

Visual Abstract

Key Words: fibrosis, Nox2, reactive oxygen species, stenosis, tripterine, valve interstitial cells
Abbreviations and Acronyms: AV, aortic valve; AVIC, aortic valvular interstitial cell; CAVD, calcific aortic valve disease; GSK3B, glycogen synthase kinase 3 beta; HC, high cholesterol; LV, left ventricular; Nox2, reduced nicotinamide adenine dinucleotide phosphate oxidase 2; OGM, osteogenic medium; OPN, osteopontin; ROS, reactive oxygen species; Runx2, runt-related transcription factor 2; vitD2, vitamin D2
Highlights
-
•
The reactive oxygen species–generating enzyme Nox2 is up-regulated in the leaflets of both rabbit and human with CAVD.
-
•
Nox2 is markedly induced in cultured porcine AVICs after osteogenic stimulation. Knockdown of endogenous Nox2 substantially suppressed AVIC calcification.
-
•
Celastrol, a natural compound capable of inhibiting Nox2 activity, significantly decreased AVIC calcification in vitro, and mitigated the severity of aortic valve fibrosis, calcification, and stenosis in a rabbit model of CAVD in vivo.
-
•
The protective effects of celastrol may, in part, involve the inhibition of Nox2-mediated glycogen synthase kinase 3 beta/β-catenin pathway.
Summary
This study sought to investigate whether reactive oxygen species (ROS)–generating reduced nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2) contributes to calcific aortic valve disease (CAVD) or whether celastrol, a natural Nox2 inhibitor, may provide potential therapeutic target for CAVD. CAVD is an active and cellular-driven fibrocalcific process characterized by differentiation of aortic valvular interstitial cells (AVICs) toward an osteogenic-like phenotype. ROS levels increase in calcified aortic valves, while the sources of ROS and their roles in the pathogenesis of CAVD are elusive. The roles of Nox2 and the effects of celastrol were studied using cultured porcine AVICs in vitro and a rabbit CAVD model in vivo. Nox2 proteins were significantly upregulated in human aortic valves with CAVD. In vitro, Nox2 was markedly induced upon stimulation of AVICs with osteogenic medium, along with the increases in ROS production and calcium nodule formation. Celastrol significantly decreased calcium deposition of AVICs by 35%, with a reduction of ROS generation. Knockdown of endogenous Nox2 substantially suppressed AVIC calcification by 39%, the inhibitory effect being similar to celastrol treatment. Mechanistically, either celastrol treatment or knockdown of Nox2 significantly inhibited glycogen synthase kinase 3 beta/β-catenin signaling, leading to attenuation of fibrogenic and osteogenic responses of AVICs. In a rabbit CAVD model, administration of celastrol significantly reduced aortic valve ROS production, fibrosis, calcification, and severity of aortic stenosis, with less left ventricular dilatation and better preserved contractile function. Upregulation of Nox2 is critically involved in CAVD. Celastrol is effective to alleviate CAVD, likely through the inhibition of Nox2-mediated glycogen synthase kinase 3 beta/β-catenin pathway in AVICs.
Calcific aortic valve disease (CAVD) is currently the third most prevalent cardiovascular disease after coronary artery disease and hypertension, affecting up to 25% of the population over the 65 years of age in developed countries (1). In CAVD, the leaflets become thickened, fibrosed, stiffened, and calcified, resulting in valve sclerosis and progressive aortic stenosis, in which hemodynamic obstruction leads to cardiac hypertrophy and eventually heart failure, if untreated, with significant morbidity and mortality (2). To date, aortic valve (AV) replacement or implantation is the only viable clinical option with no long-life guarantee, and there are no effective drug interventions to delay or retard its progression (3). There is, therefore, a major unmet clinical need to identify other therapies capable of treating this disease.
It has been widely accepted that CAVD is an active and cellular-driven fibrocalcific disease, rather than a passive degeneration and inexorable consequence of aging (4). At the microstructural level, a hallmark of CAVD is extensive fibrotic collagen accumulation and the presence of calcium-rich nodules on the valve surface and within the annulus region. Although the fibrotic and calcific mechanisms underlining CAVD are not fully understood, an increasing evidence from in vivo and in vitro studies suggests that activation of aortic valvular interstitial cells (AVICs) and their transdifferentiation into osteoblastic-like cells appears to be a central step in the disease initiation and development (5). This phenotypic switch of quiescent AVICs to osteogenic myofibroblasts is supported by gene-profiling findings of increased valvular expression of osteoblast-specific marker proteins, such as osteopontin (OPN), and transcription factor, runt-related transcription factor 2 (Runx2), which is essential for osteoblastic differentiation (1). A number of molecules and signaling pathways have been identified to be critical in the regulation of AVIC differentiation, including the Notch (6) and Wnt/β-catenin pathways (7).
The pathogenesis of CAVD is complex while previous studies suggest that reactive oxygen species (ROS) may be implicated in the cellular and extracellular alternations during the process of CAVD. Evidence of increased ROS generation and decreased antioxidants has been shown both in experimental animal models of CAVD and in calcified region of human stenotic valves (8, 9, 10, 11). The findings that increases in ROS precede AV dysfunction in a mouse CAVD model suggest that oxidative stress may be causative, not merely the readout of valve calcification and dysfunction (8). Moreover, exogenous ROS promotes calcification of AVICs by activating profibrotic and pro-osteogenic signaling such as Runx2 (11), strongly indicating that oxidative stress may precede the transdifferentiation of AVICs to an osteoblastic phenotype. However, the sources of ROS and their roles in CAVD remain elusive.
Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2) is a member of the Nox family proteins, which are major ROS sources in cardiovascular system (12). Nox enzymes produce ROS as their primary function and are therefore especially important in redox signaling. Nox2 is widely expressed in cardiac and vascular cells and is acutely activated by agonists, metabolic factors, or mechanical forces in a process that involves association with p47phox, p67phox, p40phox, and Rac1 cytosolic subunits. A large number of studies indicate that Nox2 is a pivotal player in the pathogenesis of diverse cardiovascular diseases including cardiac hypertrophy, fibrosis and remodeling (13), as well as inflammation, metabolic disorders, and atherosclerosis (14). Interestingly, it was reported that Nox2 might be involved in vascular calcification (15,16). A few studies showed that the expression levels of Nox2 were altered in calcified aortic valves (9,17). It is unknown, however, whether Nox2 contributes to CAVD or whether its involvement may provide a potential therapeutic target in the development of CAVD.
Celastrol, a pentacyclic triterpene naturally extracted from the roots of the Chinese Thunder God wine Tripterygium wilflordii, has long been used for the treatment of cancer, neurodegeneration and autoimmune diseases (18). Celastrol was recently identified as a leptin sensitizer and potential novel antiobesity drug (19,20). Celastrol also exhibits protection against inflammation (21), fibrosis (22), metabolic disorders (23), and atherosclerosis (24), all being risk factors of CAVD and characterized by an increase in ROS production. Of note, celastrol may block ROS generation as a potent Nox inhibitor with higher potency against Nox2 (25). However, whether celastrol has any beneficial effect on CAVD has not been investigated.
Here, we explore the hypothesis that Nox2 intrinsically contributes to the development of CAVD. Using cultured AVICs in vitro and a rabbit CAVD model in vivo, we found that celastrol inhibits Nox2-mediated glycogen synthase kinase 3 beta (GSK3B)/β-catenin pathway in AVICs, such that AV fibrocalcification and stenosis are alleviated and cardiac function is improved.
Methods
Detailed methods are provided in the Supplemental Appendix.
The control noncalcified AVs and tricuspid calcified AVs were explanted from patients undergoing valve replacement surgery.
Primary porcine AVICs were isolated and cultured. To induce calcification, cells were incubated with osteogenic medium (OGM). AVICs were transfected with adenoviral vectors expressing a short hairpin sequence targeted against Nox2, human Nox2 (26), green fluorescent protein (GFP), or β-galactosidase (27).
Animal experiments were conducted in accordance with institutional and national standards. Male New Zealand White rabbits were fed 0.5% cholesterol-enriched chow plus 25,000 IU/day vitamin D2 (vitD2) in drinking water (28), with or without celastrol treatment (1 mg/kg/day) for 18 weeks. Cardiac and AV function were assessed by echocardiography.
Data are mean ± SEM. Comparisons were made by unpaired Student's t-test or 1- or 2-way analysis of variance as appropriate followed by Tukey's post hoc test for all pairwise group comparisons using a familywise error rate. Correlations between continuous variables were assessed using linear regression models with goodness of fit evaluated using plots and coefficients of determination (R2). Analyses were performed on GraphPad Prism 8.0.0 for Windows (GraphPad Software, San Diego, California). A p value <0.05 is considered statistically significant.
Results
Nox2 increases in human AVs with CAVD
We first examined the changes of Nox2 expression in human AVs with CAVD. In normal valves, the expression of Nox2 was relatively low as revealed by immunohistology (Figure 1A). By contrast, the diseased valves with CAVD exhibited intense Nox2 accumulation around the calcified region throughout the valves (Figure 1A). Quantification by Western blots showed that the levels of Nox2 were significantly upregulated around 2.6-fold in CAVD compared with noncalcification valves, accompanying with higher expression of calcific marker Runx2 (Figure 1B). Moreover, the protein levels of Nox2 were positively correlated with Runx2 proteins as a surrogate marker of the severity of AV calcification (Figure 1C).
Figure 1.
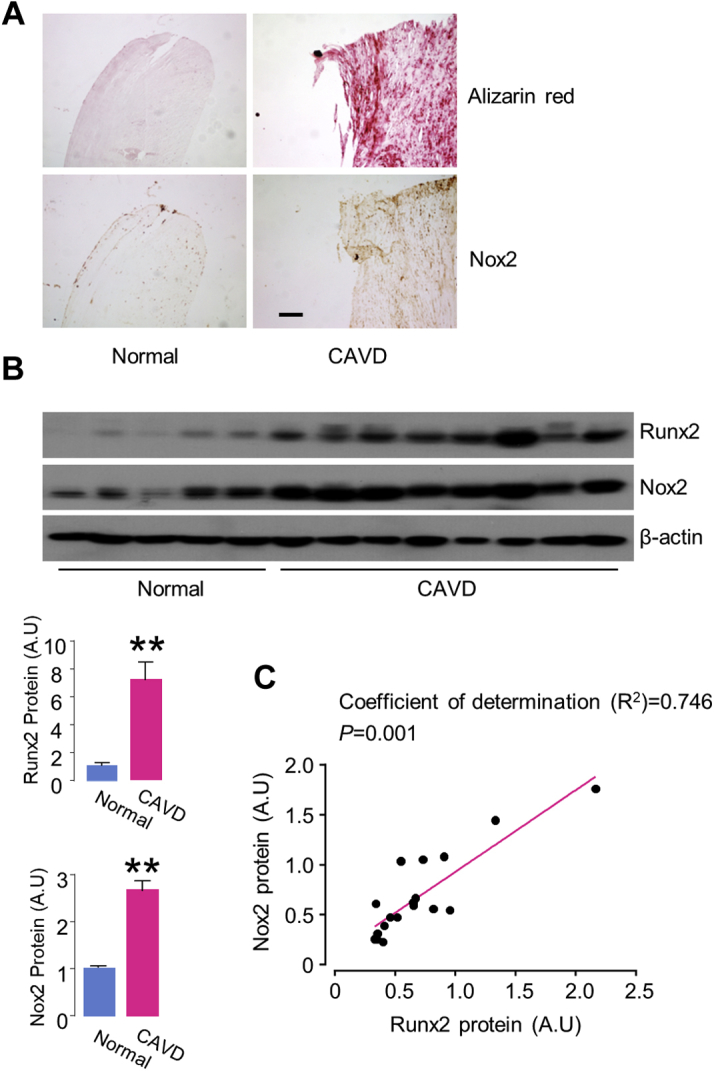
Upregulation of Nox2 Levels in Human Aortic Valve Tissue With CAVD
(A) Alizarin red staining for calcified valve leaflet and immunohistochemistry for Nox2 expression. Scale bars: 100 μm. (B) Immunoblots (upper) and relative quantification (lower) of calcification marker runt-related transcription factor 2 (Runx2) and reduced nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2) proteins. n = 5–8/group. **p < 0.01, compared with noncalcific valves, Student's unpaired t-test. All data are mean ± SEM. (C) Correlation of protein levels of Nox2 and Runx2 in aortic valve tissues with calcified aortic valve disease (CAVD). n = 18. The coefficients of determination (R2) from linear regression analysis and p value are shown on the graph. A.U = arbitrary units.
Nox2 up-regulation is associated with osteoblast differentiation of AVICs
Next, we investigate whether Nox2 up-regulation contributes to CAVD. As a phenotype transition of AVICs from fibroblast-like to osteoblast-like cells plays a major role in the development of CAVD, we isolated and cultured porcine AVICs, and investigated whether Nox2 expression was altered during the process of osteoblastic differentiation of AVICs in vitro. As shown in Figures 2A and 2C, compared with control cells cultured in Dulbecco’s modified Eagle medium, 14 days treatment of OGM resulted in markedly calcification of AVICs, as revealed by dramatic formation of calcium nodules with Alizarin red staining. Interestingly, Nox2 expression was significantly increased in AVICs by immunofluorescence staining after calcific induction (Figure 2A). The time-course changes of Nox2 proteins were further examined by Western blots. The results showed that Nox2 was markedly induced upon stimulation of AVICs with OGM for 7 days, and this up-regulation was maintained up to 2 weeks (Figures 2B and 2D). Importantly, this change profiling of Nox2 protein was paralleled with increases in a typical osteogenic marker OPN and classic calcification regulator Runx2 (Figures 2B and 2D), indicating that Nox2 up-regulation is associated with osteoblast transdifferentiation of AVICs.
Figure 2.

Nox2 Up-Regulation During AVIC Calcification
(A) Representative images of Alizarin red staining for calcium deposition (upper right) and Nox2 up-regulation in aortic valvular interstitial cell (AVIC) detected by immunofluorescence (lower right). Scale bar: 100 μm. (B) Western blots showing time-course changes of protein levels of osteogenic markers osteopontin (OPN), Runx2, and Nox2 in AVICs. (C) Quantification of calcium nodules formation of AVICs stimulated by osteogenic medium for 14 days. n = 5/group. (D) Mean data of protein immunoblots. n = 5/group. *p < 0.05, **p < 0.01 compared with respective control animals, Student's unpaired t-test. All data are mean ± SEM. Cal = calcification; Ctl = control; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; other abbreviations as in Figure 1.
Celastrol has an antiosteogenic effect in AVICs via inhibition of Nox2
We then evaluated the effect of celastrol on cultured AVIC calcification in vitro. Incubation of 10 nmol/l celastrol had no basal effect on AVIC growth and viability (Supplemental Figure 1A). However, compared with AVICs stimulated by OGM, treatment with celastrol significantly inhibited calcium deposition of AVICs by 35% (Figure 3A), together with significant decrease in Runx2 protein levels (Figure 3C). The attenuation of AVIC calcification by 10 nmol/l celastrol was accompanying with effective and significant reduction of ROS generation, as evaluated by dihydroethidium staining in situ (Figure 3B, Supplemental Figures 1B and 1C). However, simply inhibition of ROS with polyethylene glycol superoxide dismutase (SOD) did not attenuate, but rather actually promoted AVIC calcification (Supplemental Figure 2). This result suggests that the antiosteogenic effect of celastrol is likely achieved by specifically targeting ROS-generator Nox2 rather than nonspecific ROS inhibition. Indeed, the calcification-induced up-regulation of Nox2 was significantly diminished by celastrol treatment (Figure 3C).
Figure 3.

Celastrol Attenuates AVIC Calcification In Vitro
(A) Calcium deposition by Alizarin red staining with or without celastrol (Cel) (10 nmol/l) treatment. Scale bar: 100 μm. Mean data shown at the right. n = 5/group. (B) Reactive oxygen species (ROS) production in AVICs evaluated by hydroethidine (dihydroethidium [DHE]) staining. Scale bar: 100 μm. Mean data shown at the right. n = 5/group. (C) Protein levels of Runx2 and Nox2. n = 5/group. *p < 0.05, **p < 0.01, compared with respective control animals, #p < 0.05, ##p < 0.01, compared with calcification without celastrol treatment, 2-way analysis of variance with a post hoc Tukey’s test. All data are mean ± SEM. Abbreviations as in Figures 1 and 2.
To more directly investigate the role of Nox2 in AVIC calcification, we studied the effects of specific alternations of endogenous Nox2 by loss-of-function and gain-of-function approaches. Compared with GFP virus as control, Nox2 protein levels were significantly down-regulated with shNox2 adenovirus transfection both at baseline and after OGM stimulation for 2 weeks (Figure 4B). Importantly, knockdown of Nox2 substantially suppressed AVIC calcium nodule formation by 39% (Figure 4A), the inhibitory effect was similar to celastrol treatment. As expected, the protein levels of calcification-related marker Runx2 were also significantly abated by inhibition of Nox2 (Figure 4B). Conversely, overexpression of Nox2 significantly enhanced AVIC calcium deposition and Runx2 protein after OGM stimulation (Supplemental Figure 3), supporting the intrinsic contribution of Nox2 to AVIC mineralization. No significant phenotype change was observed in Nox2-overexpressing AVICs cultured in normal media even for 2 weeks (Supplemental Figure 3), consistent with the fact that Nox2 is dormant unless activated by disease stimuli.
Figure 4.

Knockdown of Endogenous Nox2 Inhibits AVIC Calcification
(A) Downregulation of Nox2 with shNox2 adenovirus decreases calcium deposition in AVICs. Scale bar: 100 μm. Mean data shown at the right. n = 5/group. (B) Immunoblots (left) and relative quantification (right) for Runx2 and Nox2 in AVICs. n = 5/group. *p < 0.05, **p < 0.01, compared with respective control animals; #p < 0.05, ##p < 0.01, compared with calcification of AVICs transfected with green fluorescent protein (GFP) virus, †p < 0.05, compared with GFP alone, 2-way analysis of variance with a post hoc Tukey’s test. All data are mean ± SEM.
Celastrol inhibits GSK3B/β-catenin signaling in AVICs
Previous works showed that GSK3B and β-catenin pathways are crucial regulators in the development of fibrogenesis and osteogenesis of AVs (7,29). We found that stimulation of AVICs with OGM caused GSK3B inactivation via phosphorylation of serine 9 (S9) and resulted in β-catenin accumulation, leading to increases in fibrotic marker fibronectin and osteogenic marker OPN (Figure 5A). Treatment with celastrol significantly inhibited GSK3B phosphorylation and β-catenin induction, and attenuated fibronectin and OPN protein levels (Figure 5A). Moreover, compared with green fluorescent protein controls, knockdown of Nox2 recapitulated the inhibitory effects of celastrol on GSK3B/β-catenin signaling, along with significant inhibition of fibrogenetic and osteogenetic response of AVICs, as evidenced by reduced levels of fibrotic marker fibronectin and calcific marker OPN (Figure 5B).
Figure 5.

The Antifibrocalcific Effect of Celastrol in AVICs Engages Inhibition of GSK3B/β-Catenin Pathway
(A) Western blots (left) and relative quantification (right) of glycogen synthase kinase 3 beta (GSK3B) inactivation, β-catenin, fibrotic marker fibronectin, and osteogenic marker OPN in AVICs with or without celastrol treatment (10 nmol/l, 14 days). n = 5/group. *p < 0.05, **p < 0.01, compared with respective control animals; #p < 0.05, ##p < 0.01, compared with calcification of AVICs without celastrol treatment. (B) The effect of knockdown endogenous Nox2 on GSK3B/β-catenin signaling. n = 5/group. *p < 0.05, **p < 0.01, compared with respective control animals; ##p < 0.01, compared with calcification of AVICs transfected with green fluorescent protein (GFP) virus, 2-way analysis of variance with a post hoc Tukey’s test. All data are mean ± SEM. p-GSK3B = phosphorylated glycogen synthase kinase 3 beta; other abbreviations as in Figures 1, 2, 3, and 4.
Celastrol mitigates aortic stenosis in a rabbit model of CAVD
To further investigate the protective role of celastrol against CAVD in vivo, we used a classical rabbit model of aortic stenosis, which is similar to human clinical condition (28,30). With safety concern regarding the clinical use of celastrol, we first examined if celastrol had any side effects in rabbits. There was no evidence of liver damage or renal dysfunction with oral administration of celastrol at the dosage of 1 mg/kg/day for 18 weeks by histology and plasma biochemical analysis (Supplemental Figures 4A and 4B). In addition, we did not observe any cardiac toxic effect of celastrol at baseline in terms of normal heart weight-to-body weight ratio, heart rates and ejection fraction (Supplemental Figure 4C).
Eighteen weeks’ high-cholesterol (HC) plus vitD2 diet resulted clearly in aortic stenosis evaluated by echocardiography. Compared with normal-diet control animals, HC+vitD2-treated rabbits displayed significant decreases in AV area and AV area indexed by body surface area, accompanied by markedly enhanced transvalvular peak and mean jet velocity (Figure 6). Rabbits fed with HC+vitD2 also exhibited ventricular dilatation (Figure 7A) and contractile impairment (Figure 7B) without changes of heart rates (Figure 7C), as well as exaggerated cardiac hypertrophy in terms of greater heart weight-to-body weight ratio (Figure 7D), increased interventricular septum and left ventricular (LV) posterior wall thickness by echocardiography (Figure 7F), and bigger cardiomyocyte size by wheat germ agglutinin staining (Figure 7G). We documented the similar body weight among groups, while rabbits actually exhibited slight but not significant body weight loss after 18 weeks’ HC+vitD2 diet compared with chow diet control animals (Figure 7E).
Figure 6.

Effect of Celastrol on Aortic Valves in a CAVD Rabbit Model Assessed by Echocardiography
(A) Representative 2-dimensional echocardiographic images of the aortic valve cusps from the parasternal long-axis (top) and short-axis (middle) views, and continuous pulsed-wave Doppler (bottom) at 18 weeks. In control rabbits fed a chow diet, the aortic valves exhibit a thin structure that is a barely visible. However, CAVD rabbits fed a high-cholesterol diet and vitamin D2 supplements exhibited stenosis evidenced by thickened and hyperechogenic valves (arrows), as well as increased aortic flow velocity. Treatment with celastrol (CAVD+Cel) alleviated aortic valve stenosis. (B) Mean data of the aortic valve area (AVA), AVA indexed by body surface area, transvalvular peak, and mean flow velocity (see Methods for calculation details). n = 6/group. **p < 0.01, compared with control animals; #p < 0.05, ##p < 0.01, compared with CAVD group, 1-way analysis of variance with a post hoc Tukey’s test. All data are mean ± SEM. Abbreviations as in Figures 1, 2, and 3.
Figure 7.

Celastrol Improves Cardiac Function in CAVD Rabbit
(A) Left ventricle internal diameter at end-diastole (LVIDd) and at end-systole (LVIDs); (B) ejection fraction (EF); (C) Heart rates; (D) heart weight-to-body weight ratio (HW/BW); (E) body weight; (F) interventricular septum thickness at end-diastole (IVSd) and at end-systole (IVSs) and LV posterior free wall during diastole (LVPWd) and systole (LVPWs). (G) Representative sections for cardiomyocyte area by wheat germ agglutinin staining. Scale bar: 50 μm. Mean data shown at the right. n = 5 to 6/group. *p < 0.05, **p < 0.01, compared with control animals; #p < 0.05, ##p < 0.01, compared with CAVD group, 1-way analysis of variance with a post hoc Tukey’s test. All data are mean ± SEM. Abbreviations as in Figures 1, 2, and 3.
Strikingly, administration of celastrol significantly mitigated the severity of aortic stenosis. Compared with HC+vitD2-fed rabbits, celastrol treatment substantially mitigated decrease in mean AV area by 12.7%, and the alleviation was still significant after adjustment for body surface area (Figure 6). Moreover, transvalvular peak and mean jet velocity were significantly decreased by 22% and 26%, respectively, compared with CAVD rabbits (Figure 6). These data therefore indicate that celastrol is effective to protect against aortic stenosis in rabbits in vivo. Consistent with this, animals treated with celastrol also developed less LV dilatation with better-preserved cardiac function than did HC+vitD2-fed rabbits (Figures 7A and 7B). However, celastrol had no significant protective effect on cardiac hypertrophy compared with CAVD hearts (Figures 7D, 7F, and 7G).
Celastrol attenuates AV fibrosis and calcification in CAVD rabbit
Histological staining demonstrated thickened valve leaflets with increased calcium deposits by Alizarin red staining in AVs of HC+vitD2 rabbits (Figures 8A and 8B), as well as enhanced calcific marker OPN expression by reverse transcriptase polymerase chain reaction analysis (Figure 8C). We also observed markedly increased fibrotic marker fibronectin by immunohistochemistry (Figures 8A and 8B). The enhanced fibrosis was further confirmed by Masson’s trichrome staining in HC+vitD2-treated rabbits (Figures 8A and 8B). Celastrol treatment markedly reduced HC+vitD2-induced calcium deposits (Figures 8A and 8B), OPN messenger RNA levels (Figure 8C), and fibronectin and fibrosis positive staining area (Figures 8A and 8B), supporting the beneficial effect of celastrol against AV fibrosis and calcification. Of note, Nox2 expression levels significantly increased in AVs of HC+vitD2-fed rabbits analyzed by both immunostaining and reverse transcriptase polymerase chain reaction, accompanied by enhanced ROS generation by in situ dihydroethidium fluorescence, all of which, however, were substantially attenuated by celastrol treatment (Figures 8A, 8C, and 8D). Compared with normal tissues, messenger RNA levels of several antioxidant enzymes including copper-zinc superoxide dismutase (SOD1), manganese superoxide dismutase (SOD2), and thioredoxin were significantly decreased in calcified valves, while extracellular superoxide dismutase (SOD3) and thioredoxin interacting protein were unchanged (Supplemental Figure 5). Interestingly, celastrol trended to improve antioxidant system in AV, but did not reach significance compared with that of HC+vitD2 rabbits (Supplemental Figure 5).
Figure 8.

Celastrol Alleviates Aortic Valve Fibrosis and Calcification in Rabbits
(A) Representative microscopy images of aortic valves. Compared with control groups, CAVD rabbits fed a high-cholesterol diet and vitamin D2 supplements develop thickened and calcified valves by Alizarin red stain, increased fibrosis by Masson trichrome stain, enhanced fibronectin and Nox2 expression by immunohistochemical staining, and elevated ROS production by DHE fluorescence. Administration of celastrol significantly alleviates these changes. Scale bar: 100 μm. (B) Quantification of percent positive areas. n = 6/group. (C) Messenger RNA levels of OPN and Nox2 in aortic valves. n = 6/group. (D) Mean data of DHE density. n = 3/group. *p < 0.05, **p < 0.01, compared with control animals; #p < 0.05, ##p < 0.01, compared with CAVD group, 1-way analysis of variance with a post hoc Tukey’s test. All data are mean ± SEM. Abbreviation as in Figures 1, 2, and 3.
Compared with chow diet–fed control animals, plasma levels of low-density lipoprotein cholesterol, total cholesterol, triglyceride, and calcium concentration were significantly elevated in HC+vitD2 rabbits (Supplemental Figure 6). Administration of celastrol significantly decreased low-density lipoprotein cholesterol, total cholesterol, and triglyceride levels compared with HC+vitD2 animals, though it had no effect on plasma calcium levels (Supplemental Figure 6). Interestingly, this beneficial effect of celastrol on cholesterol metabolism was not due to the less consumption of HC-enriched chow, as we did not find any significant effect of celastrol on food intake and body weight of rabbits fed with either normal diet or HC+vitD2 chow (Supplemental Figure 7), although it was reported that celastrol may protect against metabolic dysfunction and obesity in mice (19,31).
Discussion
In this study, we found Nox2 levels increase in human calcified AVs. An elevation of Nox2 promotes AVIC fibrocalcification by activating GSK3B/β-catenin pathway. Knockdown of endogenous Nox2 suppresses, by contrast, overexpression of Nox2 enhances AVIC calcium nodule formation. This result indicates that an increase in Nox2 may have detrimental effect on AVIC mineralization. Celastrol, a natural herb extract capable of inhibiting Nox2 activity, significantly ameliorates AVIC osteoblastic differentiation. More strikingly, using a rabbit model of CAVD, administration of celastrol in vivo before the development of aortic stenosis is effective to prevent the development of AV fibrosis, calcium deposit, stenosis, and hemodynamic obstruction, such that cardiac function is significantly improved.
It is well established that increased oxidative stress is a central pathophysiological component of numerous cardiovascular diseases including inflammation, metabolic disorder, and atherosclerosis (14), all of which are risk factors of CAVD (32). Over last decade, increasing evidence lends credence to the concept that ROS also play important roles in the development of CAVD, although the specific origin and contribution of ROS are in debate. It was reported ROS were significantly increased in the calcified and pericalcific region of valves from patients with end-stage AV stenosis (10). Although infiltration of inflammatory cells is evident within disease valves, in a rabbit model of CAVD, the topography of increased ROS signals is preferentially around calcifying foci from cells expressing osteogenic markers, but not macrophage markers (9). This strongly reflects the critical role of ROS as signaling molecular in valvular cell differentiation during the process of CAVD. We found that ROS levels significantly enhanced in cultured AVICs stimulated with osteogenic medium, further highlighting the implication of oxidative stress in osteoblast transdifferentiation of AVICs, which is the key component of pathogenesis of CAVD.
Several ROS sources may in principle contribute to ROS production and oxidative stress including mitochondria, uncoupled nitric oxide synthases, xanthine oxidases, and Nox enzymes. Among these sources, substantial studies indicate that Nox seem to play the central role in cardiovascular pathophysiology (12). Of 7 Nox homologues (Nox1 to Nox5, Duox1, and Duox2) have been identified in mammals so far, Nox2 (also termed as gp91phox) is most widely expressed in the heart and within vasculature. Previous studies from both experimental models and human showed that cardiac Nox2 activity and protein levels are elevated in adverse cardiac remodeling and heart failure (27,33, 34, 35), and vascular pathologies such as hypertension and atherosclerosis (36). However, its changing profile and biological role in CAVD remain unclear. It was reported that Nox2 messenger RNA levels increased in calcified AV of LDLr–/–/ApoB100/100 mice fed a Western-type diet (17). However, other studies did not find Nox induction in calcifying valve segments, and there was no significant difference in Nox-dependent ROS generation measured by lucigenin chemiluminescence between normal and disease human valves (10). This discrepancy could be probably related to the viability of patients who were receiving drugs such as statins which are capable of inhibiting Nox. In addition, reduced nicotinamide adenine dinucleotide phosphate–dependent lucigenin chemiluminescence is an imperfect method to examine Nox activity (37). We found that Nox2 levels in noncalcified human AVs were variable but generally low, and the variation could be explained by the effect of other comorbidities such as aging and diabetes. However, Nox2 proteins substantially increase around 2.6-fold in human calcified valves compared with noncalcification leaflets, which was further validated by immunohistology showing that the intense Nox2 accumulation are around the calcified region in diseased valves. The higher Nox2 expression was also observed in calcified AVs of rabbits given an HC+vitD2 diet, which is consistent with previous findings (9). Furthermore, our studies revealed that the protein levels of Nox2 up-regulation are in line with the severity of both cultured AVIC osteoblastic differentiation and the degree of human AV calcification. Importantly, with the use of complementary adenovirus-mediated silence and overexpression approaches, we show for the first time that Nox2 promotes AVIC calcification.
Although it has been increasingly recognized that Nox2 could be an attractive candidate as a potential therapeutic target, specific and effective Nox2 inhibitors are not clinically available. Another issue to consider with respect to targeted Nox2 inhibition is the potential to compromise neutrophil function, but previous work showed that this requires very substantial Nox2 inhibition (38), thus safe therapeutic targeting of cardiovascular Nox2 could be feasible. Celastrol, a plant-derived constituent of traditional Chinese medicine, has been shown as a preferable Nox2 inhibitor through interference with interaction between the tandem SH3 domain of p47phox and the proline-rich region of p22phox, which is essential for Nox2 activation (25). Celastrol may also inhibit Nox1 via disrupting the binding of NOXO1 and p22phox (25), but Nox1 is undetectable in AVICs (data not shown). We found that treatment of AVICs with celastrol markedly decreased ROS generation and significantly attenuated calcium nodule formation by 35%, the inhibitory effect similar to silencing endogenous Nox2. Next, we investigated whether celastrol could exert protection in vivo using an established rabbit CAVD model (28). One concern regarding the clinical use of celastrol is its relatively narrow therapeutic window of dose together with the occurrence of some adverse side effects (39). Hematoxylin and eosin–stained histological liver and kidney sections and serum chemistry analysis demonstrated the absence of any readily apparent drug toxicity in rabbits with the dosage used in this study. Strikingly, administration of celastrol for 18 weeks significantly mitigated the degree of AV calcification by Alizarin red staining, and alleviated the severity of aortic stenosis by more than 20% in terms of transvalvular jet velocity assessed by echocardiography. This finding represents to the best of our knowledge the first beneficial effect of celastrol on CAVD that has so far been identified. Accordingly, compared with CAVD rabbits, animals receiving celastrol developed less LV dilatation with better preserved contractile function without significant changes of cardiac hypertrophy. On the basis of the fact that both increased afterload and HC+vitD2 diet can activate Nox2 in the myocardium leading to chamber dilatation and heart failure, it is quite plausible that the protective effect of celastrol on the heart may likely result from either improved hemodynamics or direct inhibition of cardiac Nox2, or both. In addition, celastrol treatment decreased Nox2 levels both in AVICs with osteogenic stimulation and in calcified rabbit valves, the effect may be related to less induction of Nox2 by diminished calcification, and not by the direct action of celastrol itself, as we did not notice celastrol has any basal effect on Nox2 expression.
Celastrol has been recently put under the spotlight for its protective impacts on obesity and metabolic disorder (19,31); however, we did not document any significant changes in body weight and food intake of rabbit fed either with normal chow or HC+vitD2, which could be attributable to the differences of species, animal models, and dosages of celastrol. Interestingly, we found that rabbits treated with celastrol had lower low-density lipoprotein cholesterol, total cholesterol, and triglyceride levels compared with HC+vitD2-treated animals, the hypolipidemic effect of celastrol was also reported in other high fat diet models (31,40,41). The underlying mechanisms probably involve in the activation of HSF/PGC1a-dependent metabolic programs (31), and increase in adenosine triphosphate–binding cassette transporter A1 expression in the liver (41). The relationship between circulating cholesterol and CAVD has not been established in humans because large clinical trials failed to show any influence of lipid-lowering therapy with statins on aortic stenosis (42,43). However, statins are usually initiated after the disease has developed to advanced stage. Furthermore, as hypercholesterolemia is a causal risk factor in animal CAVD models, early intervention to lower plasma cholesterols could halt progression of AV stenosis in mice (44). Therefore, we cannot exclude the possibility that other pleiotropic or synergetic effects of celastrol such as protection against metabolic dysfunction contribute to its efficacy; however, the benefit of celastrol for the treatment of CAVD in vivo is at least in part via Nox2 inhibition in valvular cells.
There is strong evidence indicating the involvement of canonical Wnt/β-catenin signaling cascades in valve calcification. In active state, Wnt ligands bind Frizzled receptor and LDL receptor–related protein 5/6, resulting in the inactivation of GSK3B by phosphorylation and attenuation of β-catenin degradation. As a result, β-catenin is stabilized, accumulates in the cytoplasm, and translocates to the nucleus, and subsequently drives the process of osteogenic differentiation (45). Previous studies showed that canonical Wnt/β-catenin signaling is increased in calcified valves from patients, experimental models, and cultured AVICs (46, 47, 48). This was further supported by our observation that osteogenic stimulation enhanced GSK3B phosphorylation and β-catenin accumulation in cultured AVICs. Importantly, silence of endogenous Nox2 significantly activates GSK3B by inhibiting its phosphorylation and promotes β-catenin degradation, placing Nox2 the upstream in GSK3B/β-catenin signaling. Moreover, GSK3B may also mediate Ca2+-dependent noncanonical Wnt signaling, in which Wnt-Frizzled receptor binding leads to increased intracellular Ca2+ concentrations from the endoplasmic reticulum (45). Interestingly, Nox2 is critical to modulate cardiac calcium handling in disease settings such as angiotensin II stimulation (27). Although it was reported noncanonical Wnt signaling is implicated in human AV calcification (7), whether Nox2 enhances CAVD by regulating intracellular Ca2+ in AVICs as well needs further investigation.
CAVD is often termed a fibrocalcific disease because a hallmark of CAVD initiation is fibrotic collage accumulation that leads to sclerotic leaflets (49,50). Ectopic calcium deposits forming within these stiffened valves further decrease tissue compliance, eventually resulting in valve stenosis. Pathological process involved in CAVD may act in parallel to promote valvular fibrosis and calcification. We demonstrated that activation of Nox2-mdidated GSK3B/β-catenin signaling promotes fibrogenic and osteogenic responses in AVICs. Targeting this pathway by either knockdown of Nox2 expression or inhibition of Nox2 activity by celastrol in AVICs clearly blocks GSK3B/β-catenin signaling and markedly decreases fibrotic marker fibronectin and calcific marker OPN. It was reported there were sex differences in the AV phenotypes, with female patients demonstrating a more fibrotic AVs and males a more calcific phenotype (51). Therefore, effective pharmacological treatment for CAVD may be different according to sex and should ideally target both profibrotic and procalcific signaling to slow progression of valvular dysfunction (50). The capability and efficacy of celastrol in alleviating both valvular fibrosis and calcium nodule formation in vivo make this natural Nox2 inhibitor particularly interesting and encouraging to assess as a potential therapy for this debilitating disease.
Study limitations
First, the contribution of Nox is isoform-dependent and tissue and cell type–specific through complicated but different mechanisms. For example, in the heart, endothelial Nox2 promotes cardiac fibrosis and diastolic dysfunction by enhancing endothelial-mesenchymal transition (34). By contrast, myocardial Nox4 may protect against load-induced cardiac remodeling via increasing angiogenesis and activation of Nrf2 (52,53). We show here that Nox2 is critical in AVIC fibrocalcification, the contribution of Nox2 in other valvular cells and the roles of other Nox isoform such as Nox4 in CAVD warrants further investigation (Nox1 and Nox5 messenger RNA expressions were below detectable limits in AVICs). Using tissue- and isoform-specific gene-modified animals will be of help to provide valuable information. Second, both inflammation and hyperlipidemia contribute to the development of CAVD. Considering the therapeutic potential of celastrol in inflammatory diseases (18) and metabolic disorders, the systemic effect of celastrol could not be excluded. Third, as AV area and transvalvular velocity are modulated by cardiac function and afterload, comprehensive hemodynamic assessment of animal CAVD model warrants further investigation, although it was reported that blood pressure was not altered in rabbits fed with the HC+vitD2 diet (54). Fourth, compared with clinical scenario of advanced aortic stenosis, the development of the rabbit CAVD model used in this study is relatively fast and the stenosis is less severe. Therefore, off-target effects of celastrol should not be discounted if a long-term and high dose of celastrol could be used for the treatment of patients with severe stenosis. Nanoparticle-coated and cell-targeted drug delivery will be a promising strategy to improve efficacy and safety of celastrol (55).
Conclusions
In this study, we report that up-regulation of Nox2 is critically involved in AV calcification. Celastrol is effective to alleviate the osteoblastic differentiation of AVICs in vitro and mitigate AV fibrocalcification and stenosis in rabbits in vivo, likely through the inhibition of the Nox2-mediated GSK3B/β-catenin pathway in AVICs. The findings imply that targeted inhibition of Nox2 may be a possible therapeutic strategy in CAVD, and celastrol could potentially be clinically useful for early intervention to mitigate the development of CAVD or slow stenosis.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: The pathogenesis of CAVD involves complex-regulated mechanisms, including ROS-mediated redox signaling. ROS-generating enzyme Nox2 in AVICs critically contributes to fibrocalcification of the AV. Inhibition of Nox2 with a natural compound, celastrol, may be a promising pharmacological target to improve the outcome of the disease.
TRANSLATIONAL OUTLOOK: Additional studies are needed to investigate whether celastrol has therapeutic effectiveness on established CAVD, as well as the perspective of other more specific Nox2 inhibitors.
Footnotes
This study was supported by the National Natural Science Foundation of China (81470506), Key Scientific Research Project of Higher Education Institutions in Henan Province (18A320005), Key Research Project of the Heart Center of Xinxiang Medical University (2017360), Foundation and Frontier Technology Research Project in Henan Province (142300410191), and grants from British Heart Foundation (PG/17/39/33027 to Dr. Zhang, RG/13/11/30384 to Dr. Shah). The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Appendix
For expanded Methods and References sections as well as supplemental figures, please see the online version of this paper.
Contributor Information
Guoan Zhao, Email: guoanzhao@xxmu.edu.cn.
Min Zhang, Email: min.zhang@kcl.ac.uk.
Appendix
References
- 1.Lindman B.R., Clavel M.-A., Mathieu P. Calcific aortic stenosis. Nat Rev Dis Primers. 2016;2:16006. doi: 10.1038/nrdp.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawade T.A., Newby D.E., Dweck M.R. Calcification in aortic stenosis: the skeleton key. J Am Coll Cardiol. 2015;66:561–577. doi: 10.1016/j.jacc.2015.05.066. [DOI] [PubMed] [Google Scholar]
- 3.Hutcheson J.D., Aikawa E., Merryman W.D. Potential drug targets for calcific aortic valve disease. Nat Rev Cardiol. 2014;11:218–231. doi: 10.1038/nrcardio.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yutzey K.E., Demer L.L., Body S.C. Calcific aortic valve disease: a consensus summary from the alliance of investigators on calcific aortic valve disease. Arterioscler Thromb Vasc Biol. 2014;34:2387–2393. doi: 10.1161/ATVBAHA.114.302523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rutkovskiy A., Malashicheva A., Sullivan G. Valve interstitial cells: the key to understanding the pathophysiology of heart valve calcification. J Am Heart Assoc. 2017;6 doi: 10.1161/JAHA.117.006339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garg V., Muth A.N., Ransom J.F. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 7.Albanese I., Yu B., Al-Kindi H. Role of noncanonical Wnt signaling pathway in human aortic valve calcification. Arterioscler Thromb Vasc Biol. 2017;37:543–552. doi: 10.1161/ATVBAHA.116.308394. [DOI] [PubMed] [Google Scholar]
- 8.Weiss R.M., Ohashi M., Miller J.D., Young S.G., Heistad D.D. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation. 2006;114:2065–2069. doi: 10.1161/CIRCULATIONAHA.106.634139. [DOI] [PubMed] [Google Scholar]
- 9.Liberman M., Bassi Evo, Martinatti M.K. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol. 2008;28:463–470. doi: 10.1161/ATVBAHA.107.156745. [DOI] [PubMed] [Google Scholar]
- 10.Miller J.D., Chu Y., Brooks R.M., Richenbacher W.E., Peña-Silva R., Heistad D.D. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008;52:843–850. doi: 10.1016/j.jacc.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Branchetti E., Sainger R., Poggio P. Antioxidant enzymes reduce DNA damage and early activation of valvular interstitial cells in aortic valve sclerosis. Arterioscler Thromb Vasc Biol. 2013;33:e66–e74. doi: 10.1161/ATVBAHA.112.300177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lassègue B., San Martín A., Griendling K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M., Perino A., Ghigo A., Hirsch E., Shah A.M. NADPH oxidases in heart failure: poachers or gamekeepers? Antioxid Redox Signal. 2013;18:1024–1041. doi: 10.1089/ars.2012.4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forrester S.J., Kikuchi D.S., Hernandes M.S., Xu Q., Griendling K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122:877–902. doi: 10.1161/CIRCRESAHA.117.311401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agharazii M., St-Louis R., Gautier-Bastien A. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcification. Am J Hypertens. 2015;28:746–755. doi: 10.1093/ajh/hpu225. [DOI] [PubMed] [Google Scholar]
- 16.Lin C.P., Huang P.H., Lai C.F., Chen J.W., Lin S.J., Chen J.S. Simvastatin attenuates oxidative stress, NF-kappaB activation, and artery calcification in LDLR-/- mice fed with high fat diet via down-regulation of tumor necrosis factor-alpha and TNF receptor 1. PLoS One. 2015;10 doi: 10.1371/journal.pone.0143686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chu Y., Lund D.D., Weiss R.M. Pioglitazone attenuates valvular calcification induced by hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2013;33:523–532. doi: 10.1161/ATVBAHA.112.300794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S.-R., Dai Y., Zhao J., Lin L., Wang Y., Wang Y. A mechanistic overview of triptolide and celastrol, natural products from Tripterygium wilfordii Hook F. Front Pharmacol. 2018;9:104. doi: 10.3389/fphar.2018.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J., Lee J., Salazar Hernandez M.A., Mazitschek R., Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161:999–1011. doi: 10.1016/j.cell.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng X., Guan D., Auen T. IL1R1 is required for celastrol’s leptin-sensitization and antiobesity effects. Nat Med. 2019;25:575–582. doi: 10.1038/s41591-019-0358-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu M., Luo Q., Alitongbieke G. Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol Cell. 2017;66:141–153.e6. doi: 10.1016/j.molcel.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo X., Xue M., Li C.-J. Protective effects of triptolide on TLR4 mediated autoimmune and inflammatory response induced myocardial fibrosis in diabetic cardiomyopathy. J Ethnopharmacol. 2016;193:333–344. doi: 10.1016/j.jep.2016.08.029. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y., Geng C., Liu X. Celastrol ameliorates liver metabolic damage caused by a high-fat diet through Sirt1. Mol Metab. 2017;6:138–147. doi: 10.1016/j.molmet.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gu L., Bai W., Li S. Celastrol prevents atherosclerosis via inhibiting LOX-1 and oxidative stress. PLoS One. 2013;8 doi: 10.1371/journal.pone.0065477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaquet V., Marcoux J., Forest E. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br J Pharmacol. 2011;164:507–520. doi: 10.1111/j.1476-5381.2011.01439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peterson J.R., Burmeister M.A., Tian X. Genetic silencing of Nox2 and Nox4 reveals differential roles of these NADPH oxidase homologues in the vasopressor and dipsogenic effects of brain angiotensin II. Hypertension. 2009;54:1106–1114. doi: 10.1161/HYPERTENSIONAHA.109.140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang M., Prosser B.L., Bamboye M.A. Contractile function during angiotensin-II-activation: increased Nox2 activity modulates cardiac calcium handling via phospholamban phosphorylation. J Am Coll Cardiol. 2015;66:261–272. doi: 10.1016/j.jacc.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi B., Lee S., Kim S.-M. Dipeptidyl peptidase-4 induces aortic valve calcification by inhibiting insulin-like growth factor-1 signaling in valvular interstitial cells. Circulation. 2017;135:1935–1950. doi: 10.1161/CIRCULATIONAHA.116.024270. [DOI] [PubMed] [Google Scholar]
- 29.Liu F., Chu C., Wei Q., Shi J., Li H., Dong N. Metformin ameliorates TGF-β1–induced osteoblastic differentiation of human aortic valve interstitial cells by inhibiting β-catenin signaling. Biochem Biophys Res Commun. 2018;500:710–716. doi: 10.1016/j.bbrc.2018.04.141. [DOI] [PubMed] [Google Scholar]
- 30.Drolet M.C., Arsenault M., Couet J. Experimental aortic valve stenosis in rabbits. J Am Coll Cardiol. 2003;41:1211–1217. doi: 10.1016/s0735-1097(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 31.Ma X., Xu L., Alberobello A.T. Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1-PGC1α transcriptional axis. Cell Metab. 2015;22:695–708. doi: 10.1016/j.cmet.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Cho K.I., Sakuma I., Sohn I.S., Jo S.-H., Koh K.K. Inflammatory and metabolic mechanisms underlying the calcific aortic valve disease. Atherosclerosis. 2018;277:60–65. doi: 10.1016/j.atherosclerosis.2018.08.029. [DOI] [PubMed] [Google Scholar]
- 33.Heymes C., Bendall J.K., Ratajczak P. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol. 2003;41:2164–2171. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 34.Murdoch C.E., Chaubey S., Zeng L. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol. 2014;63:2734–2741. doi: 10.1016/j.jacc.2014.02.572. [DOI] [PubMed] [Google Scholar]
- 35.Sirker A., Murdoch C.E., Protti A. Cell-specific effects of Nox2 on the acute and chronic response to myocardial infarction. J Mol Cell Cardiol. 2016;98:11–17. doi: 10.1016/j.yjmcc.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konior, Schramm, Czesnikiewicz-Guzik M., Guzik T.J. NADPH oxidases in vascular pathology. Antiox Redox Signal. 2014;20:2794–2814. doi: 10.1089/ars.2013.5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rezende F., Prior K.-K., Löwe O. Cytochrome P450 enzymes but not NADPH oxidases are the source of the NADPH-dependent lucigenin chemiluminescence in membrane assays. Free Radic Biol Med. 2017;102:57–66. doi: 10.1016/j.freeradbiomed.2016.11.019. [DOI] [PubMed] [Google Scholar]
- 38.Kuhns D.B., Alvord W.G., Heller T. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600–2610. doi: 10.1056/NEJMoa1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cascão R., Fonseca J.E., Moita L.F. Celastrol: a spectrum of treatment opportunities in chronic diseases. Front Med (Lausanne) 2017;4:69. doi: 10.3389/fmed.2017.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu F., Li C., Jin X.-P. Celastrol may have an anti-atherosclerosis effect in a rabbit experimental carotid atherosclerosis model. Int J Clin Exp Med. 2014;7:1684–1691. [PMC free article] [PubMed] [Google Scholar]
- 41.Wang C., Shi C., Yang X., Yang M., Sun H., Wang C. Celastrol suppresses obesity process via increasing antioxidant capacity and improving lipid metabolism. Eur J Pharmacol. 2014;744:52–58. doi: 10.1016/j.ejphar.2014.09.043. [DOI] [PubMed] [Google Scholar]
- 42.Chan K.L., Teo K., Dumesnil J.G., Ni A., Tam J. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis. Results of the Aortic Stenosis Progression Observation: Measuring Effects of Rosuvastatin (ASTRONOMER) Trial. Circulation. 2010;121:306–314. doi: 10.1161/CIRCULATIONAHA.109.900027. [DOI] [PubMed] [Google Scholar]
- 43.Rossebø A.B., Pedersen T.R., Boman K. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 44.Miller J.D., Weiss R.M., Serrano K.M. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. 2009;119:2693–2701. doi: 10.1161/CIRCULATIONAHA.108.834614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albanese I., Khan K., Barratt B., Al-Kindi H., Schwertani A. Atherosclerotic calcification: Wnt is the hint. J Am Heart Assoc. 2018;7 doi: 10.1161/JAHA.117.007356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caira F.C., Stock S.R., Gleason T.G. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. 2006;47:1707–1712. doi: 10.1016/j.jacc.2006.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller J.D., Weiss R.M., Serrano K.M. Evidence for active regulation of pro-osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol. 2010;30:2482–2486. doi: 10.1161/ATVBAHA.110.211029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gu G.-J., Chen T., Zhou H-m, Sun K.-X., Li J. Role of Wnt/β-catenin signaling pathway in the mechanism of calcification of aortic valve. J Huazhong Univ Sci Technol Med Sci. 2014;34:33–36. doi: 10.1007/s11596-014-1228-x. [DOI] [PubMed] [Google Scholar]
- 49.Weiss R.M., Miller J.D., Heistad D.D. Fibrocalcific aortic valve disease: opportunity to understand disease mechanisms using mouse models. Circ Res. 2013;113:209–222. doi: 10.1161/CIRCRESAHA.113.300153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller J.D., Weiss R.M., Heistad D.D. Calcific aortic valve stenosis: methods, models, and mechanisms. Circ Res. 2011;108:1392–1412. doi: 10.1161/CIRCRESAHA.110.234138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simard L., Côté N., Dagenais F. Sex-related discordance between aortic valve calcification and hemodynamic severity of aortic stenosis. Circ Res. 2017;120:681–691. doi: 10.1161/CIRCRESAHA.116.309306. [DOI] [PubMed] [Google Scholar]
- 52.Zhang M., Brewer A.C., Schröder K. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smyrnias I., Zhang X., Zhang M. Nicotinamide adenine dinucleotide phosphate oxidase-4-dependent up-regulation of nuclear factor erythroid-derived 2-like 2 protects the heart during chronic pressure overload. Hypertension. 2015;65:547–553. doi: 10.1161/HYPERTENSIONAHA.114.04208. [DOI] [PubMed] [Google Scholar]
- 54.Arishiro K., Hoshiga M., Negoro N. Angiotensin receptor-1 blocker inhibits atherosclerotic changes and endothelial disruption of the aortic valve in hypercholesterolemic rabbits. J Am Coll Cardiol. 2007;49:1482–1489. doi: 10.1016/j.jacc.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 55.Guo L., Luo S., Du Z. Targeted delivery of celastrol to mesangial cells is effective against mesangioproliferative glomerulonephritis. Nat Commun. 2017;8:878. doi: 10.1038/s41467-017-00834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.