

Abstract
Breast cancer patients often suffer from disease relapse and metastasis due to the presence of breast cancer stem-like cells (BCSCs). Numerous studies have reported that high levels of inflammatory factors, including tumor necrosis factor alpha (TNF-α), promote BCSCs. However, the mechanism by which TNF-α promotes BCSCs is unclear. In this study, we demonstrate that TNF-α up-regulates TAZ, a transcriptional co-activator promoting BCSC self-renewal capacity in human breast cancer cell lines. Depletion of TAZ abrogated the increase in BCSCs mediated by TNF-α. TAZ is induced by TNF-α through the non-canonical NF-κB pathway, and our findings suggest that TAZ plays a crucial role in inflammatory factor–promoted breast cancer stemness and could serve as a promising therapeutic target.
Subject terms: Cancer stem cells, Inflammation, Breast cancer
Introduction
Breast cancer is one of the most common malignances and a serious threat to women’s health worldwide1. Inflammation, especially chronic inflammation, plays an important role in cancer initiation and progression2. Tumor cells and a variety of leukocytes attracted by tumor cells produce various cytokines and chemokines that affect cancer development3. In general, cytokines are divided into two groups. One group comprises pro-inflammatory factors, including tumor necrosis factor alpha (TNF-α), IL1β, IL-6, etc4,5. The other group is made of anti-inflammatory factors, including IL-10, IL-13, etc5. High levels of pro-inflammatory cytokines promote tumor growth and migration, enhance the survival of malignant cells, suppress adaptive immune responses, and cause resistance to hormones and chemotherapeutic agents6,7. Non-steroidal anti-inflammatory drugs decrease the risk for developing and the risk of mortality in breast cancer3. Characterization of the mechanisms by which inflammatory cytokines promote breast cancer development may offer new therapeutic opportunities.
TNF-α is a well-documented pro-inflammatory cytokine that is up-regulated in breast cancer, and high levels of TNF-α are associated with breast cancer recurrence8,9. Additionally, TNF-α levels are positively correlated with tumor grade in serous ovarian tumors10. Moreover, TNF-α knockout mice are less susceptible to DMBA- or TPA-induced skin tumors8. TNF-α binds to two different receptors, TNF-α receptor 1 and 2 (TNFR1/2), to activate the NF-κB signaling pathway11,12. TNFR1/2 activates IKK and subsequently causes IκBα phosphorylation, ubiquitination, and degradation, leading to p65, RelB or p50 translocation to the nucleus. In addition to the canonical NF-κB pathway, TNF-α is able to activate JNK, MAPKs, AKT, and the non-canonical NF-κB pathway13–15. TNF-α up-regulates over 400 inflammatory genes, including cell-adhesion molecules, anti-apoptotic proteins, inflammatory cytokines, and chemokines16,17.
Ginalu Storci et al. reported that TNF-α increases the proportion of breast cancer stem-like cells (BCSCs) through NF-κB/HIF1α/Slug18. BCSCs are a small subpopulation of the primary breast tumor with differentiation and self-renewal capacities that are resistant to chemo- and radio-therapies19,20. Aldehyde dehydrogenases positivity and CD44high CD24low are generally considered two of the most frequently used identification makers of BCSCs21. It has been reported that CD44 positive/high expression is responsible for maintenance of multipotency, proliferation, and migration22, CD24 negative/low expression is responsible for cell growth and migration23, and ALDH1 positive/high expression is responsible for cell proliferation and stemness24. In addition, mammosphere formation can also identify breast cancer stem-like cells, which is based on the ability of BCSCs to propagate as multicellular spheroids in suspension culture. Currently, BCSCs have been implicated in breast cancer relapse and metastasis due to their resistance to chemo- and radiotherapies and tumorigenic properties25,26. However, there are currently no effective therapeutic strategies to specifically eliminate BCSCs.
TAZ, a transcriptional co-activator with a PDZ binding motif, has been implicated in sustaining BCSCs. A previous study demonstrated that TAZ confers self-renewal and tumor initiation capacity to non-BCSCs27. As a main effector of the Hippo pathway, TAZ interacts with TEADs to activate transcription of target genes, including CYR61, CTGF, and BIRC528–30. High TAZ expression is associated with low survival rates in breast cancer patients27. Our previous study suggested that TAZ depletion dramatically suppresses basal type breast cancer HCC1937 growth in vivo31. While regulation of TAZ at the post-translational modification level is becoming increasingly clear, knowledge of TAZ regulation at the transcriptional level is relatively limited. To date, four transcription factors, RelA32, HIF-133, SRF and MRTF34, have been reported to promote TAZ transcription.
Herein, we illustrate that TNF-α increases the percentage of BCSCs and TAZ expression levels in human breast cancer cell lines, and depletion of TAZ abrogates this phenotype. We further demonstrate that TAZ is induced by TNF-α through the non-canonical NF-κB pathway. Our findings indicate that inflammatory factors such as TNF-α increases stemness via up-regulation of TAZ transcription through non-canonical NF-κB pathway. We suggest that TAZ plays a crucial role in TNF-α–promoted breast cancer stemness and could serve as a promising therapeutic target.
Results
TNF-α increases breast cancer stem-like cells and up-regulates TAZ transcription in breast cancer cell lines
Two breast cancer cell lines, MCF7 and MDA-MB-468, were used in this study. MCF7 is ERα+ breast cancer cell line and MDA-MB-468 is triple-negative breast cancer (TNBC) cell line. It has been reported that TNBC cells are more like cancer stem cells (CSC) in terms of gene expression signature35.
TNF-α is well known as an important effector in breast cancer36. To confirm whether TNF-α promotes breast cancer stem-like cells, we cultured MCF7 cells under non-adherent culture conditions to foster mammosphere formation and found that TNF-α addition (10 ng/ml) significantly increased the number of mammospheres (Fig. 1A,B). We further performed ALDEFLUOR assays in MCF7 and MDA-MB-468 cell lines with or without TNF-α treatment and found that TNF-α significantly increased the population of ALDH positive cells in both MCF7 (Fig. 1C,D) and MDA-MB-468 (Fig. S1A,B) cell lines. We also tested the percentage of CD44+ and CD24− cells in these two cell lines. The percentage of CD44+ cells increased significantly in MCF7 (Figs. 1E and S1C). All MCF7 cells are CD24 positive, and all MDA-MB-468 cells are both CD44 and CD24 positive (Fig. S1C,D), as previously reported37,38. We found that CD24 was increased in MCF7 cells but decreased in MDA-MB-468 cells after TNF-α treatment (Fig. S1C,D).
Figure 1.

TNF-α increases BCSCs and TAZ expression in MCF-7 breast cancer cells. (A) TNF-α increases BCSCs in MCF7 cells, as measured by mammosphere culture. MCF7 cells were treated with 10 ng/ml TNF-α or 0.1% BSA for 48 h, and cells were seeded at a density of 1,000 cells per well and cultured for 14 d. The number of primary mammospheres with diameter ≥60 μm was then quantified. (B) The number of primary mammospheres per 1,000 cells initially seeded was quantified (mean ± SEM; n = 3). **P < 0.01, t-test. (C) TNF-α increases BCSCs in MCF7 cells as measured by ALDH assays. MCF7 cells were treated with 10 ng/ml TNF-α or 0.1% BSA for 48 h. Cells were collected for aldehyde dehydrogenase assays by FACS. (D) The percentage of ALDH + cells was quantified (mean ± SEM; n = 3). *P < 0.05, t-test. (E) TNF-α increases BCSCs in MCF7 as measured by CD marker staining. MCF7 cells were treated with 10 ng/ml TNF-α or 0.1% BSA for 48 h. The cells were collected for CD marker staining by FACS. The percentage of CD44 + cells was quantified (mean ± SEM; n = 3). **P < 0.01, t-test. (F) TNF-α induces TAZ protein expression in MCF7 cells. MCF7 cells were treated with 10 ng/ml TNF-α or 0.1% BSA for 24 h and 48 h, respectively. TAZ protein levels were measured by Western blotting. (G) TNF-α induces TAZ and CYR61 mRNA expression in MCF7 cells. TAZ and CYR61 mRNA levels were analyzed by RT-qPCR. Data are normalized to untreated samples (mean ± SEM; n = 3). **P < 0.01, t-test. NS indicates not significant.
Next, we demonstrated that TNF-α promotes protein expression of TAZ, a transcriptional coactivator necessary for self-renewal and tumor initiation in BCSCs27 in MCF7 (Fig. 1F). Similar results were observed in MDA-MB-468 cells (Fig. S1E). Because YAP shares a similar regulatory mechanism with TAZ in the Hippo pathway, we tested whether TNF-α also induces YAP. However, YAP was not induced by TNF-α in either MCF7 or MDA-MB-468 cells (Fig. S2C,D).
To characterize the potential mechanism by which TNF-α induces TAZ, we first measured TAZ mRNA levels by RT-qPCR. Our results showed that TNF-α significantly up-regulates mRNA levels of both TAZ and its target gene Cyr61 in both MCF7 (Fig. 1G) and MDA-MB-468 (Fig. S1D) cells. We also measured TAZ protein half-life and found that TNF-α does not affect TAZ protein stability (Fig. S2A,B). We concluded that TNF-α up-regulates TAZ expression predominately at the transcriptional level rather than the post-transcriptional level.
TAZ mediates TNF-α-increased the proportion of BCSCs
To explore whether TNF-α promotes BCSCs via up-regulation of TAZ, we knocked down TAZ using two individual siRNAs in MCF7 cells and assessed BCSC levels. TNF-α-induced mammosphere increase was completely abolished when TAZ was knocked down (Fig. 2A–C). In agreement with this, TAZ knockdown significantly blocked TNF-α-induced ALDH positive cell increase in MCF7 cells (Fig. 2D). Similar results were observed in MDA-MB-468 cells (Fig. S3A–C). TAZ knockdown also significantly decreased the TNF-α induced increase of CD44+ cells in MCF7 (Fig. S3D,E). TAZ knockdown did not blocked the TNF-α mediated the CD24 expression changes in both cell lines (Fig. S3G). These results indicate that TAZ may be necessary for TNF-α-increased the proportion of BCSCs.
Figure 2.

TAZ mediates TNF-α-increased the proportion of breast cancer stem-like cell. (A) TAZ depletion blocks TNF-α-promoted BCSC increase, as measured by mammosphere culture. MCF7 cells were transfected with 20 nM TAZ siRNA for 48 h and then exposed to 10 ng/ml TNF-α or 0.1% BSA for 48 h. (B) Quantitative data for panel A. **P < 0.01, t-test. NS indicates significant. (C) TAZ protein levels were knocked down using siTAZ1# and siTAZ3#. Protein expression was determined by WB. (D) TAZ depletion blocks TNF-α-promoted BCSC increase, as measured by ALDH assays. MCF7 cells were transfected with 20 nM TAZ siRNA for 48 h and then exposed to 10 ng/ml TNF-α or 0.1% BSA for 48 h. Cells were collected for ALDH assays by FACS. The percentage of ALDH+ cells was quantified (mean ± SEM; n = 3). *P < 0.05, t-test. NS indicates not significant.
TNF-α induces TAZ transcription through the non-canonical NF-κB pathway
TNF-α is a well-known activator of the canonical NF-κB pathway, and RelA regulates TAZ transcription in mesenchymal stem cells32. To further characterize the mechanism by which TNF-α induces TAZ transcription, we first tested whether RelA is responsible for TNF-α induction of TAZ transcription. After RelA knockdown, TAZ was still induced by TNF-α (Fig. 3A). Next, we knocked down other transcriptional factors in the canonical NF-κB pathway, including p105 and RelB. However, knockdown of neither p105 nor RelB suppressed TAZ induction by TNF-α (Fig. 3B,C). These results indicate that TNF-α may not induce TAZ transcription via the canonical NF-κB pathway.
Figure 3.

TNF-α induces TAZ not through RelA, RelB, and p105. (A) RelA knockdown did not block TNF-α induced TAZ protein expression in both MCF7 and MDA-MB-468. Cells were treated with TNF-α or 0.1% BSA for 48 h and TAZ and RelA proteins were detected by WB. (B) p105 knockdown did not block TNF-α induced TAZ protein expression in both MCF7 and MDA-MB-468. Cells were treated with TNF-α or 0.1% BSA for 48 h. The protein level of TAZ were detected by WB. The p105 knockdown was measured by RT-qPCR. (C) RelB knockdown did not block TNF-α induced TAZ protein expression in both MCF7 and MDA-MB-468. Cells were treated with TNF-α or 0.1% BSA for 48 h and TAZ and RelB protein was detection by WB.
Subsequently, we knocked down IKKα and found that this manipulation suppressed TNF-α-induced TAZ and CYR61 up-regulation (Figs. 4A and S4A). IKKα plays a crucial role in the non-canonical NF-κB pathway. Next, we knocked down transcription factor p100 in MCF7 and MDA-MB-468 cells and found that p100 depletion also inhibited TNF-α-induced TAZ and CYR61 up-regulation at both the protein and mRNA level (Figs. 4B,E and S4B,D). When the non-canonical NF-κB pathway is activated, p100 is processed through the proteasome39. We used MG132, a proteasome inhibitor, to pretreat cells before TNF-α stimulation. As expected, MG132 stabilized TAZ protein but suppressed TNFα-induced TAZ up-regulation in both MCF7 and MDA-MB-468 cells (Figs. 4C and S4C). We also pretreated cells with the IKKα inhibitor BAY11-7082 after TNF-α stimulation. Similar to MG132, BAY11-7082 inhibited TNF-α-induced TAZ and CYR61 increase in both MCF7 and MDA-MB-468 cells (Figs. 4D and S4E).Then we preformed ALDEFLUOR assays and found that IKKα silencing suppressed TNF-α-induced ALDH positive cell increase in both MCF7 and MDA-MB-468 cells (Fig. S4F,G). At the same time, we also detected CD44 and CD24 expression levels in these cells. Knockdown of either IKKα or p100 decreased the TNF-α induced increase of CD44+ cells in MCF7 (Fig. S4G,H). In MDA-MB-468 cells, IKKα and p100 knockdown had no significant effect on the expression of CD24 (Fig. S4G,I).
Figure 4.

TNF-α induces TAZ through the non-canonical NF-κB pathway. (A) IKKα knockdown blocks TNF-α induced TAZ and CYR61 protein expression in MCF7. Cells were treated with TNF-α or BSA for 48 h and TAZ and CYR61 proteins were detected by WB. Band intensities were quantatified by IMAGE J. (B) p100 knockdown blocks TNF-α induced TAZ and CYR61 protein expression in MCF7 cells. (C) MG132 blocks TNF-α induced TAZ and CYR61 protein expression in MCF7 cells. s.e. indicates the abbreviation of short exposure time. (D) BAY11-7082 blocks TNF-α induced TAZ and CYR61 protein expression in MCF7 cells. BAY11-7082 is an NF-κB inhibitor, which inhibits TNF-α and induces IκBα phosphorylation. MCF7 cells were treated with BAY11-7082 (2 μM) and then exposed to TNF-α. (E) IKKα knockdown, p100 knockdown, and MG132 block TNF-α induced TAZ and CYR61 mRNA expression in MCF7 cells. Cells were treated with TNF-α or BSA for 48 h. qPCR was performed to detect TAZ, p100, and CYR61 mRNA expression levels. **P < 0.01, t-test. (F) p100 knockdown blocks TNF-α-induced mammosphere increase in MCF7 cells. Cells were transfected with 20 nM p100 siRNA for 48 h and then exposed to 10 ng/ml or 0.1% BSA TNF-α for 48 h. (G) Quantitative data for panel F. **P < 0.01, t-test. p100 knockdown blocks TNF-α-induced p52 protein accumulation in MCF7 cells.
We next silenced p100 in MCF7 cells and found significant suppression of TNF-α-increased the proportion of mammosphere (Fig. 4F–G). Furthermore, TNF-α promoted not only p100 protein expression, but also its processing (Fig. 4B). p100 knockdown suppressed p100 processing into p52, as well as inhibiting TAZ and CYR61 induction by TNF-α (Fig. 4B). These results further support the notion that TNF-α up-regulates TAZ through the non-canonical NF-κB pathway.
p52 binds to the TAZ promoter to initiate TAZ transcription through TNF-α-induced p100 expression and processing
To further confirm that TNF-α induces TAZ through non-canonical NF-κB signaling, we treated MCF7 and MDA-MB-468 cells with TNF-α and examined p100 processing. We observed that p100 processing was enhanced with time as p52 gradually increased (Fig. 5A). Additionally, we tested whether RANKL, a typical non-canonical NF-κB pathway activator, can induce TAZ. Similar to TNF-α, RANKL also up-regulates TAZ and CYR61 through enhancing p100 processing in MDA-MB-468 cells (Fig. S5A). Furthermore, we performed the ALDEFLUOR assay and found that RANKL significantly increased ALDH positive cells in MDA-MB-468 (Fig. S5B). MCF7 cells are not sensitive to RANKL (100 ng/ml, data not shown).
Figure 5.

TNF-α induces generation of transcription factor p52 and TAZ transcription. (A) TNF-α induces generation of transcription factor p52 from p100 in both MCF7 and MDA-MB-468 cells. Cells were treated with 10 ng/ml TNF-α or 0.1% BSA for 12, 24 and 48 h. TAZ, CYR61 and p100/p52 protein expression levels were determined by WB. (B) The TAZ promoter sequence contains a p52 binding site, as highlighted in the shaded box. (C) TNF-α induces p52 binding to the TAZ promoter in both MCF7 and MDA-MB-468 cells, as determined by chromatin Immunoprecipitation (ChIP) assays. Cells were treated with vehicle (PBS) or TNF-α for 3 h. Immunoprecipitation was performed using anti-p52 antibody. PCR or qPCR were performed to quantify precipitated TAZ promoter using specific primers flanking the p52 binding site. (D).
Then, we tested whether p52 directly promotes TAZ transcription. We cloned the TAZ promoter region (−5000 to ATG) based on the Eukaryotic Promoter Database (http://epd.vital-it.ch/index.php) and searched potential p52 binding sites in the TAZ gene promoter region, identifying a p52 binding site from −861 to −851 (Fig. 5B). To determine whether p52 promotes TAZ transcription directly through this site, we performed a chromatin immunoprecipitation (ChIP) assay using an anti-p100/p52 antibody. As expected, the p52 binding site-containing region was immunoprecipitated by the antibody in both MCF7 and MDA-MB-468 cells. Furthermore, p52 binding was increased by TNF-α stimulation (Fig. 5C).
Discussion
TNF-α triggers immune reaction and promotes the initiation9,40,41, proliferation42,43, survival44, invasion45,46 and metastasis of tumor cells. However, the mechanism by which TNF-α promotes BCSCs has not been fully elucidated. In this study, we demonstrate that TNF-α increases BCSCs in MCF7 and MDA-MB-468 breast cancer cell lines through induction of TAZ (but not YAP) transcription. Furthermore, TNF-α induces TAZ expression through the non-canonical NF-κB pathway. Activated p100 is processed into p52 by the proteasome, at which time p52 forms homodimers or heterodimers with another transcriptional factor and translocate to the nucleus, binding to the p52 binding site at the TAZ promoter to induce TAZ transcription (Fig. 6). Our findings suggest that TAZ plays a crucial role in inflammatory factor TNF-α–increased BCSCs and could serve as a promising therapeutic target.
Figure 6.
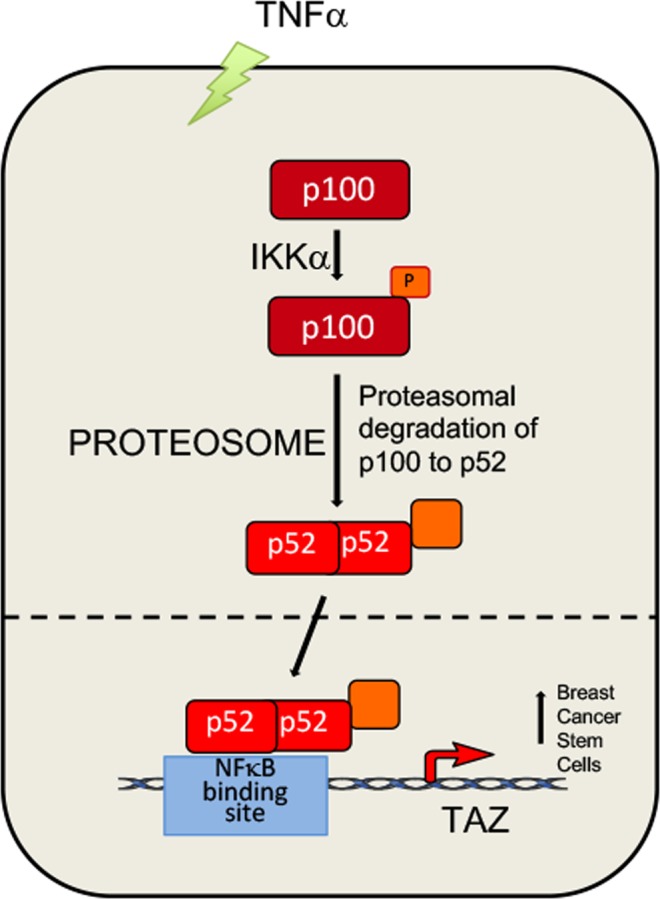
Schematic of the hypothesis that TNF-α increases BCSCs through up-regulation of TAZ expression via the non-canonical NF-κB pathway. TNF-α induces expression of p100 and activates p100 processing into p52 by IKKα and the proteasome. p52 subsequently binds to a p52 binding site within the TAZ promoter, subsequently up-regulating the latter’s transcription. TAZ in turn promotes BCSCs in human breast cancer cell lines.
For the first time, we discovered there is crosstalk between the non-classical NF-κB pathway and the Hippo pathway. Several lines of evidence support that TNF-α induces IKKα-mediated p100 processing into p52, which is translocated to the nucleus and binds to the TAZ promoter, inducing its transcription. First, TNF-α induces p100, TAZ, and its downstream target gene CYR61 expression at both the mRNA and protein level. Next, TNF-α increases expression of p52 that is processed from p100 by the proteasome, and knockdown of p100 or administration of the proteasome inhibitor MG132 abrogated induction of TAZ by TNF-α. Furthermore, knockdown of IKKα blocked the induction of TAZ and CYR61 although the exact functional mechanism of IKKα is unknown. On the one hand, IKKα could phosphorylate p100 to activate its procession. On the other hand, IKKα could phosphorylate H3 in the nucleus47 to promote TAZ transcription. Finally, RANKL, as a typical non-classical NF-κB pathway activator, also induced expression of TAZ and CYR61.
The correlation between inflammation and cancer has been confirmed by numerous studies. In 1863, Virchow hypothesized the association between cancer and inflammation48. To some extent, cancer is a kind of chronic inflammation, and the inflammatory environment supports the development of cancer. In 2008, Frances Balkwill and Alberto Mantovani reported that there were various similarities between tumor and chronic inflammation in the microenvironment49. Both immunocytes and inflammatory factors play important roles in tumor development and immunosuppression. TNF-α, a well characterized inflammatory factor, has strong tumor-initiating effects at a low and constant dose. In animal models, blockage of TNF-α and its receptor are strongly antitumor49.
It appears that TNF-α increases BCSCs through different mechanisms. In 2010, TNF-α was reported to induce Slug expression by up-regulating HIF1α via the canonical NF-κB pathway, thus promoting BCSCs18. In 2012, Li reported that TNF-α up-regulates Twist1 and induces epithelial-mesenchymal transition via the NF-κB pathway50. In normal mammary epithelial cells, TNF-α expression levels are very low, whereas TNF-α is over-expressed in most breast tumors18. In addition, TNF-α expression levels are significantly positively correlated with recurrence and malignance in these breast cancer patients. In this study under low dose, long-term stimulation, TNF-α increases BCSCs in both MCF7 and MDA-MB-468 cell lines. Importantly, the mechanism of TNF-α-induced BCSCs appears to occur through up-regulation of TAZ expression via the non-canonical NF-κB pathway. TAZ is necessary for TNF-α to increase BCSCs. It has been shown that activated TAZS89A is sufficient to endow CSC-like properties to non-CSCs27.
TAZ is an important transcriptional coactivator in the Hippo pathway18. High expression levels of TAZ are positively correlated with occurrence and development of cancer51,52. TAZ is tightly regulated in response to a wide range of extracellular and intrinsic signals53. Estrogen was reported to activate TAZ through G protein-coupled estrogen receptor 1 (GPER, also known as GPR30) and re-organization of F-actin cytoskeleton54. Transcriptional regulation of TAZ has not been well studied. It is reported that up-regulation of TAZ expression occurs during osteogenic differentiation of stromal cells derived from human adipose tissue under hypoxic stress conditions32. During bone differentiation of human adipose tissue, TNF-α initiates transcription of TAZ by activating the classical NF-κB pathway, which induces p65 translocation to the nucleus, where it binds to the TAZ promoter. Under hypoxic conditions, HIF-1 directly binds to the HRE site between the second and third exons of TAZ and promotes TAZ mRNA transcription of various breast cancer cell lines33. Additionally, the survival rate of breast cancer is negatively correlated with concurrent high-expression of TAZ and HIF-133. Moreover, MRTF/SRF was reported to induce TAZ transcription in breast cancer cells in response to Heregulin β134. We found that the p52 transcription factor, part of the non-canonical NF-κB pathway55, directly induces TAZ transcription in both MCF7 and MDA-MB-468 breast cancer cells in response to TNF-α. Taken together, multiple transcription factors, including HIF-1, SRF, p65, and p52, promote TAZ gene transcription in different contexts.
Several questions remain to be addressed in this study. First, since p52 only contains a DNA binding domain but not a transcription activation domain, p52 must be integrated with other factors that have transcriptional activation functionality to form heterodimers. It has been reported that p52 forms heterodimers with RelB or Bcl3 to initiate expression of target genes. In this study, RelB depletion did not influence expression of TAZ induced by TNF-α. Therefore, p52 may activate expression of TAZ by integrating with other distinct transcription factors. Further investigation is required to identify p52 transcriptional factor partners for TAZ induction. In addition, TNF-α significantly induces the expression of p100 in several breast cancer cell lines; however, the molecular mechanism for this is unclear.
In conclusion, we demonstrate that TNF-α launches the cells through a stemness differentiation via up-regulation of TAZ transcription through a non-canonical NF-κB pathway in human breast cancer cell lines. Our findings implicate TAZ as a crucial component in inflammatory factor-promoted breast cancer cells stemness differentiation and suggest that TAZ could serve as a promising therapeutic target in breast cancer.
Methods
Cell culture
Human breast cancer cell lines MCF7 and MDA-MB-468 were purchased from American Type Culture Collection (ATCC). These cell lines have been authenticated with STR assays in 2015 by Conservation Genetics CAS Kunming Cell Bank. These cells were cultured in MEM medium supplemented with 10% fetal bovine serum. All cells were maintained at 37 °C in an incubator with 5% CO2. The presence of mycoplasma was routinely tested by PCR to eliminate contamination.
Drugs, reagents and antibodies
TNF-α was purchased from PEPRO TECH (Rocky Hill, USA). RANKL was purchased from R&D Systems (Minneapolis, MN). Cycloheximide (CHX) was purchased from MP Biomedicals (Irvine, CA, USA). BAY 11-7082, LPS and MG132 were purchased from Sigma-Aldrich (St Louis, MO, USA). The anti-TAZ V386 (25937), anti-CYR61 (3491), anti-p100/p52 (4882), anti-RelA (8242) and anti-RelB (4954) antibodies used for WB were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-tubulin (F7425) and anti-YAP (WH0010413 M1) antibodies were purchased from Sigma-Aldrich. Anti-GAPDH (sc-25778) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-p100/p52 (ab7972) antibody used for ChIP assays was purchased from Abcam (Cambridge, MA).
ALDEFLUOR assay
We performed ALDH assays using an ALDEFLUOR Assay Kit (no. 01700; Stem cell Technologies, Vancouver, BC, Canada) according to the standard protocol. In brief, 25,000 cells were collected and resuspended in 1 ml assay buffer. Next, 5 μl activated reagent was added. Half of the samples (0.5 ml) were immediately put into control tubes with 5 μl DEAB buffer. All samples were incubated for 40 min at 37 °C, after which time they were centrifuged for 5 min at 250 g. Cells were resuspended in 0.5 ml assay buffer and subjected to flow cytometry analysis.
CD marker staining assays
1 × 106 cells were collected and resuspended in 1 ml 2% fetal bovine serum PBS. Combinations of fluorochrome-conjugated monoclonal antibodies against human CD44 (FITC; cat. #555478) and CD24 (PE; cat. #555428) were obtained from BD Biosciences (San Diego, California, USA). Primary antibodies or the respective isotype controls (BD Biosciences) were added to the cell suspension and incubated at 4 °C in the dark for 30 min. The cells were washed with clod PBS for three times, were resuspended in 0.5 ml 2% fetal bovine serum PBS and were subjected to flow cytometry analysis.
Mammosphere culture
We performed mammosphere assays using a Mammosphere Culture Kit (no. 05620; Stem cell Technologies). MCF7 cells were first treated with TNF-α for 48 hours and then plated in ultra-low attachment plates (no. 3473, Corning Inc., Corning, NY, USA) at a density of 500 cells per well. The cells were cultured in complete MammoCult™ Medium (Basal Medium + Proliferation Supplement + heparin + hydrocortisone) without TNF-α. Preparation of the complete MammoCult™ Medium was carried out according to the method instructions in Mammosphere Culture Kit (no. 05620; Stem cell Technologies). The number of mammospheres with a diameter >60 μm after 14 days in culture was then quantified.
Transfection
We used Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for siRNA transfection according to the manufacturer’s recommended protocols. Control siRNA was purchased from RiboBio Co., Ltd. (Guangzhou, Guangdong, China). Other siRNA sequences are listed in Supplementary Table S1.
RT-qPCR
RNA was extracted using TRIzol reagent (Invitrogen Corp., Carlsbad, CA). Reverse transcription was performed using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA), and RNA levels were quantified using SYBR Green Select Mastermix (no. 4472908, Applied Biosystems, Foster, CA, USA) on the ABI-7900HT System (Applied Biosystems). Primer sequences are listed in Supplementary Table S2.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed using MCF7 and MDA-MB-468 cells, which were cultured in 10-cm dishes and treated with TNF-α (10 ng/ml) for 48 h. Cells were then treated with formaldehyde for crosslinking. Cells were scraped into Eppendorf tubes and centrifuged at 1000 g, 4 °C for 10 min. Cell lysates were sonicated to shear DNA to an average fragment size of 150–500 bp. After sonication, cell debris were removed by centrifugation at 10,000 g at 4 °C for 1 min. DNA-protein complexes were mixed with the antibody-A/G-beads and incubated at 4 °C for 10 h. Chromosomal DNA was purified and analyzed by quantitative PCR. Primers for the TAZ gene promoter p52 binding site were as follows: 5′-TCTACTTCCAGCCACCTGC-3′ (forward) and 5′-GCAACATCCGTGAGGGTTG-3′ (reverse).
Supplementary information
Acknowledgements
This study was supported in part by grants from the National Nature Science Foundation of China (81830087, U1602221 and 31771516 to Chen, C., 81672639 to Zhou, Z., 81802671 and 81872414 to Jiang, D.), Yunnan Fundamental Research Projects (2019FB112 to Jiang, D.) and Diagnosis, Treatment and Transformation Engineering Technology Research Center for Renal Cell Carcinoma of Yunnan Province (2016DNH001 to Li, W.).
Author contributions
J.D., M.X. and C.C. designed the experiments and provided financial support. L.W., L.X. and S.P. carried out most of the experiments and analyzed the data. Y.G., Z.Z. and L.W. provided technical support. L.W. and C.C. wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Wenjing Liu, Xiaoqing Lu and Peiguo Shi.
Contributor Information
Xiaoyun Mao, Email: maoxiaoyun@126.com.
Dewei Jiang, Email: jiangdewei@mail.kiz.ac.cn.
Ceshi Chen, Email: chenc@mail.kiz.ac.cn.
Supplementary information
is available for this paper at 10.1038/s41598-020-58642-y.
References
- 1.Kyu HH, et al. Physical activity and risk of breast cancer, colon cancer, diabetes, ischemic heart disease, and ischemic stroke events: systematic review and dose-response meta-analysis for the Global Burden of Disease Study 2013. Bmj. 2016;354:i3857. doi: 10.1136/bmj.i3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balkwill F, Coussens LM. An inflammatory link. Nat. 2004;431:405. doi: 10.1038/431405a. [DOI] [PubMed] [Google Scholar]
- 4.Sethi G, Sung B, Aggarwal BB. TNF: a master switch for inflammation to cancer. Front. bioscience: a J. virtual library. 2008;13:5094–5107. doi: 10.2741/3066. [DOI] [PubMed] [Google Scholar]
- 5.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Zhu P, et al. Macrophage/Cancer Cell Interactions Mediate Hormone Resistance by a Nuclear Receptor Derepression Pathway. Cell. 2006;124:615–629. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 7.Naugler WE, et al. Gender Disparity in Liver Cancer Due to Sex Differences in MyD88-Dependent IL-6 Production. Sci. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 8.Moore RJ, et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med. 1999;5:828–831. doi: 10.1038/10552. [DOI] [PubMed] [Google Scholar]
- 9.Aggarwal BB, et al. TNF blockade: an inflammatory issue. Ernst Scher. Res. Found. Workshop. 2006;56:161–186. doi: 10.1007/3-540-37673-9_10. [DOI] [PubMed] [Google Scholar]
- 10.Maylor, M. Tumor necrosis factor and its receptors in human ovarian cancer. Journal of Clinical Investigation91 (1993). [DOI] [PMC free article] [PubMed]
- 11.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat. reviews. Immunology. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 12.Aggarwal BB, Shishodia S, Ashikawa K, Bharti AC. The role of TNF and its family members in inflammation and cancer: lessons from gene deletion. Curr. drug. targets. Inflamm. allergy. 2002;1:327–341. doi: 10.2174/1568010023344571. [DOI] [PubMed] [Google Scholar]
- 13.Darnay BG, Aggarwal BB. Early events in TNF signaling: a story of associations and dissociations. J. Leukoc. Biol. 1997;61:559–566. doi: 10.1002/jlb.61.5.559. [DOI] [PubMed] [Google Scholar]
- 14.Bhardwaj A, Aggarwal BB. Receptor-mediated choreography of life and death. J. Clin. Immunol. 2003;23:317–332. doi: 10.1023/A:1025319031417. [DOI] [PubMed] [Google Scholar]
- 15.Aggarwal BB, Takada Y. Pro-apototic and anti-apoptotic effects of tumor necrosis factor in tumor cells. Role of nuclear transcription factor NF-kappaB. Cancer Treat. Res. 2005;126:103–127. doi: 10.1007/0-387-24361-5_5. [DOI] [PubMed] [Google Scholar]
- 16.Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Yadav VR, Prasad S, Sung B, Kannappan R, Aggarwal BB. Targeting inflammatory pathways by triterpenoids for prevention and treatment of cancer. Toxins. 2010;2:2428–2466. doi: 10.3390/toxins2102428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Storci G, et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J. Cell. Physiol. 2010;225:682–691. doi: 10.1002/jcp.22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owens, T. & Naylor, M. Breast cancer stem cells. Frontiers in Physiology4, 10.3389/fphys.2013.00225 (2013). [DOI] [PMC free article] [PubMed]
- 20.Conley SJ, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl Acad. Sci. U S Am. 2012;109:2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kao J, et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One. 2009;4:e6146. doi: 10.1371/journal.pone.0006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nam K, Oh S, Lee KM, Yoo SA, Shin I. CD44 regulates cell proliferation, migration, and invasion via modulation of c-Src transcription in human breast cancer cells. Cell. Signal. 2015;27:1882–1894. doi: 10.1016/j.cellsig.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Schabath H, Runz S, Joumaa S, Altevogt P. CD24 affects CXCR4 function in pre-B lymphocytes and breast carcinoma cells. J. Cell. Sci. 2006;119:314–325. doi: 10.1242/jcs.02741. [DOI] [PubMed] [Google Scholar]
- 24.Giovannelli P, et al. Breast cancer stem cells: The role of sex steroid receptors. World J. Stem Cell. 2019;11:594–603. doi: 10.4252/wjsc.v11.i9.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu F, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 26.Zielske SP, Spalding AC, Wicha MS, Lawrence TS. Ablation of breast cancer stem cells with radiation. Transl. Oncol. 2011;4:227–233. doi: 10.1593/tlo.10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cordenonsi M, et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147:759–772. doi: 10.1016/j.cell.2011.09.048. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem. 2009;284:13355–13362. doi: 10.1074/jbc.M900843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai D, Ho KC, Hao Y, Yang X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011;71:2728–2738. doi: 10.1158/0008-5472.CAN-10-2711. [DOI] [PubMed] [Google Scholar]
- 30.Babic AM, Kireeva ML, Kolesnikova TV, Lau LF. CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc. Natl Acad. Sci. 1998;95:6355–6360. doi: 10.1073/pnas.95.11.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao D, Zhi X, Zhou Z, Chen C. TAZ antagonizes the WWP1-mediated KLF5 degradation and promotes breast cell proliferation and tumorigenesis. Carcinogenesis. 2012;33:59–67. doi: 10.1093/carcin/bgr242. [DOI] [PubMed] [Google Scholar]
- 32.Cho HH, et al. NF-kappaB activation stimulates osteogenic differentiation of mesenchymal stem cells derived from human adipose tissue by increasing TAZ expression. J. Cell Physiol. 2010;223:168–177. doi: 10.1002/jcp.22024. [DOI] [PubMed] [Google Scholar]
- 33.Xiang L, et al. Hypoxia-inducible factor 1 mediates TAZ expression and nuclear localization to induce the breast cancer stem cell phenotype. Oncotarget. 2014;5:12509–12527. doi: 10.18632/oncotarget.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu CY, et al. MRTF/SRF dependent transcriptional regulation of TAZ in breast cancer cells. Oncotarget. 2016;7:13706–13716. doi: 10.18632/oncotarget.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park, Choi, Nam Targeting Cancer Stem Cells in Triple-Negative Breast Cancer. Cancers. 2019;11(7):965. doi: 10.3390/cancers11070965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soria G, et al. Inflammatory mediators in breast cancer: Coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer. 2011;11:130–130. doi: 10.1186/1471-2407-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ricardo S, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011;64:937–946. doi: 10.1136/jcp.2011.090456. [DOI] [PubMed] [Google Scholar]
- 38.Maycotte Paola, Jones Kenneth L., Goodall Megan L., Thorburn Jacqueline, Thorburn Andrew. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Molecular Cancer Research. 2015;13(4):651–658. doi: 10.1158/1541-7786.MCR-14-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilmore TD. Introduction to NF-κB: players, pathways, perspectives. Oncogene. 2006;25:6680. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 40.Ashikawa, K. The role of TNF and its family members in inflammation and cancer: lessons from gene deletion. Current Drug Targets - Inflammation & Allergy1, - (2002). [DOI] [PubMed]
- 41.Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: How hot is the link? Biochemical Pharmacology. 2006;72:1605–1621. doi: 10.1016/j.bcp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 42.Goillot E, et al. Tumor Necrosis Factor as an Autocrine Growth Factor for Neuroblastoma. Cancer Res. 1992;52:3194. [PubMed] [Google Scholar]
- 43.Tsukasaki K, et al. Tumor necrosis factor alpha polymorphism associated with increased susceptibility to development of adult T-cell leukemia/lymphoma in human T-lymphotropic virus type 1 carriers. Cancer Res. 2001;61:3770–3774. [PubMed] [Google Scholar]
- 44.Montesano R, Soulié P, Eble JA, Carrozzino F. Tumour necrosis factor alpha confers an invasive, transformed phenotype on mammary epithelial cells. J. Cell. Sci. 2005;118:3487. doi: 10.1242/jcs.02467. [DOI] [PubMed] [Google Scholar]
- 45.Nabors LB, et al. Tumor necrosis factor alpha induces angiogenic factor up-regulation in malignant glioma cells: a role for RNA stabilization and HuR. Cancer Res. 2003;63:4181. [PubMed] [Google Scholar]
- 46.Yoshida S, et al. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol. Cell. Biol. 1997;17:4015–4023. doi: 10.1128/MCB.17.7.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nat. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- 48.Casbon AJ, Lohela M, Plaks V, Werb Z. Abstract IA24: How tumors regulate their microenvironment in primary sites and metastasis. Mol. Cancer Res. 2013;11:IA24–IA24. doi: 10.1158/1557-3125.Advbc-ia24. [DOI] [Google Scholar]
- 49.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nat. 2008;454:436. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 50.Li CW, et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012;72:1290–1300. doi: 10.1158/0008-5472.Can-11-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29:783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moroishi T, Hansen CG, Guan K-L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer. 2015;15:73. doi: 10.1038/nrc3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou X, Lei QY. Regulation of TAZ in cancer. Protein Cell. 2016;7:548–561. doi: 10.1007/s13238-016-0288-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z, et al. GPER stabilizes F-actin cytoskeleton and activates TAZ via PLCbeta-PKC and Rho/ROCK-LIMK-Cofilin pathway. Biochem. Biophys. Res. Commun. 2019;516:976–982. doi: 10.1016/j.bbrc.2019.06.132. [DOI] [PubMed] [Google Scholar]
- 55.Miranda MZ, et al. TGF-beta1 regulates the expression and transcriptional activity of TAZ protein via a Smad3-independent, myocardin-related transcription factor-mediated mechanism. J. Biol. Chem. 2017;292:14902–14920. doi: 10.1074/jbc.M117.780502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.