in 100 mM buffers of varying pH (4–9) at 30 °C for 4 h and 24 h, then assaying enzyme activity. Thermostability was evaluated by incubating the enzyme both in plain phosphate buffer (20 mM, pH 6.4) and with the addition of 100 mM NaCl for 4 h and 24 h at increasing temperatures from 20–60 °C.
Solvent Tolerance of P. pini GDH
To evaluate the effect of solvents on P. pini GDH stability, the enzyme was incubated with either 10% v/v or 50% v/v of water-miscible and biphasic solvents. The residual GDH activity was assayed after 1 h treatment at 30 °C. Relative activity was calculated as % based on a control where water was added in place of solvent.
Substrate Specificity
Paenibacillus pini GDH activity was assayed with different mono- and di-saccharide substrates at 200 mM concentration with 2 mM NAD+ in 100 mM phosphate buffer of pH 6.4.
Steady-State Kinetics
Apparent km and Vmax values of P. pini GDH were estimated by determining GDH specific activity at various concentrations of glucose (2.5–200 mM) at a fixed saturating concentration of NAD+ (2 mM) acceptor. In another assay, NAD+ (0.05–2 mM) or NADP+ (0.05–4 mM) concentrations were varied at fixed glucose concentration (200 mM). Initial rates were measured at 37 °C and pH 6.4 as described earlier. The kinetic constants of the enzyme were determined using non-linear fitting of the Michaelis–Menten equation.
Application of P. pini GDH as NADH Regenerator in Biotransformation
Alcohol dehydrogenase (ADH) from A. aceti [13] was prepared using shake flask cultivation as described earlier in Sect. 2.2. Escherichia coli cells expressing recombinant A. aceti ADH or P. pini GDH were lysed by sonication and the cell-free extracts were lyophilized with trehalose as cryoprotectant (1 mg trehalose for 1 mg protein). For the biotransformation reaction (200 µl), 2 U each of ADH and GDH in phosphate buffer pH 7 were used. The substrate (100 mM acetophenone or 200 mM ethyl-4-chloro-3-oxobutanoate) was added to start the reaction, and the mixture was incubated at 32 °C with 200 rpm shaking up to 4 h. Glucose at 1.25 times the substrate concentration and 0.1 mM NAD+ were also included in the reaction. The alcohol products were extracted with equal volume of ethyl acetate and quantified using GC-FID analysis as described earlier [3, 13]. Alternatively, a biotransformation reaction with ADH and isopropanol (1.25 times the substrate concentration) as co-substrate was also set up for comparison.
Results and Discussion
Identification and Sequence Analysis of P. pini GDH
GDH sequences were mined from genome databases using the B. subtilis GDH (GenBank Accession WP_043858127.1) as a query. Sequences with identity between 40 and 80% were chosen and analysed with respect to the amino acids in the catalytic tetrad and nucleotide binding site. Both P. pini GDH and B. firmus GDH showed 72% sequence homology with B. subtilis GDH, while GDH from Oscillatoria sp. PCC10802 was 46% similar to the query sequence. Multiple sequence alignment of the mined GDHs with reported enzymes (Fig. 1) showed the conservation of key features of short chain dehydrogenases (SDRs) including the coenzyme binding site GxxxGxG [20] and the catalytic tetrad Asn116-Ser145-Tyr158-Lys162 [21]. The codon-optimized GDH genes were synthesized and cloned in pET21a vector for expression in E. coli. The pET system along with E. coli BL21 host strain is widely used for the production of recombinant enzymes and offers several biotechnological advantages including high rates of protein synthesis and reduced proteolytic degradation [22]. The GDH from P. pini showed soluble expression at 30% of total protein, while the enzymes from Oscillatoria sp. and B. firmus were largely accumulated as inclusion bodies with significantly lower enzyme activity in the soluble fraction compared to that for P. pini GDH. Paenibacillus pini GDH displayed an activity of 40 U/ml in cell-free lysate at 25 °C with 10 mM glucose and 0.25 mM NAD+. Considering its higher expression and activity, P. pini GDH was chosen for further characterization. In addition, P. pini GDH showed a ninefold specific activity with NAD+ over NADP+ as a cofactor. Analysis of the GDH sequence motifs using the Cofactory [23] web server predicted a score of 0.750/0.170 for NAD+/NADP+. The Cofactory algorithm uses HMM models based on structural data of enzyme-cofactor complexes, to study the cofactor binding Rossmann fold domains and predict specificity for NADP, NAD or FAD for the enzyme sequence [23]. In comparison, B. subtilis GDH with a dual cofactor specificity scored 0.873/0.700 for NAD+/NADP+. Increased cofactor preference of P. pini GDH for NAD+ augurs well for application of the enzyme in combination with NADH-dependent ADH, such as from A. aceti [13].
Fig. 1.

Multiple sequence alignment of the protein sequences of glucose-1-dehydrogenase (GDH) genes from Oscillatoria sp. (WP_017717521), B. firmus (WP_035330588), P. pini (WP_036650715), L. sphaericus (ACR78513), B. megaterium GDH-III (WP_013082700), GDH-IV (WP_129542354.1) B. subtilis (KIU10883) and B. amyloliquefaciens (AWM50557) using Clustal Omega. Nucleotide binding domain (11-35 amino acids) with residues in green indicates the Rossmann fold GxxxGxG. Catalytic tetrad is shown in yellow (Asn116, Ser145, Tyr158, Lys162) and substrate binding domain from amino acids 192-218
Expression and Purification of P. pini GDH
Usually, the presence of a hexa-histidine tag to a recombinant protein sequence aids in enhancing protein solubility and purification. However, the addition of a His-tag at the N-terminal or the C-terminal drastically reduced the soluble expression in the case of P. pini GDH, indicating that the His-tag probably interfered with the protein structure or folding. Therefore the enzyme was cloned in pET21a vector without any tag. The cultivation of E. coli cells expressing P. pini GDH was done in a shake flask employing additional doses of glycerol and yeast extract in the culture medium [16]. This protocol increased both cell growth (OD600 = 20) and protein synthesis, resulting in a GDH productivity of 3.3 × 105 U/l in the lysate. The GDH enzyme was purified using a combination of ammonium sulphate fractionation, anion exchange and size exclusion chromatography, with a protein yield of 192 mg/l. The specific activity of purified P. pini GDH was 247.5 ± 3.5 U/mg protein at 37 °C with saturating concentrations of d-glucose (200 mM) and NAD+ (2 mM). The specific activity was twice that of B. amyloliquefaciens GDH [7] and 31% that of B. megaterium GDH-IV [24]. The purified P. pini GDH enzyme was stored at 4 °C in phosphate buffer pH 6.4 with 100 mM NaCl till 3 months with minimal loss of activity. SDS-PAGE analysis showed a clear single protein band corresponding to 28 kDa for the purified GDH (Fig. 2). The native molecular weight of P. pini GDH was estimated using size exclusion chromatography to be about 112.5 kDa, confirming the homotetrameric subunit composition of the enzyme similar to that reported for other GDHs [5, 8].
Fig. 2.

SDS-PAGE showing expression of P. pini GDH in E. coli BL21 (DE3). Lane M: prestained protein marker (PAGEmark Tricolor Plus), Lane 1: cell-free lysate, Lane 2: dialysed fraction, Lane 3: fraction after anion exchange chromatography, Lane 4: 15 µg of protein eluted from size exclusion column
Effect of pH and Temperature on Activity and Stability of P. pini GDH
Paenibacillus pini GDH was active over a pH range of 4–9, with maximum specific activity of 287 U/mg at pH 7.5 in phosphate buffer at 37 °C (Fig. 3a). The enzyme displayed at least 70% of its maximum specific activity in the pH range 6–8.5, which reduces the need for strict maintenance of pH during biotransformation. The optimal pH range was closer to GDHs from B. megaterium [24] and B. thuringiensis [9], in contrast to GDHs from Lysinibacillus sphaericus [8] and B. amyloliquefaciens [7] that were more active at pH 9–10. While P. pini GDH was most stable at pH 7, the enzyme was also highly stable over a wide pH range from 4 to 8.5, retaining 80–100% activity even after 24 h incubation at 30 °C (Fig. 3b). Paenibacillus pini GDH was more stable in acidic pH than the wild-type L. sphaericus GDH which showed 40% loss of activity within 1 h at pH 4 [8]. The enzyme also retained 50% activity at pH 9 after 24 h, compared to GDHs from other Bacillus species which are generally inactivated at pH 9 due to the dissociation of the tetrameric structure [7, 24].
Fig. 3.

a Effect of pH on the activity of P. pini GDH. GDH activity was assayed at 37 °C in different buffer systems (100 mM) with 200 mM glucose and 2 mM NAD+. b pH stability of P. pini GDH. GDH activity was assayed in 100 mM phosphate buffer pH 6.4 after incubation in buffers of different pH for 4 h and 24 h at 30 °C. GDH activity in phosphate buffer pH 6.4 at the start of incubation was used as 100%. c Effect of temperature on the activity of P. pini GDH. GDH activity was assayed using Phosphate buffer (100 mM) pH 6.4 at different temperatures. Glucose and NAD+ concentrations were 200 mM and 2 mM respectively. d Thermostability of P. pini GDH. The enzyme activity was assayed at 37 °C after incubation at different temperatures in the range 20–60 °C. The enzyme was incubated either in plain phosphate buffer (20 mM) pH 6.4 or with the addition of 100 mM NaCl. GDH activity at the start of incubation was used as 100%
Paenibacillus pini GDH activity was enhanced considerably with increase in temperature, with a maximum specific activity of 360.2 U/mg at 65 °C. The activity decreased gradually with further increase in temperature to 75 °C, after which there was a significant loss of activity (Fig. 3c). Although the enzyme activity was maximum at 65 °C, the stability of the enzyme was affected at temperatures above 50 °C in absence of NaCl (Fig. 3d). Nevertheless, the enzyme was able to maintain over 75% activity on incubation at 40 °C till 24 h and at 50 °C till 4 h. With the addition of 100 mM NaCl, P. pini GDH was able to retain more than 90% activity till 24 h at 60 °C. In comparison, the DS255 variant of L. sphaericus GDH [25] engineered with inter-subunit disulfide bonds exhibited a half-life of 9900 min at 50 °C, while the wild-type enzyme was completely inactivated within 1 h when incubated without 20% glycerol or 2 M NaCl. GDH from B. amyloliquefaciens [7] has been reported to withstand up to 40 °C for 6 h, whereas B. megaterium GDH-III [6] was inactivated within 20 min in absence of 2 M NaCl above 40 °C. Although GDH from Bacillus subtilis and B. megaterium exhibit a higher specificity over P. pini GDH, these enzymes require the inclusion of 20% (w/v) glycerol for the maintenance of GDH structure and activity [26]. The GDH from thermophilic Sulfolobus solfataricus [7] is active and stable at 70 °C, but its low turnover and inhibition by glucose above 25 mM could hinder its biocatalytic application.
Paenibacillus pini GDH Tolerance to Organic Solvents
Paenibacillus pini GDH showed high solvent tolerance, retaining 90–100% activity with 10% v/v concentration of many water-miscible and organic solvents tested (Fig. 4). Similar to GDHs reported from L. sphaericus [8] and Bacillus amyloliquefaciens [7], P. pini GDH was inactivated at higher concentration (50% v/v) of alcohols and acetone. However, the enzyme retained 60% activity in the presence of n-butanol, whereas reported GDHs are usually inactivated by butanol even at 10%. Paenibacillus pini GDH was also able to tolerate up to 50% of DMSO and non-polar solvents such as n-hexane, toluene and ethyl acetate. Such solvents are often used in biphasic systems to improve substrate solubility, suppress side reactions or enhance enantioselectivity [13, 27]. The solvent tolerance of P. pini GDH would be beneficial in designing processes for oxidoreductase biocatalysis involving high concentrations of non-polar substrates.
Fig. 4.

Effect of solvents on the activity of P. pini GDH. GDH activity was assayed following incubation of the enzyme in different solvents at 50% v/v and 10% v/v concentration for 1 h at 30 °C. Buffer was added in place of solvent in the control
Substrate Specificity and Kinetic Constants
Different mono- and disaccharides were examined as possible substrates for P. pini GDH enzyme using NAD+ as the cofactor. Paenibacillus pini GDH exhibited relative activities of 2–8% for aldose sugars and disaccharides when compared to d-glucose as substrate (Table 2). A similar substrate spectrum was observed in GDH-III and GDH-IV from B. megaterium at pH 8 and 37 °C [24]. However, P. pini GDH displayed higher activity on D-galactose compared to d-mannose, similar to GDH from L. sphaericus [10].
Table 2.
Substrate specificity of P. pini GDH. GDH activity was assayed at 37 °C in phosphate buffer (100 mM), pH 6.4 using different substrates at 200 mM concentration and 2 mM NAD+
| Substrate | Relative activity (%) | |||
|---|---|---|---|---|
| P. pini | L. sphaericus | B. megaterium GDH-IIIa | B. megaterium GDH-IVa | |
| d-Glucose | 100 | 100 | 100 | 100 |
| d-Mannose | 2.5 | 7.6 | 1.7 | 6.1 |
| d-Galactose | 4.9 | 17.3 | 1.1 | 3.4 |
| Maltose | 8.0 | 22.4 | NA | NA |
| Sucrose | 2.0 | 6.3 | NA | NA |
| Lactose | 0.3 | 4.2 | NA | NA |
| Reference | This study | [10] | [22] | [22] |
NA data not available
aGDH activity at pH 8 with substrate concentration of 148 mM
Kinetic constants of P. pini GDH were estimated using the non-linear curve-fitting method (Table 3). The enzyme had a lower Km of 0.25 mM for NAD+ compared to 2.16 mM for NADP+, underlining its preference for NAD+. GDHs usually employed in combination with oxidoreductases either prefer NADP+ such as the isoforms GDHIWG3, GDHII from B. megaterium [5]; or as in the case of B. subtilis [24] and L. sphaericus [10], exhibit equal preference for both cofactors. A cofactor preference for NAD+ similar to P. pini has been observed in GDH-III and GDH-IV from B. megaterium [5, 24]; however, they are rarely used in biocatalytic processes.
Table 3.
Kinetic constants for P. pini GDH enzyme. Initial rates were measured under conditions as described in Sect. 2.8. Results are expressed as mean ± SD
| Km (mM) | Vmax (μmol/min/mg) | kcat (s−1)a | kcat/Km | |
|---|---|---|---|---|
| NAD | 0.25 ± 0.01 | 247.5 ± 3.5 | 115.4 ± 2.5 | 462.1 ± 16.3 |
| NADP | 2.16 ± 0.2 | 333 ± 5.7 | 155.4 ± 3.9 | 143.5 ± 16.6 |
| Glucose (with NAD) | 26.75 ± 1.1 | – |
akcat values were calculated for single subunit of the enzyme
NADH Regeneration by P. pini GDH in Biotransformation
Paenibacillus pini GDH was further evaluated as an NADH-regenerating enzyme in the enantioselective bioreduction of acetophenone to (R)-phenyl-1-ethanol and ethyl-4-chloro-3-oxobutanoate (COBE) to (S)-ethyl-4-chloro-3-hydroxybutyrate (CHBE). The reduction of both these ketone substrates by alcohol dehydrogenases or carbonyl reductases has been well-established in literature [28, 29]. Reduction of acetophenone derivatives provides chiral phenylethanols, which are important pharmaceutical precursors [28]; while (S)-CHBE is a key chiral intermediate in the synthesis of HMG-coA inhibitors like lipitor and other cholesterol lowering drugs [14]. We used the NADH-dependent anti-Prelog alcohol dehydrogenase from A. aceti [13] in combination with P. pini GDH for the biotransformation reaction. Both enzymes were added as lyophilized crude lysate at 2 U loading; with only NAD added as the cofactor. A conversion of 95% with 100 mM acetophenone and 98% with 200 mM COBE was achieved within 2 h (Fig. 5) when the two-enzyme system was used, demonstrating the efficiency of P. pini GDH to regenerate NADH. A recent report [14] has demonstrated 95% conversion of 100 mM COBE in 2 h using the combination of ADH from Barnotella apis and GDH from Bacillus sp. (50 μg/ml). In comparison, the use of isopropanol for cofactor regeneration resulted in only 35% conversion especially in the case of acetophenone, probably caused by equilibrium constraints [30]. Although the conversion of COBE with the use of isopropanol was comparable to GDH, the requirement of excess solvent might lead to enzyme inactivation and waste accumulation. Moreover, scale up studies with other ketoreductases [31] have shown that isopropanol mediated reactions suffered from lower substrate loading and longer time for completion. Superior conversion efficiency could be achieved using GDH with glucose addition and pH control [31].
Fig. 5.
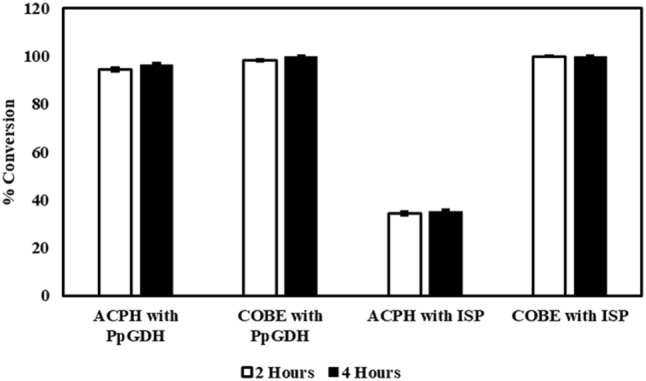
Demonstrating the potential of P. pini GDH as a co-factor recycling enzyme. Paenibacillus pini GDH was coupled with A. aceti ADH for the bioreduction of 100 mM acetophenone (ACPH) or 200 mM ethyl-4-chloro-3-oxobutanoate (COBE). 0.1 mM NAD+ was used as a cofactor. Isopropanol (ISP) or glucose was added at 1.25 times the substrate concentration
In conclusion, we have identified and characterized a robust GDH from P. pini that shows preference for NAD+ over NADP+. The enzyme showed a high level of soluble expression in E. coli and exhibits excellent operational stability till 24 h in a wide pH range and at temperatures up to 50 °C, without any need for salt or glycerol protection. The enzyme was also used as lyophilized crude lysate in combination with P. pini GDH therefore has significant potential for application in industrial biocatalysis as a cost-effective cofactor regeneration system, especially in combination with NADH-dependent oxidoreductases.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
AVS acknowledges the award of National Postdoctoral Fellowship by the Science and Engineering Research Board (SERB) (Grant No. PDF_2016_003685), Government of India. The authors acknowledge financial support from the Wadhwani Research Center for Bioengineering at Indian Institute of Technology Bombay.
Author’s Contribution
AVS and PW designed the research. SS, PS and AVS performed the research and analysed the data. SS, AVS and PW wrote the paper. All authors read and approved the final manuscript.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Shikha Shah and Avinash Vellore Sunder have equally contributed to this manuscript.
References
- 1.Prier CK, Kosjek B. Recent preparative applications of redox enzymes. Curr Opin Chem Biol. 2019;49:105–112. doi: 10.1016/j.cbpa.2018.11.011. [DOI] [PubMed] [Google Scholar]
- 2.Wu H, Tian C, Song X, Liu C, Yang D, Jiang Z. Methods for the regeneration of nicotinamide coenzymes. Green Chem. 2013;15:1773–1789. doi: 10.1039/C3GC37129H. [DOI] [Google Scholar]
- 3.Sengupta A, Sunder AV, Sohoni SV, Wangikar PP. The effect of CO2 in enhancing photosynthetic cofactor recycling for alcohol dehydrogenase mediated chiral synthesis in cyanobacteria. J Biotechnol. 2018;289:1–6. doi: 10.1016/j.jbiotec.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Han L, Liang B. New approaches to NAD(P)H regeneration in the biosynthesis systems. World J Microbiol Biotechnol. 2018;34:141. doi: 10.1007/s11274-018-2530-8. [DOI] [PubMed] [Google Scholar]
- 5.Nagao T, Mitamura T, Wang XH, Negoro S, Yomo T, Urabe I, Okada H. Cloning, nucleotide-sequences, and enzymatic-properties of glucose-dehydrogenase isozymes from Bacillus megaterium IAM1030. J Bacteriol. 1992;174:5013–5020. doi: 10.1128/jb.174.15.5013-5020.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diederichs S, Linn K, Luckgen J, Klement T, Grosch JH, Honda K, Ohtake H, Buchs J. High-level production of (5S)-hydroxyhexane-2-one by two thermostable oxidoreductases in a whole-cell catalytic approach. J Mol Catal B Enzym. 2015;121:37–44. doi: 10.1016/j.molcatb.2015.08.001. [DOI] [Google Scholar]
- 7.Pongtharangkul T, Chuekitkumchorn P, Suwanampa N, Payongsri P, Honda K, Panbangred W. Kinetic properties and stability of glucose dehydrogenase from Bacillus amyloliquefaciens SB5 and its potential for cofactor regeneration. AMB Expr. 2015;5:68. doi: 10.1186/s13568-015-0157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding HT, Du YQ, Liu DF, Li ZL, Chen XJ, Zhao YH. Cloning and expression in E. coli of an organic solvent-tolerant and alkali-resistant glucose 1-dehydrogenase from Lysinibacillus sphaericus G10. Bioresour Technol. 2011;102:1528–1536. doi: 10.1016/j.biortech.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 9.Hyun J, Abigail M, Choo JW, Ryu J, Kim HK. Effects of N-/C-terminal extra tags on the optimal reaction conditions, activity, and quaternary structure of Bacillus thuringiensis glucose-1-dehydrogenase. J Microbiol Biotechnol. 2016;26:1708–1716. doi: 10.4014/jmb.1603.03021. [DOI] [PubMed] [Google Scholar]
- 10.Smith DL, Budgen N, Bungard JS, Danson JM, Hough WD. Purification and characterization of glucose dehydrogenase from the thermoacidophilic archaebacterium Thermoplasma acidophilum. Biochem J. 1989;261:973–977. doi: 10.1042/bj2610973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naeem M, Rehman AU, Shen B, Ye L, Yu H. Semi-rational engineering of carbonyl reductase YueD for efficient biosynthesis of halogenated alcohols with in situ cofactor regeneration. Biochem Eng J. 2018;137:62–70. doi: 10.1016/j.bej.2018.05.008. [DOI] [Google Scholar]
- 12.Cui ZM, Zhang JD, Fan XJ, Zheng GW, Chang HH, Wei WL. Highly efficient bioreduction of 2-hydroxyacetophenone to (S)- and (R)-1-phenyl-1,2-ethanediol by two substrate tolerance carbonyl reductases with cofactor regeneration. J Biotechnol. 2017;243:1–9. doi: 10.1016/j.jbiotec.2016.12.016. [DOI] [PubMed] [Google Scholar]
- 13.Shah S, Agera R, Sharma P, Sunder AV, Bajwa H, James HM, Gaikaiwari RP, Wangikar PP. Development of biotransformation process for asymmetric reduction with novel anti-Prelog NADH-dependent alcohol dehydrogenases. Proc Biochem. 2018;70:71–78. doi: 10.1016/j.procbio.2018.04.016. [DOI] [Google Scholar]
- 14.Zhu YH, Liu CY, Cai S, Guo LB, Kim IW, Kalia VC, Lee JK, Zhang YW. Cloning, expression and characterization of a highly active alcohol dehydrogenase for production of ethyl-(S)-4-chloro-3-hydroxybutyrate. Ind J Microbiol. 2019;59:225–233. doi: 10.1007/s12088-019-00795-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chanique AM, Parra LP. Protein engineering for nicotinamide coenzyme specificity in oxidoreductases: attempts and challenges. Front Microbiol. 2018;9:194. doi: 10.3389/fmicb.2018.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kratzer R, Woodley JM, Nidetzky B. Rules for biocatalyst and reaction engineering to implement effective NAD(P)H-dependent, whole cell bioreductions. Biotechnol Adv. 2015;33:1641–1652. doi: 10.1016/j.biotechadv.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sohoni SV, Nelapati D, Sathe S, Javadekar-Subhedar V, Gaikaiwari RP, Wangikar PP. Optimization of high cell density fermentation process for recombinant nitrilase production in E. coli. Bioresour Technol. 2015;188:202–208. doi: 10.1016/j.biortech.2015.02.038. [DOI] [PubMed] [Google Scholar]
- 18.Bradford MM. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 19.Laemmli UK. Cleavage of structural proteins during assembly of head of bacteriophage-T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.Vidal LS, Kelly CL, Mordaka PM, Heap JT. Review of NAD(P)H dependent oxidoreductases: properties, engineering and application. BBA Prot Prot. 2018;1866:327–347. doi: 10.1016/j.bbapap.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Oppermann U, Filling C, Hult M, Shafqat N, Wu XQ, Lindh M, Shafqat J, Nordling E, Kallberg Y, Persson B, Jornvall H. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem Biol Interact. 2003;143:247–253. doi: 10.1016/S0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 22.Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol. 2014;5:172. doi: 10.3389/fmicb.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geertz-Hansen HM, Blom N, Feist AM, Brunak S, Petersen TN. Cofactory: sequence-based prediction of cofactor specificity of Rossmann folds. Prot Struct Func Bioinform. 2014;82:1819–1828. doi: 10.1002/prot.24536. [DOI] [PubMed] [Google Scholar]
- 24.Nishioka T, Yasutake Y, Nishiya Y, Tamura T. Structure-guided mutagenesis for the improvement of substrate specificity of Bacillus megaterium glucose-1-dehydrogenase IV. FEBS J. 2012;279:3264–3275. doi: 10.1111/j.1742-4658.2012.08713.x. [DOI] [PubMed] [Google Scholar]
- 25.Ding H, Gao F, Liu D, Li Z, Xu X, Wu M, Xhao Y. Significant improvement of thermostability of glucose-1-dehydrogenase by introducing disulfide bonds at the tetramer interface. Enzym Microb Technol. 2013;53:365–372. doi: 10.1016/j.enzmictec.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Ramaley RF, Vasantha N. Glycerol protection and purification of Bacillus subtilis glucose dehydrogenase. J Biol Chem. 1983;258:12558–12565. [PubMed] [Google Scholar]
- 27.Kumar A, Dhar K, Kanwar SS, Arora PK. Lipase catalysis in organic solvents: advantages and applications. Biol Proced Online. 2016;18:2. doi: 10.1186/s12575-016-0033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li BJ, Li YX, Bai DM, Zhang X, Yang HY, Wang J, Liu G, Yue JJ, Ling Y, Zhou DS, Chen HP. Whole-cell biotransformation systems for reduction of prochiral carbonyl compounds to chiral alcohol in Escherichia coli. Sci Rep. 2014;4:5. doi: 10.1038/srep06750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He YC, Tao ZC, Zhang X, Yang ZX, Xu JH. Highly efficient synthesis of ethyl (S)-4-chloro-3-hydroxybutanoate and its derivatives by a robust NADH-dependent reductase from E. coli CCZU-K14. Bioresour Technol. 2014;161:461–464. doi: 10.1016/j.biortech.2014.03.133. [DOI] [PubMed] [Google Scholar]
- 30.Jakoblinnert A, Mladenov R, Paul A, Sibilla F, Schwaneberg U, Ansorge-Schumacher MB, De María PD. Asymmetric reduction of ketones with recombinant E. coli whole cells in neat substrates. Chem Commun. 2011;47:12230–12232. doi: 10.1039/c1cc14097c. [DOI] [PubMed] [Google Scholar]
- 31.Dascier D, Kambourakis S, Hua L, Rozzell JD, Stewart JD. Influence of cofactor regeneration strategies on preparative scale, asymmetric carbonyl reductions by engineered Escherichia coli. Org Process Res Dev. 2014;18:793–800. doi: 10.1021/op400312n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.