

In this study, Chen et al. set out to describe how kinetochores are restructured from a mitotic form to a meiotic form when a cell enters meiosis. Using in vitro and in vivo approaches in budding yeast, the author elucidates the mechanism by which meiotic cells modulate their kinetochore composition through regulated Ndc80 degradation and demonstrate that Aurora B-dependent regulation of kinetochores extends beyond altering microtubule attachments.
Keywords: meiosis, kinetochore, Aurora B, Ndc80, chromosome, proteolysis, APC
Abstract
The kinetochore complex is a conserved machinery that connects chromosomes to spindle microtubules. During meiosis, the kinetochore is restructured to accommodate a specialized chromosome segregation pattern. In budding yeast, meiotic kinetochore remodeling is mediated by the temporal changes in the abundance of a single subunit called Ndc80. We previously described the regulatory events that control the timely synthesis of Ndc80. Here, we report that Ndc80 turnover is also tightly regulated in meiosis: Ndc80 degradation is active in meiotic prophase, but not in metaphase I. Ndc80 degradation depends on the ubiquitin ligase APCAma1 and is mediated by the proteasome. Importantly, Aurora B-dependent Ndc80 phosphorylation, a mark that has been previously implicated in correcting erroneous microtubule–kinetochore attachments, is essential for Ndc80 degradation in a microtubule-independent manner. The N terminus of Ndc80, including a 27-residue sequence and Aurora B phosphorylation sites, is both necessary and sufficient for kinetochore protein degradation. Finally, defects in Ndc80 turnover predispose meiotic cells to chromosome mis-segregation. Our study elucidates the mechanism by which meiotic cells modulate their kinetochore composition through regulated Ndc80 degradation, and demonstrates that Aurora B-dependent regulation of kinetochores extends beyond altering microtubule attachments.
Reproduction is a fundamental feature of life and depends on the accurate segregation of chromosomes from one generation to the next. In eukaryotes, a conserved protein complex known as the kinetochore mediates chromosome segregation. Research over the past three decades has identified at least 40 different proteins that constitute the core of this essential machinery (reviewed extensively in Biggins 2013). While the function of individual kinetochore components has been well established, much less is understood about how the levels of specific subunits are regulated under varying cellular states and how these changes affect kinetochore function.
The kinetochore is composed of two distinct parts: inner and outer kinetochore. The inner kinetochore subunits associate with the chromosome at the centromere, while the outer kinetochore components interact with spindle microtubules. It has been shown that changes in either part can have a profound impact on kinetochore activity and genome inheritance, with potentially deleterious consequences. For example, overexpression of the centromeric histone CENP-A, a component of the inner kinetochore, in yeast, flies, and human cells causes chromosome mis-segregation and genomic instability (Heun et al. 2006; Au et al. 2008; Shrestha et al. 2017). Additionally, overexpression of the outer kinetochore subunits, such as Hec1 (also known as Ndc80) or SKA1, has been observed in many types of cancers and implicated in tumorigenesis (Chen et al. 1997, 2018; Hayama et al. 2006; Li et al. 2014; Shen et al. 2016).
Aside from these pathological states, changes to kinetochore composition also occur in physiological contexts. In various organisms, the kinetochore undergoes extensive remodeling during meiotic differentiation (Asakawa et al. 2005; Sun et al. 2011; Miller et al. 2012; Kim et al. 2013; Meyer et al. 2015), which is the developmental program that generates reproductive cells through two consecutive nuclear divisions. Specifically in budding yeast, the Ndc80 complex disassembles in early meiosis and reassembles during the meiotic divisions, thereby restricting kinetochore activity in a temporal fashion (Asakawa et al. 2005; Miller et al. 2012; Meyer et al. 2015; Chen et al. 2017). This dynamic kinetochore behavior is driven by the fluctuating Ndc80 levels, which are barely detectable in meiotic prophase but become highly abundant during the meiotic divisions. Failure to temporally regulate Ndc80 protein levels and kinetochore activity causes defects in meiotic chromosome segregation and gamete inviability (Miller et al. 2012; Chen et al. 2017), highlighting the importance of Ndc80 regulation.
One way to regulate Ndc80 protein levels occurs through controlling Ndc80 synthesis. Ndc80 production is relatively high during the meiotic divisions, but is completely shut down in meiotic prophase (Fig. 1A; Chen et al. 2017; Chia et al. 2017). This repression in synthesis requires the expression of a meiosis-specific, 5′ extended mRNA expressed from an alternate NDC80 promoter. This transcript, called LUTI (long undecoded transcript isoform), is induced by the transcription factor complex Ime1–Ume6 after meiotic entry and cannot be translated into Ndc80 protein. Instead, NDC80LUTI expression acts to interfere with the transcription of the canonical, protein-coding NDC80 mRNA isoform. As a result, in meiotic prophase, a stage when NDC80LUTI is highly expressed, Ndc80 protein synthesis is turned off. After cells exit from meiotic prophase, transcription of the coding NDC80 isoform is induced by another transcription factor called Ndt80, leading to resynthesis of Ndc80 and kinetochore activation (Chen et al. 2017). Thus, the developmentally coordinated toggling between these two functionally distinct mRNA isoforms controls Ndc80 production in meiosis.
Figure 1.
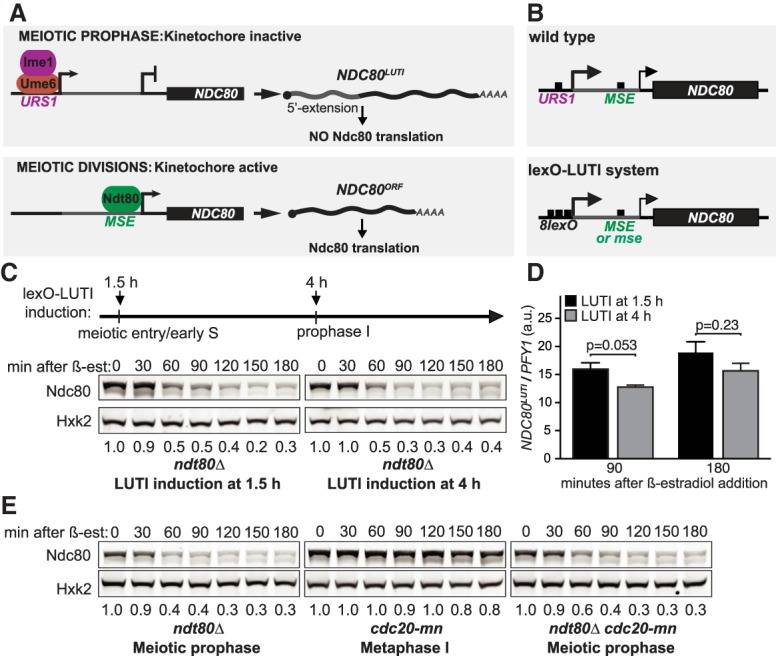
Ndc80 degradation is temporally regulated during meiosis. (A) LUTI-based regulation of Ndc80 protein synthesis in budding yeast meiosis. In meiotic prophase, the Ime1–Ume6 transcription factor complex induces a long undecoded transcript isoform of NDC80 called NDC80LUTI, which cannot produce Ndc80 protein due to the upstream open reading frames in its 5′ extension. NDC80LUTI represses transcription of a protein-coding isoform of NDC80, NDC80ORF. Through this combined act of transcriptional and translational repression, NDC80LUTI inhibits Ndc80 protein synthesis. In the meiotic divisions, NDC80ORF is induced by a second meiotic transcription factor, Ndt80. URS1 (upstream regulatory sequence 1), a DNA-binding motif for Ume6. MSE (mid-sporulation element), a DNA-binding motif for Ndt80. (B) The lexO-LUTI system induces NDC80LUTI expression upon β-estradiol addition, thus conditionally inhibiting NDC80ORF expression and Ndc80 protein synthesis. (Top) Regulatory elements of the NDC80 gene. (Bottom) The lexO-LUTI system. mse, a mutant MSE site defective in Ndt80 binding. (C) Ndc80 turnover in early or late meiotic prophase. The strain carrying the lexO-LUTI (UB14883) was transferred to the sporulation medium (SPO) at 0 h to induce meiosis, and β-estradiol was added at either 1.5 or at 4 h after meiosis induction. The strain was halted in meiotic prophase using an ndt80Δ block. Here and throughout, Ndc80 levels were determined by anti-V5 immunoblot. Hxk2, loading control. Unless specified, the numbers below the immunoblots were calculated by first normalizing Ndc80 levels to Hxk2 levels in each lane, and then dividing the ratio to the 0-h time point. All the experiments in this study were performed at least twice, and one representative biological replicate is shown. (D) Induction levels of NDC80LUTI mRNA for the experiment in C, measured by reverse transcription followed by quantitative PCR (RT-qPCR). For all RT-qPCR experiments, NDC80LUTI signals were normalized to that of PFY1. (a.u.) Arbitrary unit. The mean from three independent experiments, along with the standard error of the mean, is displayed. The P-values were calculated by a two-tailed Student's t-test. (E) Ndc80 turnover in late meiotic prophase or in metaphase I arrest. The ndt80Δ (UB19616) and the ndt80Δ cdc20-mn (UB19618) strains were cultured in SPO for 4 h before β-estradiol addition. Both strains were halted in meiotic prophase with an ndt80Δ block. The cdc20 meiotic null mutant (cdc20-mn, UB19678) was cultured in SPO for 5 h before β-estradiol addition and subsequently halted in metaphase I for 3 h.
The LUTI-based regulation explains how meiotic cells can effectively repress Ndc80 protein synthesis. However, since Ndc80 is clearly detected at meiotic entry (Asakawa et al. 2005; Miller et al. 2012; Meyer et al. 2015; Chen et al. 2017), regulated Ndc80 synthesis alone cannot fully explain kinetochore inactivation in meiotic prophase. Additional mechanisms must be in place to clear the existing pool of Ndc80 such that the kinetochores can disassemble in a timely manner. Interestingly, the human homolog of Ndc80, Hec1, undergoes degradation in a cell-cycle-dependent manner, but the turnover mechanism remains elusive (Ferretti et al. 2010). More generally, little is known about the factors that mediate kinetochore subunit degradation in a developmental context.
Here, we describe the mechanism by which Ndc80 degradation is controlled in budding yeast meiosis. We found that the degradation of Ndc80 is temporally regulated. Its proteolysis in meiotic prophase requires Aurora B/Ipl1 kinase-dependent phosphorylation, which has been previously linked to correcting erroneous microtubule–kinetochore attachments (for review, see Biggins 2013). The N terminus of Ndc80, including a 27-residue sequence and Ipl1 phosphorylation sites, is both necessary and sufficient for kinetochore protein degradation. In addition to phosphorylation, Ndc80 degradation depends on the ubiquitin ligase APCAma1 and proteasome activity. Failure to degrade Ndc80 causes premature kinetochore assembly in meiotic prophase and predisposes cells to meiotic chromosome segregation defects. Our results provide mechanistic insight into how cells can developmentally modulate kinetochore composition through subunit proteolysis and highlight the importance of timely Ndc80 degradation in promoting accurate meiotic chromosome segregation.
Results
Ndc80 degradation is temporally regulated in meiosis
In meiotic prophase, the residual Ndc80 protein from the premeiotic cell cycle is turned over by an unknown mechanism (Chen et al. 2017). To study Ndc80 degradation without a confounding effect from its synthesis regulation, we took advantage of a previously established method, which allowed us to turn off Ndc80 synthesis in a conditional manner (Chia et al. 2017). Specifically, we used a strain in which the endogenous NDC80LUTI promoter was replaced with an inducible promoter controlled by an array of eight lex operators (8lexO) (Fig. 1B). The same strain carries a chimeric lexA-B112 transcription factor fused to an estradiol-binding domain (lexA-B112-ER), which allows inducible transcription from the 8lexO promoter in the presence of β-estradiol (Ottoz et al. 2014). Without β-estradiol (uninduced), the coding NDC80 transcript (hereafter referred to as NDC80ORF) is expressed and Ndc80 is synthesized. After β-estradiol addition, NDC80LUTI is expressed, resulting in repression of Ndc80 synthesis. In comparison with wild-type cells, this induction system led to similar kinetics of Ndc80 degradation following meiotic entry (Supplemental Fig. S1A).
Using this system, we examined Ndc80 turnover at different stages of meiosis to determine the specific time window of Ndc80 degradation. We treated cells with β-estradiol either close to meiotic entry (1.5 h after meiotic induction) or later (4 h after meiotic induction). Meanwhile, the cells were held in meiotic prophase by deletion of NDT80, which encodes a transcription factor required for meiotic progression. We found that Ndc80 was degraded with similar kinetics in either condition (Fig. 1C). The levels of NDC80LUTI induction were also similar, as measured by reverse transcription followed by quantitative polymerase chain reaction (RT-qPCR) (Fig. 1D), suggesting that Ndc80 synthesis was successfully repressed. This result suggests that Ndc80 turnover can occur throughout meiotic prophase.
To determine whether Ndc80 is degraded beyond meiotic prophase, we monitored Ndc80 levels during a metaphase I arrest induced by Cdc20 depletion (cdc20-mn). Cdc20 is an activator of the anaphase-promoting complex, APC/C, a ubiquitin ligase necessary for metaphase-to-anaphase transition (Visintin et al. 1997; Hwang et al. 1998; Yu 2007). For this experiment, we mutated the Ndt80 binding site (also known as mid-sporulation element [MSE]) at the NDC80 promoter (Chen et al. 2017). This alteration is required because the second burst of Ndc80 synthesis, which depends on the MSE site, occurs after cells exit meiotic prophase. Mutating this site ensures that Ndc80 synthesis can be repressed by β-estradiol addition even after meiotic prophase. We found that while Ndc80 was degraded in meiotic prophase, it remained remarkably stable during the metaphase I arrest induced by cdc20-mn (Fig. 1E). The level of NDC80LUTI induction was ∼40% lower in cdc20-mn cells than in wild type (Supplemental Fig. S1B). In principle, this reduction of NDC80LUTI could cause an increase in Ndc80 synthesis, leading to higher protein levels. To exclude this possibility, we used cycloheximide to globally inhibit protein synthesis. Ndc80 was still stable during the metaphase I arrest and degraded in late prophase I under these conditions (Supplemental Fig. S1C), suggesting that the stability of Ndc80 protein differed between the two states. While it is possible that APCCdc20 may regulate Ndc80 degradation in metaphase I, we found that Cdc20 was dispensable for Ndc80 degradation in meiotic prophase (ndt80Δ cdc20-mn) (Fig. 1E). We conclude that Ndc80 degradation is temporally regulated, occurring in a meiotic prophase-specific manner.
Proteasome activity and Aurora B/Ipl1 regulate Ndc80 degradation
To identify the regulators of Ndc80 degradation, we surveyed the key cellular and proteolytic events in meiotic prophase. We found that synapsis, recombination, and DNA replication were all dispensable for Ndc80 degradation. Ndc80 degradation was normal in spo11Δ cells despite the lack of recombination and synapsis (Fig. 2A; Giroux et al. 1989; Cao et al. 1990; Keeney et al. 1997). Ndc80 degradation also occurred normally in cells depleted of the DNA replication factor Cdc6 (cdc6-mn) (Hochwagen et al. 2005; Brar et al. 2009; Blitzblau et al. 2012), consistent with a previous report (Fig. 2A; Meyer et al. 2015). These results suggest that Ndc80 degradation, and thus kinetochore remodeling, is independent of major meiosis-specific changes to chromosomes.
Figure 2.
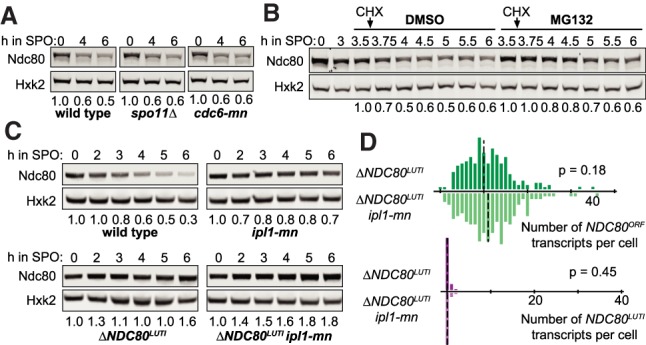
Proteasome and the Aurora B/Ipl1 kinase regulate Ndc80 degradation. (A) Dependency of Ndc80 protein degradation on recombination, synapsis, or DNA replication. The wild-type (UB1338), spo11Δ (UB11793), and cdc6 meiotic null (cdc6-mn, UB13656) strains were transferred to SPO at 0 h and halted in meiotic prophase using the pGAL-NDT80 GAL4.ER system until 6 h in SPO. The numbers below the immunoblots were calculated by first normalizing Ndc80 levels to Hxk2 levels in each lane, and then dividing the ratio to the 0-h time point. (B) Dependency of Ndc80 degradation on active proteasomes. Cells (pdr5Δ, UB2405) were induced to sporulate at 0 h. After 3 h in SPO, cells were split and treated with either DMSO or the proteasome inhibitor MG132 (100 µM). Thirty minutes later, cycloheximide (CHX) was added (0.2 mg/mL). For either condition (DMSO or MG132), all of the time points were normalized to the time point immediately before the cycloheximide addition (3.5 h). (C) The effects on Ndc80 levels when the meiotic depletion of IPL1 (ipl1-mn) was combined with a mutant that fails to repress Ndc80 synthesis (ΔNDC80LUTI). The wild-type (UB1338), ipl1-mn (UB1013), ΔNDC80LUTI (UB2932), and ΔNDC80LUTI ipl1-mn (UB3948) cells were sporulated as in A. (D) smFISH quantification of the NDC80ORF and NDC80LUTI mRNA levels in the ΔNDC80LUTI cells (UB2932) and ΔNDC80LUTI ipl1-mn (UB3948) cells in meiotic prophase. Samples were taken at 5 h in SPO. The relative frequency histograms of the cells with a given number of NDC80LUTI and NDC80ORF transcripts per cell were graphed using the data pooled from two independent experiments. (Dashed line) The median number of transcripts per cell. A total of 218 cells was counted for ΔNDC80LUTI and 208 cells for ΔNDC80LUTI ipl1-mn. Two-tailed Wilcoxon rank sum test was performed.
Next, we tested the ubiquitin-proteasome system, a key protein degradation pathway in the cell (Finley et al. 2012). In meiotic prophase, proteasomes are localized to chromosomes (Ahuja et al. 2017) and could mediate Ndc80 degradation. We treated meiotic prophase cells with the proteasome inhibitor MG132. Compared with the vehicle control, Ndc80 levels were modestly stabilized within 1 h of MG132 treatment (Supplemental Fig. S2A). During this time, the expression of NDC80LUTI did not change significantly, but it had decreased by ∼50% by the second hour of treatment (Supplemental Fig. S2B). To exclude the confounding effects from the differences in Ndc80 synthesis, we performed a cycloheximide chase experiment. The levels of Ndc80 were stabilized in the first hour of cycloheximide treatment although they eventually declined (Fig. 2B). This result indicates that the proteasome contributes, at least partially, to Ndc80 degradation.
Previously, it has been shown that the decline of Ndc80 levels in meiotic prophase requires the kinase Aurora B/Ipl1 (Meyer et al. 2015), raising the possibility that Ipl1 regulates Ndc80 degradation. We confirmed that Ndc80 abundance was increased upon meiotic depletion of Ipl1 (ipl1-mn) (Fig. 2C). This increase was not the result of elevated Ndc80 synthesis, as shown by three observations. First, the level of NDC80LUTI mRNA was not altered in ipl1-mn mutants (Supplemental Fig. S2C). Second, Ipl1 depletion increased Ndc80 levels additively with a mutant that fails to repress Ndc80 synthesis (ΔNDC80LUTI) (Fig. 2C). Finally, the expression of the protein-coding NDC80ORF isoform did not significantly change in the double mutant (ΔNDC80LUTI ipl1-mn) compared with the single mutant (ΔNDC80LUTI), as shown by single molecule RNA fluorescence in situ hybridization (smFISH) (Fig. 2D; Supplemental Fig. S2D). Based on these data, we conclude that Ipl1 regulates Ndc80 turnover rather than synthesis.
Ndc80 degradation requires Ipl1-mediated phosphorylation
How does Ipl1 regulate Ndc80 abundance mechanistically? In one model, Ipl1 depletion may alter Ndc80 abundance indirectly by affecting microtubule behavior in meiotic prophase. At this meiotic stage, the yeast centrosomes, known as the spindle pole bodies (SPBs), are duplicated but prevented from separating to form spindle microtubules. Meanwhile, the kinetochores become dispersed from the SPBs, presumably due to the lack of microtubule–kinetochore interactions (Kim et al. 2013; Meyer et al. 2013, 2015). Both kinetochore dispersion and inhibition of spindle formation in meiotic prophase require Aurora B/Ipl1 activity (Kim et al. 2013; Meyer et al. 2013, 2015). ipl1-mn mutants prematurely separate the duplicated SPBs and form long microtubules (Shirk et al. 2011; Kim et al. 2013). These microtubules interact with the kinetochores, leading to kinetochore reclustering (Meyer et al. 2013, 2015). In this context, microtubules could shield Ndc80 from degradation. Accordingly, the effect of ipl1-mn would depend on the presence of microtubules. However, when we depolymerized microtubules in ipl1-mn cells using a microtubule poison cocktail (benomyl and nocodazole), Ndc80 remained stable in meiotic prophase (Supplemental Fig. S3A–C). Thus, it is unlikely that Ipl1 indirectly regulates Ndc80 degradation by affecting microtubule behavior.
Since Ipl1 is a kinase, it may promote Ndc80 turnover by phosphorylating factors that turn over Ndc80, or by phosphorylating Ndc80 itself. Previous work has shown that Ndc80 is a direct substrate of Ipl1 (Cheeseman et al. 2002; Akiyoshi et al. 2009). Ndc80 has seven known Ipl1 consensus sites ([KR]-X-[ST]-[ILVST]) (Cheeseman et al. 2002), which are required for Ipl1 to phosphorylate Ndc80 in vitro (Akiyoshi et al. 2009). To test whether Ndc80 is degraded through a phosphorylation-dependent mechanism, we first asked whether Ndc80 is phosphorylated at the time of its degradation. We immunoprecipitated Ndc80 from meiotic prophase cells 1 h after MG132 treatment and analyzed the posttranslational modifications by mass spectrometry. We found two recurring, high-confidence phosphorylation sites T54 and T248 on Ndc80 (Fig. 3A). The first site has been previously characterized and contains an Ipl1 consensus site (Cheeseman et al. 2002; Akiyoshi et al. 2009), whereas the second site lacks the Ipl1 consensus sequence.
Figure 3.
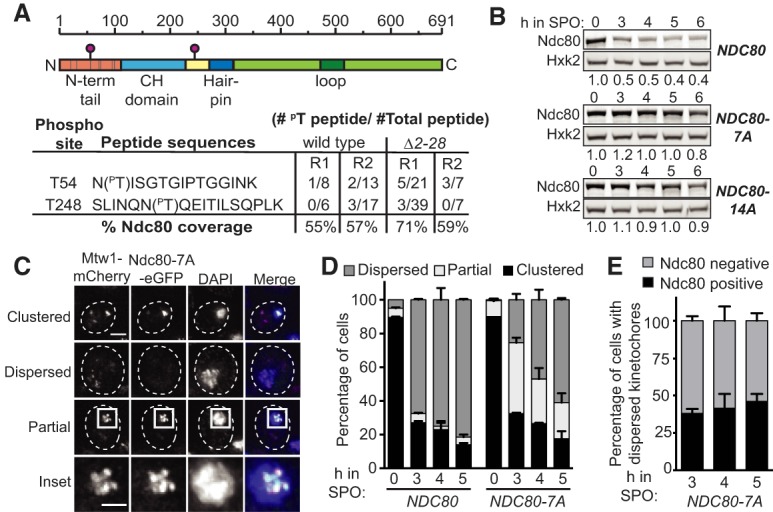
Ndc80 degradation requires Ipl1-mediated phosphorylation. (A) Phosphorylation sites detected on wild-type Ndc80 proteins and Ndc80(Δ2–28) proteins in meiotic prophase by mass spectrometry (MS). The wild-type (pdr5Δ, UB2405) cells were treated with MG132 at 3 h after transfer to SPO, and samples were collected 1 h after treatment. For the Δ2–28 cells (UB5662), samples were collected after 5 h in SPO. (Top) Schematic of Ndc80 and the in vivo phosphorylation sites detected by MS (pink circles). (Gray bars) The 7 Ipl1 consensus sites; (N-term) N-terminal; (CH) calponin homology. (Bottom) Detected Ndc80 phosphopeptides. The detected number of phosphopeptides and total peptides (phosphorylated and unmodified combined), as well as the overall sequence coverage of Ndc80, are reported for two biological replicates. (R1) repeat 1; (R2) repeat 2. (B) Ndc80 protein level in meiotic prophase for the wild-type (UB4074), NDC80-7A (UB13658), and NDC80-14A (UB17707) cells. Samples were taken and processed as in Figure 2A. The numbers below the immunoblots were calculated by first normalizing Ndc80 levels to Hxk2 levels in each lane, and then dividing the ratio to the 0-h time point. (C) Representative images of the clustered, dispersed, and partially clustered kinetochores in NDC80-7A (UB15701) cells, which were fixed at 5 h after transfer to SPO. Mtw1 was tagged with mCherry and Ndc80-7A tagged with eGFP. DNA was stained with DAPI. Scale bar, 2 µm; inset scale bar, 1 µm. (D) The percentage of cells with clustered, partially clustered, or dispersed kinetochores in meiotic prophase. The wild-type (UB1083) and NDC80-7A (UB15701) cells were fixed immediately after transfer to SPO (0 h) and at 3, 4, and 5 h later. One-hundred cells were counted per time point. The mean and the range of the percentage for two biological replicates are graphed. (E) The percentage of cells with dispersed kinetochores that contained Ndc80-7A-eGFP signal on at least one kinetochore at the indicated time points.
Next, we mutated the serine or threonine in all seven Ipl1 consensus sites, generating the allele (NDC80-7A) known to greatly reduce Ndc80 phosphorylation by Ipl1 in vitro (Akiyoshi et al. 2009). The mutations did not affect NDC80LUTI expression (Supplemental Fig. S3D), ruling out the possibility that the synthesis repression of Ndc80 was disrupted. We found that Ndc80-7A was highly stable in meiotic prophase (Fig. 3B). Mutating additional serine and threonine residues around T54 and T248 (NDC80-14A) did not enhance Ndc80 stabilization (Fig. 3B). Thus, we conclude that the seven Ipl1 consensus sites at the N-terminal region of Ndc80 are required for its turnover in meiotic prophase.
We further asked whether the stabilized Ndc80-7A proteins localized to the kinetochores and affected kinetochore behavior. We fused Ndc80-7A to the enhanced green fluorescent protein (eGFP), and generated a mCherry-tagged allele for the inner kinetochore protein Mtw1, which remains chromosome-associated throughout meiotic prophase. For both the wild-type and NDC80-7A mutant, over 85% of the cells had clustered kinetochores at meiotic entry (0 h in SPO), and all of the kinetochore clusters had Ndc80-eGFP signal (Fig. 3C,D). When the wild-type cells progressed into meiotic prophase (3–5 h in SPO), the kinetochores became dispersed in >70% of the cells, and none of the dispersed kinetochores had Ndc80-eGFP signal (Fig. 3D). In contrast, fewer than 30% of the NDC80-7A cells had dispersed kinetochores after 3 h in SPO (Fig. 3D). Instead, ∼40% of the cells had partially clustered kinetochores (Fig. 3C, partial and inset), all of which had Ndc80-7A-eGFP on them. By 5 h, >50% of the NDC80-7A cells had dispersed kinetochores (Fig. 3D), and about half of these cells had Ndc80-7A-eGFP on at least one kinetochore (Fig. 3E). These results demonstrate that the stabilized Ndc80-7A proteins localize to the kinetochore and affect the timing of kinetochore dispersion in meiotic prophase.
Ndc80 degradation requires a specific sequence at its N terminus
Since the seven Ipl1 consensus sites are located in the 112-residue N-terminal tail of Ndc80, we systematically truncated this region to narrow down the sites necessary for Ndc80 degradation. We found residues 2–28 to be necessary for the decline of Ndc80 levels (Fig. 4A, top panel). Within this segment, residues 11–19 were the most critical (Fig. 4A, bottom panel). To our surprise, the decline of Ndc80 levels was not altered when the 11 serines and threonines in the first 30 residues of Ndc80 were mutated (NDC80-11A) (underlined in Fig. 4B), indicating that Ndc80 degradation does not depend on the potential phosphorylation sites within this region. Ndc80 turnover was also normal when the four histidines in the 11–19 region (green letters in Fig. 4B) were mutated to either alanines (NDC80-4A) or leucines (NDC80-4L) (Supplemental Fig. S4A).
Figure 4.
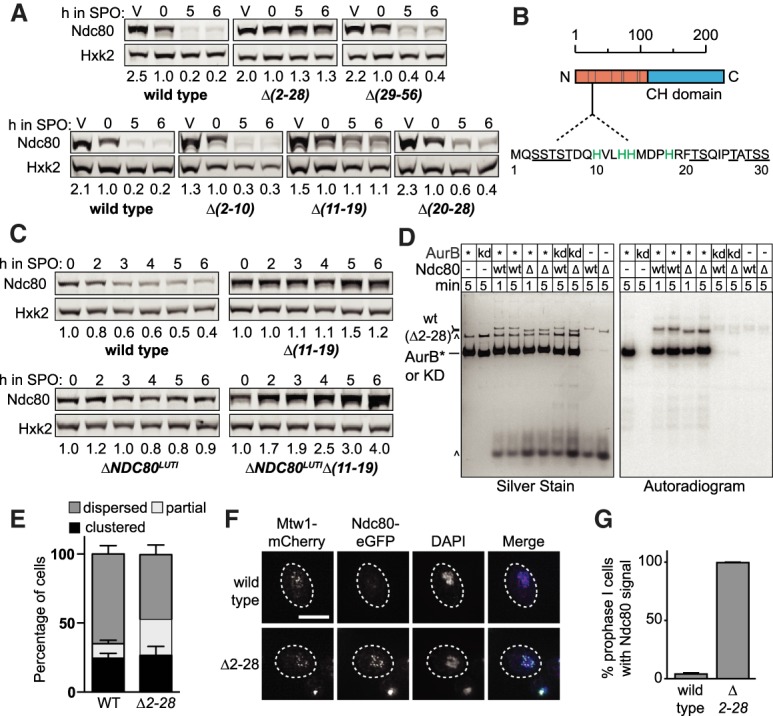
Ndc80 degradation requires a short sequence at its N terminus. (A) Truncation analysis of Ndc80 to identify the residues necessary for Ndc80 degradation. Strains harboring the deletions of residues 2–28 (Δ2–28, UB5662), residues 29–56 (Δ29–56, UB15972), residues 2–10 (Δ2–10, UB7039), residues 11–19 (Δ11–19, UB7029), and residues 20–28 (Δ20–28, UB7031) were sporulated along with wild-type cells (UB4074) as in Figure 2A. The vegetative samples (V) were taken while each strain was growing exponentially in rich medium. The numbers below the immunoblots were calculated by first normalizing Ndc80 levels to Hxk2 levels in each lane, and then dividing the ratio to the 0-h time point. Note: Hxk2 level declined slightly as meiotic prophase progressed. Thus, the normalized level of Ndc80 became >1.0 in the later stages of meiotic prophase. (B) An abridged schematic of Ndc80. The sequence of the first 30 residues is displayed. (Gray bars) The seven Ipl1 consensus sites. The underlined residues are the 11 serines and threonines mutated in the 11A mutant, and the green residues are the four histidines mutated in the 4(H to A) and 4(H to L) mutants. (C) The effects on Ndc80 levels when the Δ11–19 mutant was combined with a mutant that fails to repress Ndc80 synthesis (ΔNDC80LUTI). The wild-type (UB4074), Δ11–19 (UB7029), ΔNDC80LUTI (UB11797), and ΔNDC80LUTI Δ11–19 (UB11799) cells were sporulated as described in Figure 2A. (D) In vitro kinase assay for the wild-type Ndc80 and Ndc80(Δ2–28) proteins purified from UB16284 and UB19957, respectively. The Ndc80 proteins were phosphorylated in vitro by either 1 µM recombinant AurB* (fusion of the C-terminal activation box of INCEPSli15 to Aurora BIpl1) or 1 µM kinase-dead AurB* (KD) and γ-32P-ATP. All reactions were performed for either 1 or 5 min at room temperature, analyzed by SDS-PAGE, and then visualized by silver staining and autoradiography. (wt) Wild-type Ndc80; (Δ) Ndc80(Δ2–28); (*) AurB*; (^) background bands. (E) The percentage of the wild-type or Δ2–28 cells with dispersed, partially clustered, and clustered kinetochores after 3 h in SPO. The average, as well as the range, of two independent biological replicates are displayed. Over 100 meiotic cells were counted. (F) Representative images of the wild-type Ndc80 and Ndc80(Δ2–28) proteins in meiotic prophase. The wild-type (UB1083) and Δ2–28 (UB15619) cells were fixed after 5 h in SPO. Mtw1 was tagged with mCherry and Ndc80(Δ2–28) tagged with eGFP. DNA was stained with DAPI. Scale bar, 5 µm. (G) The average percentages of meiotic prophase cells (identified by the pachytene DAPI morphology) with colocalized Ndc80-eGFP and Mtw1-mCherry signals, as well as the range, of two independent biological replicates are displayed. At least 70 meiotic prophase cells were counted.
How does the 2–28 region regulate Ndc80 abundance? We first confirmed that the 2–28 region regulates Ndc80 degradation rather than Ndc80 synthesis. The wild-type, Δ2–28, and Δ11–19 cells had similar NDC80LUTI levels in meiotic prophase (Supplemental Fig. S4B). Also, the Ndc80 protein levels were additively increased when the Δ11–19 mutant was combined with the mutant that fails to inhibit Ndc80 synthesis (ΔNDC80LUTI) (Fig. 4C). Despite the difference in protein levels, the expression of the coding NDC80ORF mRNA was not significantly different between the double mutant (ΔNDC80LUTI Δ11–19) and the single mutant (ΔNDC80LUTI) (Supplemental Fig. S4C,D), suggesting that the 2–28 residues regulate Ndc80 stability.
Since Ndc80 degradation requires Ipl1-dependent phosphorylation, we next asked whether such phosphorylation depends on the 2–28 residues. We immunoprecipitated Ndc80(Δ2–28) protein from meiotic prophase and performed mass spectrometry. Both phosphorylation sites (T54 and T248) observed in the wild-type Ndc80 protein were detected (Fig. 3A). In addition, we performed an in vitro kinase assay using a recombinant Ipl1 protein variant (AurB*) (de Regt et al. 2018) and found no noticeable difference in the degree of Ndc80 phosphorylation between the wild-type and Ndc80(Δ2–28) protein (Fig. 4D). These results strongly suggest that the Ipl1-depedent Ndc80 phosphorylation occurs normally in the Δ2–28 mutant. Thus, we conclude that the 2–28 region is required for Ndc80 degradation at a step downstream from or in parallel to Ipl1-dependent phosphorylation.
We attempted to understand the role of the 2–28 residues in Ndc80 degradation by looking for changes in the binding partners of the wild-type or Ndc80(Δ2–28) protein during meiotic prophase. By quantitative mass spectrometry using tandem mass tags, we found that Sis1, a J domain protein that regulates heat-shock protein activity (Kampinga and Craig 2010), interacted with the wild-type but not the Ndc80(Δ2–28) protein (Supplemental Fig. S4E). However, Sis1 did not appear to be required for Ndc80 degradation since even the mutants that displayed normal Ndc80 degradation (e.g., Δ29–56) were still defective in Sis1 binding (Supplemental Fig. S4F). It remains unclear how the 2–28 segment mediates Ndc80 degradation.
Despite acting through an unknown mechanism, the 2–28 residues of Ndc80 are evidently important for clearing Ndc80 from the kinetochores and facilitating kinetochore dispersion in meiotic prophase. For the Δ2–28 mutant, we observed an increased proportion of cells with partially clustered kinetochores after 3 h in SPO (∼25%) (Fig. 4E), although the percentage was lower than that of the NDC80-7A mutant (∼40%) (Fig. 3D). By 5 h in SPO, most of the Δ2–28 cells had dispersed kinetochores in meiotic prophase, and Ndc80(Δ2–28)-eGFP signal was detected on all the dispersed kinetochores (Fig. 4F,G), demonstrating that the stabilized Ndc80(Δ2–28) protein localizes to the kinetochore. In addition, our observations suggest that while Ndc80 degradation is not required for kinetochore dispersion, it can affect dispersion kinetics (given the accumulation of cells with partially clustered kinetochores), potentially by acting in conjunction with Ipl1-dependent phosphorylation.
The N terminus of Ndc80 is sufficient to induce proteolysis
Both the 2–28 region of Ndc80 and Ipl1-mediated phosphorylation are required for Ndc80 degradation. Are these two features sufficient to induce protein degradation in meiotic prophase? We tested this idea by fusing the N-terminal region of Ndc80 (residues 2–112), including both the 2–28 region and the Ipl1 consensus sites, to an inner kinetochore protein, Ame1 (Fig. 5A). The protein levels of Ame1 were stable in meiotic prophase, as shown by a cycloheximide chase experiment (Supplemental Fig. S5). We placed this AME1-tail construct under the promoter of NDC80, which leads to synthesis repression in meiotic prophase. Addition of the N-terminal tail of Ndc80 caused Ame1 to become unstable in meiotic prophase in an Ipl1-dependent manner (Fig. 5B). Therefore, the N-terminal tail of Ndc80 is sufficient to induce proteolysis in meiotic prophase.
Figure 5.
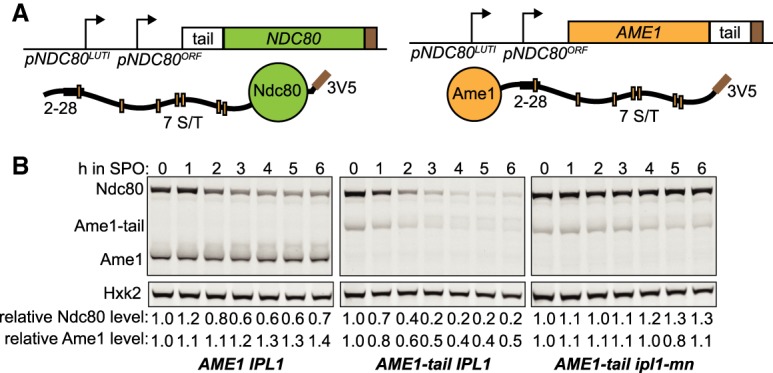
The N terminus of Ndc80 is sufficient to induce proteolysis. (A) Schematic of the Ndc80 tail-Ame1 fusion. The tail of Ndc80 (residues 2–112), including both the 27-residue sequence and the Ipl1 phosphorylation sites, is fused to the C terminus of Ame1. The fusion construct is controlled by the NDC80 promoter, which allows synthesis repression in meiotic prophase. (B) Ame1-tail is degraded in meiotic prophase in an Ipl1-dependent manner. The IPL1 AME1 (UB20358), IPL1 AME1-tail (UB20354), and ipl1-mn AME1-tail (UB22397) cells were sporulated as described in Figure 2A. In all of the strains, Ndc80 was tagged with 3V5, the same epitope tag for Ame1, as an internal control. The relative Ndc80 levels were calculated as described in Figure 1C. The relative Ame1 levels were calculated by normalizing Ame1 levels to Hxk2 levels in each lane, and then dividing the normalized values to that of time 0 h.
APCAma1 regulates Ndc80 degradation
Since proteasome activity contributes to Ndc80 degradation, we posited that Ndc80 is degraded via a system mediated by ubiquitin/proteasome, which requires one or more E3 ligases to ubiquitinate the substrate for proteasome recognition (Finley et al. 2012). Thus, we surveyed a candidate list of E3 ubiquitin ligases for their roles in Ndc80 degradation. We found the meiosis-specific activator of the APC/C, Ama1, to be required for Ndc80 turnover (Fig. 6A). APCAma1 regulates Ndc80 degradation rather than synthesis, because the levels of NDC80LUTI were not significantly altered in the ama1Δ mutant (Supplemental Fig. S6A). A cycloheximide chase experiment further supported that APCAma1 acts at a posttranslational step in regulating Ndc80 levels (Fig. 6C,D). In addition to Ndc80, the Ame1-tail fusion was also stabilized in an APCAma1-dependent manner (Fig. 6B).
Figure 6.
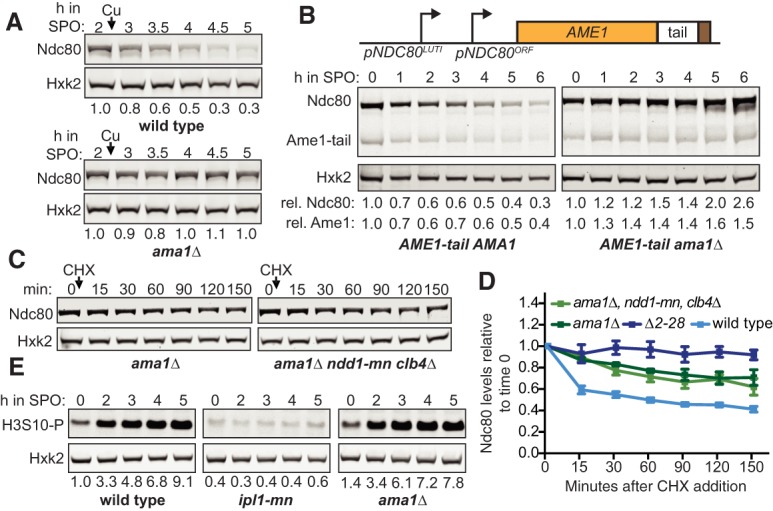
APCAma1 regulates Ndc80 degradation. (A) Dependency of Ndc80 levels on Ama1 in meiotic prophase. Sporulation of the wild-type (UB6598) and ama1Δ (UB22499) cells was synchronized with the pCUP-IME1/pCUP-IME4 system. Meiotic entry was induced by CuSO4 addition after cells were incubated in SPO for 2 h. The numbers below the immunoblots were calculated by first normalizing Ndc80 levels to Hxk2 levels in each lane, and then dividing the ratio to the 0-h time point. (B) Dependency of Ame1-tail fusion levels on Ama1 in meiotic prophase. The AME1-tail AMA1 (UB20354) and AME1-tail ama1Δ (UB23001) strains were sporulated as in Figure 2A. Quantification of Ndc80 and Ame1 were performed as in Figure 5B. (rel) Relative. (C) Assessing the requirement of Ndd1 and Clb4 in the Ndc80 stabilization that was induced by ama1Δ. The ama1Δ (UB20223) and pCLB2-NDD1 (ndd1-mn) clb4Δ ama1Δ (UB22850) cells were cultured in SPO for 3 h before a final concentration of 0.2 mg/mL cycloheximide was added. During the entire CHX experiment, both strains were halted in meiotic prophase. (D) The Ndc80 protein levels relative to the time point immediately before CHX addition. The average and the standard errors of the mean for five independent experiments are graphed. (E) Ipl1 activity in the ama1Δ mutant. The wild-type (UB3954), ipl1-mn (UB1013), and ama1Δ (UB20223) cells were sporulated as in Figure 2A. The levels of H3 serine 10 phosphorylation (H3S10-P) were detected by a phospho-specific antibody. The number below each lane was first normalized to Hxk2 levels and then to the 0-h time point of the wild-type strain.
It has been shown previously that AME1 deletion causes a mitotic-like cellular state in meiotic prophase (Okaz et al. 2012). This occurs due to stabilization of two APCAma1 substrates: Ndd1, a transcription factor required for the mitotic expression of B-type cyclins and Clb4, a B-type cyclin itself. Therefore, it is possible that such a mitotic-like state in the ama1Δ mutant could mimic a condition in which Ndc80 is stable, such as metaphase I. However, we found no evidence in support of this model. In our strains, premature spindle formation, a phenotype associated with elevated cyclin/CDK activity, was not apparent in the ama1Δ mutant (Supplemental Fig. S6B). As an additional test, we removed Ndd1 and Clb4 from the ama1Δ mutant and found that Ndc80 was still stable (Fig. 6C,D; Supplemental Fig. S6C), suggesting that AME1 deletion does not stabilize Ndc80 levels through creating an alternative cellular state in meiotic prophase.
Next, we tested whether Aurora B/Ipl1 activity is disrupted in the ama1Δ mutant, as Ipl1 down-regulation would lead to Ndc80 stabilization. We found that the wild-type and ama1Δ cells had comparable levels of serine 10 phosphorylated histone H3 (Fig. 6E), which is an established Aurora B/Ipl1 substrate (Hsu et al. 2000). Thus, Ipl1 activity is unaffected by AME1 deletion. These observations led us to conclude that APCAma1 acts downstream from Ipl1-dependent phosphorylation to mediate Ndc80 degradation.
Defects in Ndc80 degradation predispose meiotic cells to chromosome segregation errors
How does Ndc80 degradation impact kinetochore activity and function? We used the Δ2–28 mutant to address this question. We first tested whether this mutant had growth defects at a higher temperature (37°C) or on plates containing the microtubule depolymerizing drug benomyl. These two phenotypes are often exhibited by mutants with defects in kinetochore function (Spencer et al. 1990; Wigge et al. 1998; Hyland et al. 1999) and spindle assembly checkpoint (e.g., mad2Δ) (Li and Murray 1991). We found that the Δ2–28 mutant grew similarly to the wild-type cells in both conditions (Fig. 7A), demonstrating that these amino acids in Ndc80 are dispensable for normal kinetochore function in mitotic cells.
Figure 7.
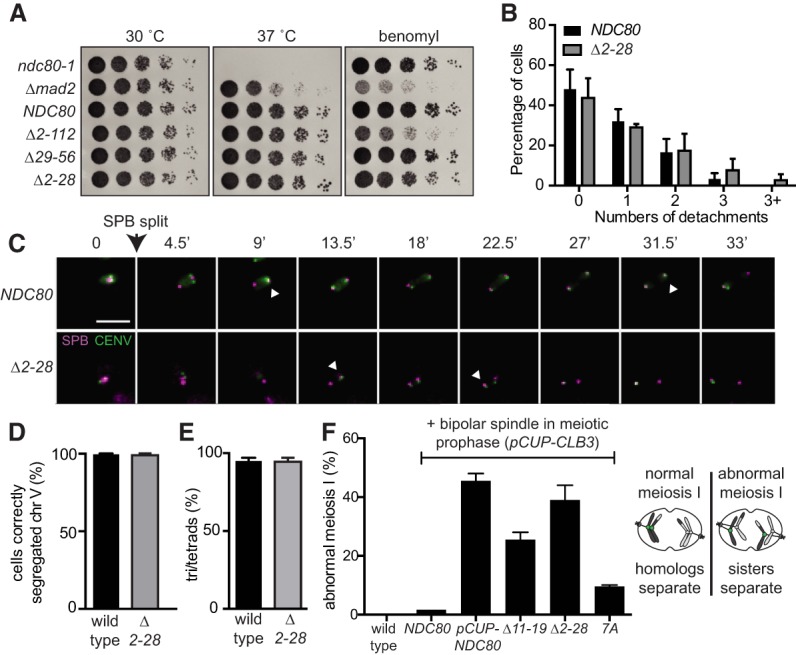
Ndc80 degradation regulates kinetochore composition in meiosis. (A) Growth phenotype associated with the various truncations of the Ndc80 N-terminal tail. Tested strains included the temperature-sensitive NDC80 allele (ndc80-1, UB494), the spindle assembly checkpoint mutant Δmad2 (UB700), wild-type NDC80 (UB3262), and the NDC80 mutants that carry the following deletion: residues 2–112 (Δ2–112, UB3275), residues 29–56 (Δ29–56, UB4695), and residues 2–28 (Δ2–28, UB5015). Cells were serially diluted and grown on plates containing nutrient-rich medium (YPD) at 30°C or 37°C, as well as on a benomyl plate (15 µg/mL) at 23°C. (B) Frequency distribution of the cells with a given number of kinetochore detachments and re-attachments for the experiment in C. The mean percentage and the standard deviation for three biological replicates are displayed. (C) Kinetochore attachments and detachments for the NDC80 (UB15905) and Δ2–28 (UB15873) strains in the spo11Δ background. Each strain carries the TetR-GFP fusion protein, and both homologs of chromosome V have the centromeric TetO array (homozygous CENV-GFP, marked in green). (Magenta) Spc42 fused with mCherry, which marks the spindle pole body (SPB). White arrowhead marks the kinetochore that would undergo detachment. The time when the SPBs were split is marked as 0. Scale bar, 5 µm. (D) The percentage of cells correctly segregated chromosome V for the wild-type (UB21757) and Δ2–28 (UB21758) strains. These strains carry the TetR-GFP fusion proteins, and both homologs of chromosome V are marked by the centromeric TetO repeats (CENV-GFP). Strains were sporulated for 7 h before formaldehyde fixation. At least 90 cells were counted for each strain. For D and E, the mean and the range of two biological replicates are graphed. (E) Spore formation for the meiosis I of wild-type (UB21756) and Δ2–28 (UB21758) strains. (F) Sister chromatid segregation in wild-type (UB2942), pCUP-CLB3 NDC80 (UB877), pCUP-CLB3 pCUP-NDC80 (UB880), pCUP-CLB3 NDC80(Δ11-19) (UB16561), pCUP-CLB3 NDC80(Δ2-28) (UB16565), and pCUP-CLB3 NDC80-7A (UB16872). All strains carry pGAL-NDT80 and GAL4.ER, which allows reversible arrest in meiotic prophase. A pair of the chromosome V sister chromatids is marked by the centromeric TetO repeats. A schematic for normal and abnormal meiosis I is shown at the right. In an abnormal meiosis I, two separated GFP dots were observed in binucleates, one in each nucleus. Cells were sporulated for 5 h before addition of CuSO4 to induce cyclin Clb3 expression. Immediately after the induction, cells were released from the prophase arrest using β-estradiol, which induces NDT80. Samples were taken 1 h 45 min after the release. The average fraction of binucleates that displayed sister segregation in meiosis I, as well as the range of two biological replicates, is graphed. One-hundred cells were counted per strain per experiment.
To examine whether Ndc80 degradation affects kinetochore function in meiosis, we monitored chromosome movements using the centromeric TetR/TetO GFP dot assay (Michaelis et al. 1997), where both homologs of chromosome V contained TetO arrays that were integrated near the centromere and were bound by the TetR-GFP fusion protein (CENV-GFP). We performed our experiment in the spo11Δ background because (1) these cells have long spindles that help resolve chromosome movements (Shonn et al. 2000) and (2) the homologous chromosomes fail to recombine (Klapholz et al. 1985), which renders the spo11Δ cells unable to establish biorientation. As a result, multiple rounds of microtubule–kinetochore attachments and detachments occur for an extended period, as part of the error correction mechanism. Ipl1 inactivation is known to cause defects in error correction, which is manifested by a reduced frequency of kinetochore detachments in the spo11Δ cells (Meyer et al. 2013). We found a similar distribution of kinetochore detachments between the wild-type and Δ2–28 cells (Fig. 7B,C), suggesting that the frequency of error correction (and, by inference, the kinetochore function) is unaltered upon Ndc80 stabilization. Consistently, >95% of the wild-type and the Δ2–28 tetrads correctly segregated their chromosomes, as shown by tracking the segregation of the homozygous CENV-GFP dots (Fig. 7D; Supplemental Fig. S7A). Both the wild-type and Δ2–28 cells had >95% sporulation efficiency (Fig. 7E) and >96% spore viability (Supplemental Fig. S7B,C), a metric that would be lowered if chromosomes missegregated. These results suggest Ndc80 degradation is dispensable for correcting erroneous microtubule–kinetochore interactions in meiosis.
Instead, we found that Ndc80 degradation modulated kinetochore activity by changing kinetochore composition. Remodeling kinetochore composition is crucial for proper meiosis I. In meiotic prophase, the outer kinetochore dissociates from the inner kinetochore to down-regulate kinetochore activity, which prevents premature microtubule–kinetochore interactions and is crucial to establish a meiosis I-specific chromosome segregation pattern (Asakawa et al. 2005; Miller et al. 2012; Meyer et al. 2015; Chen et al. 2017). When the Δ2–28 or Δ11–19 mutant were combined with a mutant that prematurely forms spindle microtubules in meiotic prophase (CUP-CLB3), we observed that sister chromatids, instead of homologous chromosomes, segregated in meiosis I (Fig. 7E). In this sensitized background, the NDC80-7A mutant also caused precocious sister chromatid segregation, albeit modestly (Fig. 7E), likely because the level of Ndc80-7A eventually reduced in prolonged prophase arrest (Fig. 3B, 6 h). The same phenotype occurs when Ndc80 is overexpressed in meiotic prophase or when the LUTI-based repression of Ndc80 synthesis is disrupted (Miller et al. 2012; Chen et al. 2017). Therefore, the lack of Ndc80 turnover in meiotic prophase prematurely activates the kinetochore by providing a high abundance of Ndc80, which is the limiting subunit of the kinetochore activity in meiotic prophase, and predisposes cells to meiotic chromosome segregation errors. Lastly, disrupting both Ndc80 synthesis repression (ΔLUTI) and Ndc80 degradation (Δ11–19) synergistically enhanced this segregation phenotype (Supplemental Fig. S7D), consistent with the idea that both modes of regulation contribute to kinetochore inactivation in meiotic prophase.
Discussion
In this study, we showed that Ndc80 degradation in meiosis is a temporally regulated process. The proteolysis of Ndc80 in meiotic prophase is independent of major chromosome remodeling events, but requires the ubiquitin ligase APCAma1 and proteasome activity. Furthermore, Ndc80 degradation is coupled to Aurora B/Ipl1-mediated phosphorylation of Ndc80, a posttranslational modification known for correcting erroneous microtubule–kinetochore attachments. The N terminus of Ndc80, which includes a 27-residue sequence and the Aurora B/Ipl1 consensus sites, is both necessary and sufficient to drive the degradation of kinetochore proteins. The failure to degrade Ndc80 in meiotic prophase alters the kinetics of kinetochore dispersion, causes premature activation of kinetochores, and predisposes meiotic cells to chromosome segregation defects. All of these observations highlight the importance of regulating Ndc80 turnover in meiosis.
Phosphorylation-mediated Ndc80 degradation in meiotic prophase
Multiple lines of evidence support a model in which Aurora B/Ipl1-dependent phosphorylation of Ndc80 triggers its degradation in meiotic prophase. First, Ipl1 is required for Ndc80 degradation. Second, phosphorylation of an Ipl1 consensus site on Ndc80 is detected by mass spectrometry in meiotic prophase, a stage when Ndc80 degradation occurs. Third, mutating the seven Ipl1 consensus sites leads to Ndc80 stabilization in meiotic prophase, suggesting that these sites are necessary for Ndc80 degradation. Altogether, these results are consistent with a phosphorylation-dependent degradation mechanism.
Given that Ndc80 phosphorylation is not disrupted when the 2–28 residues are deleted, we posit that this 2–28 segment may act in parallel to or downstream from the Ipl1-dependent phosphorylation to promote Ndc80 degradation. These residues may serve as the binding site for protein factors that mediate Ndc80 proteolysis. It is possible that such factors are recruited by Ndc80 phosphorylation, or that phosphorylation alters the local conformation around the 2–28 residues to expose this segment for protein factor binding.
Our data are consistent with the idea that the ubiquitin ligase APCAma1 and the proteasome act downstream from the Ipl1-dependent phosphorylation to mediate Ndc80 proteolysis. We excluded the possibility that the ama1Δ mutation stabilizes Ndc80 by increasing cyclin/CDK activity or by down-regulating Ipl1 activity (Fig. 6C–E). It is currently unknown whether APCAma1 directly ubiquitinates Ndc80 or regulates other player(s) in the degradation pathway to promote Ndc80 turnover. Interestingly, the APC/C degron repository (Davey laboratory and Morgan laboratory) predicts that Ndc80 has two D-box motifs and three KEN motifs, which are known recognition sites for the APC/C (Glotzer et al. 1991). However, these sites are located outside of the 112-residue N-terminal region, which is both necessary and sufficient for Ndc80 degradation in meiotic prophase. Furthermore, mutating the D-boxes does not block Ndc80 degradation (Supplemental Fig. S7E). The D-box motif is also not required for the APCAma1-dependent proteolysis of Ssp1 (Maier et al. 2007; Diamond et al. 2009). Future work to characterize the substrates of APCAma1, as well as the mechanism of substrate recognition, will be important to fully grasp the meiotic regulation of Ndc80 turnover.
Interplay between error correction and Ndc80 degradation
It is well established that Ndc80 phosphorylation by Aurora B/Ipl1 helps correct microtubule–kinetochore attachments that do not result in tension (Biggins 2013). Ndc80 phosphorylation is thought to weaken microtubule binding, resulting in microtubule detachment and thus providing the opportunity for the kinetochore to reattach in the correct orientation. How is the error correction process interrelated with Ndc80 degradation given that both processes require Ndc80 phosphorylation by Aurora B? We found that disrupting Ndc80 degradation alone (Δ2–28) does not appear to affect meiotic chromosome segregation (Fig. 7B–E; Supplemental Fig. S7A–C). This observation suggests that Ndc80 degradation is not required for error correction.
Instead, we propose that the completion of error correction may repress Ndc80 degradation in metaphase I, a stage when Ndc80 is stable (Fig. 1E; Supplemental Fig. S1B,C). Once the chromosomes attach to spindles properly, Ndc80 phosphorylation is removed by phosphatases to stabilize the microtubule attachments (Biggins 2013). Thus, it is possible that the Ndc80 levels become stable after the removal of Ndc80 phosphorylation in metaphase I. We note that it is unlikely that microtubule binding directly prevents Ndc80 degradation through steric effect because, at least in meiotic prophase, microtubule depolymerization cannot destabilize Ndc80 in ipl1-mn cells.
Our model presents a puzzle, however. As the major microtubule-binding site, Ndc80 is required to build new attachments during error correction. In our model, Ndc80 phosphorylation would cause its degradation at a time when the presence of Ndc80 is essential. This problem could be reconciled by the fact that Ndc80 protein is up-regulated as cells exit from meiotic prophase (Chen et al. 2017), which may provide extra proteins to compensate for the loss due to degradation. In addition, the activity of the ubiquitin ligase APCAma1 is repressed by Clb-CDK activity, which rises in prometaphase I (Oelschlaegel et al. 2005; Tsuchiya et al. 2011). Since APCAma1 is required for Ndc80 degradation, the degradation mechanism of Ndc80 could be turned off after meiotic prophase, even before error correction is completed.
Protein turnover as a mechanism to create meiosis-specific kinetochores
In vegetative growth, the yeast kinetochores transiently disassemble during DNA replication when the centromeric DNA replicates, but they remain bound to centromeres for the rest of the cell cycle. It has been shown that the subunit stoichiometry of the kinetochore is regulated during the cell cycle (Dhatchinamoorthy et al. 2017, 2019). However, it is unclear whether the protein levels of the kinetochore subunits are regulated in vegetative growth. Degradation of a few kinetochore proteins has been reported in yeast. These include Dsn1 (Akiyoshi et al. 2013), Cbf13 (Kaplan et al. 1997), and the centromeric histone Cse4 in budding yeast (Herrero and Thorpe 2016; Ohkuni et al. 2016; Cheng et al. 2017), as well as Spc7 in fission yeast (Kriegenburg et al. 2014). The degradation pathways of these subunits have been proposed to be quality controls that remove nonfunctional or excess proteins, rather than direct means to alter kinetochore function.
In contrast, regulating protein abundance is the key mechanism of controlling kinetochore activity in meiosis. The outer kinetochore disassembles in meiotic prophase and reassembles in prometaphase I. This disassembly is triggered by a reduction in Ndc80 levels, which occurs as a result of two separate mechanisms: LUTI-based repression of new Ndc80 synthesis and degradation of the existing Ndc80 pool. In meiosis, Ndc80 degradation is not a passive consequence of DNA replication but a targeted process mediated by Aurora B/Ipl1, which has also been implicated in preventing untimely spindle formation in meiotic prophase (Shirk et al. 2011; Kim et al. 2013). We propose that Ipl1 prevents premature microtubule–kinetochore interactions through two independent pathways: one by altering microtubule behavior, and the other by triggering Ndc80 proteolysis. This dual mechanism ensures that the kinetochore interacts with spindles only after meiotic prophase. This delay in microtubule–kinetochore interaction is required for proper meiotic chromosome segregation (Miller et al. 2012).
Cells customize kinetochore activity by controlling kinetochore assembly or disassembly
Examples from various organisms indicate that regulating kinetochore assembly or disassembly is a common way to customize kinetochore activity based on the specific cell cycle stage or cell type. During mitotic exit in human cells, the outer kinetochore components, known as the KMN network, dissociate from the inner kinetochore in every cell cycle. A failure to do so causes chromosome missegregation in the next cell cycle (Gascoigne and Cheeseman 2013). In C. elegans oocytes, kinetochores facilitate homolog biorientation and error sensing in meiosis I, but they do not segregate the chromosomes. Instead, the ring-complex and microtubule motors, such as dynein, segregate the chromosomes, while the kinetochore proteins disappear from the chromosomes in anaphase I (Dumont et al. 2010; Muscat et al. 2015; Davis-Roca et al. 2017; Laband et al. 2017). Lastly, in C. elegans, Drosophila, and iPS-derived human motor neurons, some or all of the core kinetochore proteins localize to the regions of synaptic neuropil and axons that are devoid of nuclei. There they control axon outgrowth and dendritic extension in the developing sensory nervous system (Cheerambathur et al. 2019; Zhao et al. 2019). All of these examples highlight that the timing and location of kinetochore assembly are tuned to the cell cycle and cell type.
Based on our finding that the Aurora B kinase regulates the turnover of the kinetochore subunit Ndc80, it will be interesting to test whether other kinetochore restructuring events also depend on Aurora B. For example, the protein levels of Ndc80/Hec1 decline at the mitotic exit in human cells (Ferretti et al. 2010), but the mechanism is unknown. We posit that Aurora B and/or Aurora A may trigger Ndc80/Hec1 degradation at the mitotic exit in human cells, contributing to the KMN dissociation. In addition, the C. elegans Aurora B kinase, AIR-2, localizes to spindle microtubules during anaphase I (Schumacher et al. 1998; Speliotes et al. 2000; Romano et al. 2003). Thus, it is possible that AIR-2 phosphorylation causes the dissociation or degradation of one or more kinetochore subunits, leading to kinetochore disassembly at that time. Our discovery that Aurora B phosphorylation can cause the turnover of kinetochore proteins provides a new means by which Aurora B can influence kinetochore composition and activity: Aurora B regulates not only the construction but also the destruction of the kinetochore.
Materials and methods
Yeast strains and plasmids
All strains used in this study were derivatives of SK1 unless specified. The strain genotypes are listed in Supplemental Table S1. The centromeric TetR/TetO GFP dot assay was first described in Michaelis et al. (1997), the ndc80-1 temperature-sensitive mutant was first described in Wigge et al. (1998), pCUP-NDC80 and pCUP-CLB3 were first described in Lee and Amon (2003); Miller et al. (2012), the meiotic depletion alleles pCLB2-3HA-CDC20 were first described in Lee and Amon (2003), pSCC1-3HA-CDC6 was first described in Hochwagen et al. (2005), and SPC24-6HIS-3Flag was first described in Miller et al. (2016). The following alleles were generated at the endogenous gene loci using PCR-based methods (Longtine et al. 1998): NDC80-3V5, pSCC1-3HA-IPL1, ndt80Δ, spo11Δ, pdr5Δ, mad2Δ, mad3Δ, ama1Δ, pCLB2-3HA-NDD1, clb4Δ, NDC80-eGFP, MTW1-mCherry, and ΔNDC80LUTI (deletion of the 400–600 bp upstream of the NDC80 coding region). The V5 tagging plasmid was kindly provided by Vincent Guacci (University of California at Berkeley).
Previously, an NDC80 single integration plasmid was generated (Chen et al. 2017). This plasmid includes the SK1 genomic sequence spanning from 1000 bp upstream of to 357 bp downstream from the NDC80 coding region, as well as a C-terminal fusion of the 3V5 epitope to NDC80 referred to as NDC80-3V5 (Chen et al. 2017). All the single integration plasmids used in this study were derived from this NDC80-3V5 parent vector.
We cloned the following mutant NDC80 alleles by Gibson assembly (Gibson et al. 2009) using gBlocks gene fragments (IDT): NDC80-7A-3V5, NDC80-14A-3V5, NDC80(4H to A)-3V5, NDC80(4H to L)-3V5, NDC80-11A-3V5, and NDC80-4D-box-3V5. The 8lexO-LUTI-NDC80-3V5 construct, also cloned by Gibson assembly, has eight lex operators (lexO) and the promoter from CYC1 inserted at 536 bp upstream of the translation start site of NDC80 ORF. This insertion allows lexA-B112-ER to control the expression of NDC80LUTI mRNA in a dose-dependent manner (Chia et al. 2017). The 8lexO-LUTI-mse-NDC80-3V5 construct has a mutation in the Ndt80 binding site (mse), which diminishes the Ndt80-dependent expression of NDC80ORF mRNA (Chen et al. 2017). The pNDC80-AME1-3V5 and pNDC80-AME1-tail-3V5 (referred to as AME1-tail in the Results) constructs are controlled by the NDC80 promoter (1000 bp upstream of the NDC80 coding region), which is sufficient to repress protein synthesis of the coding region in meiotic prophase (Chen et al. 2017). For the pNDC80-AME1-tail-3V5 construct, the 2–112 residues of Ndc80 were fused to the C terminus of Ame1.
In addition, systematic deletions of the N-terminal residues of Ndc80 (2–28, 29–56, 2–10, 11–19, 20–28, and 57–112) were made on the NDC80-3V5 parent vector using the Q5 site-directed mutagenesis kit (New England Biolabs). All of the single integration plasmids described above were digested with PmeI, transformed into yeast, and selected for integration at the LEU2 locus.
To generate the Δ2–28, Δ11–19, and NDC80-7A alleles at the endogenous locus of NDC80, the CRISPR/Cas9 method was used (Anand et al. 2017). Oligonucleotides encoding the guide RNA (5′-TACATCACATGGACCCTCATCGG-3′) were cloned into a centromeric plasmid carrying a URA3 maker and pPGK1-Cas9 (a gift from Gavin Schlissel, University of California at Berkeley). This plasmid was cotransformed into yeast along with the repair templates amplified from the respective LEU2 single integration plasmids by PCR (Δ2–28, Δ11–19, and NDC80-7A plasmids). For NDC80-7A, the following primers were used to amplify the repair template while introducing synonomous mutations to the PAM sequence to prevent re-editing: 5′-ACATGTGCTACATCACATGGACCCTCATCGGTTTGCTTCaCAAATACCAACTGCAACATC-3′ and 5′-CTCTTGAATAGCGCTTTGGAAGTTTTTGTCTCTTAGTGGtCTTGGATCTCTATTGCTCAG-3′. The lowercase letters correspond to the synonymous mutations that abolish the PAM site: a C-to-A mutation at the 66th nucleotide from the translation start site of Ndc80, as well as a G-to-A at the 351st nucleotide.
Yeast growth condition, synchronous sporulation, and media
To prepare for sporulation, diploid cells were grown in YPD (1% yeast extract, 2% peptone, 2% glucose, and supplemented with 22.4 mg/L uracil and 80 mg/L tryptophan) for 20–24 h at room temperature or 30°C. For optimal aeration, the total volume of the flask exceeded the volume of the medium by 10-fold. Subsequently, cells were transferred to BYTA (1% yeast extract, 2% bacto tryptone, 1% potassium acetate, 50 mM potassium phthalate) at 0.25–0.3 OD600 cells/mL and grown for another 15–17 h at 30°C. The cells were then pelleted, washed with sterile milliQ water, and resuspended at 1.85 OD600 cells/mL in sporulation (SPO) medium (0.5% [w/v] potassium acetate at pH 7, 0.02% [w/v] raffinose) at 30°C unless specified.
Two methods were used to synchronize sporulation in this study. The pCUP1-IME1 pCUP1-IME4 method was described in Berchowitz et al. (2013), in which the endogenous promoters of IME1 and IME4 were replaced with the inducible CUP1 promoter. To initiate synchronous sporulation, the expression of IME1 and IME4 was induced by adding 50 µM copper (II) sulphate 2 h after cells were transferred to SPO. The pGAL-NDT80 GAL4.ER system was used to allow cells to undergo synchronized meiotic divisions (Carlile and Amon 2008). After incubating the sporulating diploids for 5–6 h in SPO (see the figure legend for the specific arrest duration for each experiment), cells were released from pachytene arrest by inducing NDT80 expression with the addition of 1 µM β-estradiol.
RNA extraction and RT-qPCR
A previously described RNA extraction protocol was modified as described below (Koster and Timmers 2015). RNA was extracted with acid phenol:chloroform:isoamyl alcohol (125:24:1, pH 4.7) and then precipitated by isopropanol. Briefly, 1.85–3.7 OD600 of cells were harvested by centrifugation at 21,000 rcf for 1 min, and frozen in liquid nitrogen. Pellets were thawed on ice and vigorously shaken for 30 min at 65°C in 400 µL of TES buffer (10 mM Tris at pH 7.5, 10 mM EDTA, 0.5% SDS), 200 µL of acid-washed glass beads (Sigma), and 400 µL of acid phenol. The lysates were clarified by centrifugation at 21,000 rcf for 10 min at 4°C. The aqueous phase was transferred to 300 µL of chloroform, vortexed, and centrifuged at 21,000 rcf for 5 min at room temperature. To precipitate the RNA, the aqueous phase was added to 400 µL isopropanol supplemented with 0.33 M NaOAc, incubated for >2 h at −20°C, and centrifuged at 21,000 rcf for 20 min at 4°C. The precipitated RNA pellets were washed with 80% ethanol, air-dried, and resuspended in nuclease-free water.
A total of 5 µg of RNA was treated with DNase using the TURBO DNA-free kit (Thermo Fisher) and then reverse-transcribed into cDNA using SuperScript II or III (Thermo Fisher) following the standard protocol. The single-stranded cDNA was quantified by 7500 Fast real-time PCR machine (Thermo Fisher) using SYBR Green mix (Thermo Fisher). The primers used for the RT-qPCR reactions were NDC80LUTI (GGTTGAGAGCCCCGTTAAGT and TTGGCACTTTCAGTATGGGT) and PFY1 (ACGGTAGACATGATGCTGAGG and ACGGTTGGTGGATAATGAGC). The Ct value for each sample was determined by taking the average Ct of triplicate reactions, and the levels of the NDC80LUTI transcript were normalized to those of the PFY1 transcript.
Single-molecule RNA fluorescence in situ hybridization (smFISH)
Single-molecule RNA FISH was performed exactly as described previously (Chen et al. 2017). To quantify smFISH spots, the maximum-intensity projection of Z-stacks was generated in FIJI (Schindelin et al. 2012), different channels were split, and the images were analyzed by the StarSearch Program (Raj et al. 2008). Cell boundaries were hand-drawn. The thresholds were adjusted such that they fell within “plateaus,” as described previously (Raj et al. 2008). The same threshold was used for a given probe set in each experimental replicate. An NDC80LUTI transcript was identified as a colocalized spot where the two spots found in either channel have pixels overlapped for >50%. The number of NDC80ORF transcripts in each cell was calculated by subtracting the number of CF 590 (LUIT-only) spots from the total number of spots with the Q 670 (common region) signal. To compare between different strains, data were pooled from two biological replicates, and the nonparametric Wilcoxon Rank Sum test was performed with Prism 6 (Graphpad). The relative frequency histograms were normalized so that the maximum bin height is the same across all the histograms in the same figure.
Fluorescence microscopy
To fix cells, formaldehyde was added to approximately one OD600 cells to a final concentration of 3.7%, incubated for 15 min at room temperature, washed once with KPi/sorbitol buffer (100 mM potassium phosphate at pH 7.5, 1.2 M sorbitol), and resuspended in the VectaShield antifade mounting medium with DAPI overnight before imaging (Vector Laboratories). The antifade medium preserved the samples from photobleaching during imaging. All images were acquired as Z-stacks of 20 slices (0.3-µm step size) using a DeltaVision Elite wide-field fluorescence microscope, a PCO Edge sCMOS camera (DeltaVision, GE Healthcare), a 100×/1.40 oil immersion plan apochromat objective, and the following filters: DAPI (EX390/18 and EM435/48), GFP/FITC (EX475/28 and EM525/48), and mCherry (EX575/25 and EM625/45). Images were deconvolved in softWoRx software (softWoRx, GE Healthcare) with a 3D iterative constrained deconvolution algorithm (enhanced ratio) with 15 iterations.
Kinetochore classification and quantification
The three classes of kinetochores were classified as follows: The “clustered” class was defined by the presence of a single Mtw1-mCherry focus in a cell, or the presence of less than three distinct Mtw1-mCherry foci. For this class, Ndc80 signal was always detected and colocalized with the Mtw1-mCherry signal in the cells, and the DAPI signal was often compacted and round, without thread-like structures. The “dispersed” class was defined by more than six distinct Mtw1-mCherry foci located throughout the projected DAPI area of the cell. For this class, the DAPI signal was almost always thread-like, suggesting a pachytene state. The “partially clustered” class was defined by the presence of at least three distinct Mtw1-mCherry foci and all foci were located within the same half of the projected DAPI boundary of the cell.
Live-cell imaging
Time-lapse microscopy was performed in an environmental chamber heated to 30°C. Strains were induced to sporulate by transferring to SPO and halted in meiotic prophase using the pGAL-NDT80 GAL4-ER block for 5 h in SPO. Cells were then released with 1 µM β-estradiol and, 30 min later, immobilized on concanavalin A-coated, glass-bottom 96-well plates (Corning) and cultured in 100 µL of SPO (with 1 µM β-estradiol) medium in the prewarmed environmental chamber. Z-stacks of five slices (1-µm step size) were acquired using a 60×/NA1.42 oil immersion plan apochromat objective, mCherry filter (32% T, 25-msec exposure) and FITC filter (10% T, 25-msec exposure), at a frequency of 45 sec for 90 min.
The following pipeline was used to quantify kinetochore detachment events. First, the movie was maximum-projected using FIJI (Schindelin et al. 2012). Next, the time of spindle pole body (SPB) separation and each kinetochore detachment event were tracked. The time of SPB separation is defined as the first frame when two distinct mCherry foci were detected. Kinetochore attachment is defined as a CENV-GFP focus staying in the proximity of one SPB (less than a third of the pole-to-pole distance) for at least three consecutive time frames. A detachment event occurred when a CENV-GFP focus switched SPB or crossed the midline between the two poles (the CENV-GFP focus often stayed in the midzone for more than three consecutive frames before being recaptured by the original SPB). The total number of detachment events occurred after the splitting of the SPBs was quantified for each cell that maintained in the focal plane for the entire movie. The percentage of cells that underwent a given number of detachment events was calculated and graphed.
Denaturing immunoprecipitation (IP) and mass spectrometry (MS)
Denatured protein extracts were prepared as described in Sawyer et al. (2019), with the following modifications. After cells were harvested and dried completely, 150 µL of zirconia beads and 150 µL of lysis buffer (50 mM Tris at pH 7.5, 1 mM EDTA, 2.75 mM DTT, 1× PhosSTOP [Roche], 0.48 µg/mL Pefabloc SC [Sigma], 2 mM pepstatin A, 2× cOmplete EDTA Free [Roche]) were added to each tube. Cells were lysed mechanically on a Mini-Beadbeater-96 (BioSpec Products) for 5 min at room temperature. SDS was added to a final concentration of 1%, the extracts were boiled for 5 min at 95°C, and NP-40 lysis buffer (50 mM Tris at pH 7.5, 150 mM NaCl, 1% NP-40, 5% glycerol, 1× PhosSTOP, 0.64 µg/mL Pefabloc SC [Sigma], 2.67 mM pepstatin A, 3× cOmplete EDTA Free) was added to a final volume of 1.5 mL (i.e., diluting SDS to 0.1%). Lysates were clarified by centrifuging at 15,000 rcf for 15 min at 4°C. The total cleared lysate for each sample was pooled from three tubes (3825 µL total), added to 80 µL of pre-equilibrated anti-V5 agarose beads (Sigma), and incubated in loBind tubes (Eppendorf) for 2 h at 4°C with rotation. Using loBind tubes and low-adhesion tubes (USA Scientific) improved the amount of protein recovered from the IP. In each MS experiment, the cleared lysates were pooled from six tubes for each sample; therefore, two IP reactions were set up in parallel. The beads of the parallel IP reactions were washed once with the 3 mL of high-salt wash buffer (50 mM Tris at pH 7.5, 0.5 M NaCl, 1 mM EDTA, 1% NP-40), combined into one low-adhesion tube (i.e., 160 µL of beads final), and then washed once with 950 µL of high-salt wash buffer. Next, the beads were washed twice with each of the following: (1) 950 µL of buffer 2 (50 mM Tris at pH 7.5, 150 mM NaCl, 10 mM MgCl2, 0.05% NP-40, 5% glycerol) and (2) 950 µL of buffer 3 (50 mM Tris at pH 7.5, 150 mM NaCl, 10 mM MgCl2, 5% glycerol). After the last wash, the wash buffer was aspirated completely, and the beads were resuspended in 80 µL of trypsin buffer (2 M urea, 50 mM Tris at pH 7.5, 5 µg/mL trypsin) to digest the bound proteins for 1 h at 37°C with agitation. The beads were centrifuged at 100 rcf for 30 sec, and the digested peptides (the supernatant) were collected. The beads were then washed twice with 60 µL of urea buffer (2 M urea, 50 mM Tris at pH 7.5). The supernatant of both washes was collected and combined with the digested peptides (i.e., the final volume is 200 µL). After brief centrifugation, the combined digested peptides were cleared from residual beads and frozen in liquid nitrogen.
MS was performed by the Vincent J. Coates Proteomics/Mass Spectrometry Laboratory at the University of California at Berkeley, as described in Sawyer et al. (2019). The mudPIT method was used. A nano LC column (packed in a 100-µm inner diameter glass capillary with an emitter tip) was composed of 10 cm of Polaris c185-µm packing material (Varian), followed by 4 cm of Partisphere 5 SCX (Whatman). The column was loaded with a pressure bomb, washed extensively with buffer A, and then directly coupled to an electrospray ionization source mounted on a LTQ XL linear ion trap mass spectrometer (Thermo Fisher). Chromatography was performed on an Agilent 1200 HPLC equipped with a split line to deliver a flow rate of 300 nL/min. Peptides were eluted using either a four-step or an eight-step mudPIT procedure (Washburn et al. 2001). Buffer A was 5% acetonitrile and 0.02% heptafluorobutyric acid (HFBA). Buffer B was 80% acetonitrile and 0.02% HFBA. Buffer C was 250 mM ammonium acetate, 5% acetonitrile, and 0.02% HFBA. Buffer D was the same as buffer C, but with 500 mM ammonium acetate. Proteins were identified with the Integrated Proteomics Pipeline software (IP2; Integrated Proteomics Applications) using the parameters and cutoffs exactly as described by Sawyer et al. (2019). Information about the Ndc80 phosphorylation sites are provided in Supplemental Table S2.
Native IP, co-IP, and TMT quantitative MS
For the TMT quantitative MS and co-IP experiments, cells were sporulated in 2% SPO (2% [w/v] potassium acetate at pH 7, 0.02% [w/v] raffinose, supplemented with 40 mg/L uracil, 20 mg/L histidine, 20 mg/L leucine, 20 mg/L tryptophan) at 30°C. Increasing the concentration of acetate and supplementing amino acids improved sporulation efficiency. Four hours after the cells were transferred to 2% SPO, 200 mM PMSF (dissolved in ethanol) was added to cultures to a final concentration of 2 mM. For the MS and the co-IP experiments, 462 or 92 OD600 of cells, respectively, were harvested by centrifuging at 1700 rcf, 4°C, for 2 min. The supernatant was discarded, and the cell pellets were transferred to 2-mL tubes, centrifuged at 21,000 rcf for 1 min at 4°C. The supernatant was aspirated completely, and the pellets were snap-frozen in liquid nitrogen.
To extract proteins, pellets were thawed on ice, and 500 µL of NP-40 native IP buffer was added (50 mM Tris at pH 7.5, 150 mM NaCl, 2 mM MgCl2, 1% NP-40, supplemented with PhosSTOP tablet [one tablet used in 10 mL of buffer] [Sigma], cOmplete ULTRA protease inhibitor cocktail [one tablet used in 20 mL of buffer] [Sigma], 0.64 µg/mL Pefabloc SC [Sigma], 2.7 µM pepstatin A). Cells were lysed with 500 µL of zirconia beads (BioSpec) in a Fast-Prep-24 5G (MP Biomedicals LLC) using the “S. cerevisiae” setting for 40 sec at room temperature. Next, the cell lysates were chilled in an ice water bath for 2 min. The tubes were punctured with a 20.5-guage needle, and the lysates were collected in 15-mL tubes (precooled to −20°C) by centrifuging at 3000 rpm for 20–40 sec. Lysates were clarified by centrifugation at 21,000 rcf for 20 min at 4°C and snap-frozen in liquid nitrogen. The protein concentration of each lysate was determined by Bradford assay (Bio-Rad).
Ndc80-3V5 IP was performed at a protein concentration of 10 mg/mL in NP-40 native IP buffer. For the TMT quantitative MS experiment, a total of 38–40 mg of protein, pooled from 10 tubes of lysate, was added to 167 µL of pre-equilibrated anti-V5 agarose beads (Sigma) in a total volume of 4 mL (i.e., beads constituted 1/24 of the total IP volume), incubated for 2.5 h at 4°C with rotation, and then washed with each of the following: (1) once with 3.5 mL of NP-40 native IP buffer, (2) once with 950 µL of NP-40 native IP buffer, (3) twice with 950 µL of buffer 2, and (4) twice with 950 µL of buffer 3. After the last wash, the proteins on the beads were digested in trypsin buffer, washed with urea buffer, and frozen in liquid nitrogen as described in the “Denaturing immunoprecipitation (IP) and mass spectrometry (MS)”. The TMT quantitative MS was performed as described in Cheng et al. (2018). The raw data can be accessed at MassIVE MSV000084664 (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp).
The co-IP experiments were performed similarly, except for the following changes: A total of 4 mg of protein was incubated with 27 µL of anti-V5 agarose beads in a total volume of 400 µL and washed twice with 950 µL of NP-40 native IP buffer before washing with buffer 2 and buffer 3. After the last wash, the wash buffer was aspirated until 100 µL was left, and 50 µL of 3× SDS sample buffer (187.5 mM Tris at pH 6.8, 6% mercaptoethanol, 30% glycerol, 9% SDS, 0.05% bromophenol blue) was added to the beads before boiling for 5 min. After brief centrifugation, the eluted proteins were separated by SDS-PAGE as described in the immunoblotting section. A detailed protocol for the IP/TMT mass spectrometry is provided in Supplemental Table S3.
Purification of the Ndc80 complex
Native Ndc80 complex was purified from asynchronously growing S. cerevisiae cells (UB16284 and UB19957) through anti-Flag immunoprecipitation of Spc24-6His-3Flag as described in Miller et al. (2016). Cells were grown in YPD. Protein lysates were prepared by lysing cells in a Freezer/Mill (SPEX SamplePrep) submerged in liquid nitrogen (Sarangapani et al. 2014). Lysed cells were resuspended in buffer H (BH) (25 mM HEPES at pH 8.0, 2 mM MgCl2, 0.1 mM EDTA, 0.5 mM EGTA, 0.1% NP-40, 15% glycerol, 750 mM KCl), supplemented with the protease inhibitors (20 µg/mL leupeptin, 20 µg/mL pepstatin A, 20 µg/mL chymostatin, 200 µM phenylmethylsulfonyl fluoride) and the phosphatase inhibitors (0.1 mM Na-orthovanadate, 0.2 µM microcystin, 2 mM β-glycerophosphate, 1 mM Na pyrophosphate, 5 mM NaF). The resuspended cell lysates were ultracentrifuged at 98,500g for 90 min at 4°C. Dynabeads conjugated with anti-Flag antibody (Sigma) were incubated with extract for 3 h at 4°C with constant rotation, followed by three washes with BH containing the protease inhibitors, phosphatase inhibitors, 2 mM DTT, and 1 M KCl. The beads were further washed twice with BH containing 150 mM KCl and the protease inhibitors. Associated proteins were eluted from the beads by gently agitating the beads in elution buffer (0.5 mg/mL 3Flag peptide in BH with 150 mM KCl and the protease inhibitors) for 30 min at room temperature.
In vitro kinase assay
The kinase assay was performed using a recombinant variant of Ipl1 (AurB*) that was created by fusing the C-terminal activation box of Sli15 to Ipl1 (a kind gift from S. Biggins's laboratory, the Fred Hutchinson Cancer Research Center). The kinase-dead (KD) mutant of AurB* contains the K133R mutation. These two constructs were first reported in de Regt et al. (2018). To begin the reaction, in a volume of 6 µL, 6.63 ng of the purified WT-Ndc80 or Ndc80(Δ2–28) was incubated with 1 µM AurB* or KD in BH0.15 (same ingredients as BH in the previous section except with 150 mM KCl) for 4 min at room temperature. Next, 6 µL of hot ATP mix (1.1 µL of 100 mM ATP, 3 µL of 6000 Ci/mmol 10 mCi/mL g-32P ATP, 275 µL of BH0.15) was added to the kinase/substrate mix. After 1 or 5 min of incubation at 25°C (see the figure legend for details), reactions were stopped by adding 6 µL of 3× SDS sample buffer, and boiling for 5–10 min at 95°C. Proteins were separated by SDS-PAGE as described in “Immunoblotting.” The gel was fixed and silver stained based on the manufacturer's instructions (Thermo Scientific). After the staining was stopped by incubating the gel in 5% acetic acid for 10 min, the gel was dried by vacuum for 1 h at 80°C. The dried gel was exposed to a phosphor storage screen for over 24 h. Screens were imaged with a Typhoon Scanner (GE Healthcare).
Immunoblotting
To extract proteins from cells, approximately four OD600 units of cells were treated with 5% trichloroacetic acid for at least 15 min at 4°C. After an acetone wash, the cell pellet was air dried, lysed with acid-washed glass beads (Sigma) in 100 µL of lysis buffer (50 mM Tris at pH 7.5, 1 mM EDTA, 2.75 mM DTT, cOmplete EDTA Free protease inhibitor cocktail [Roche]) using a Mini-Beadbeater-96 (Biospec Products). Next, 3× SDS sample buffer (187.5 mM Tris at pH 6.8, 6% 2-mercaptoethanol, 30% glycerol, 9% SDS, 0.05% bromophenol blue) was added and the lysate was boiled for 5 min. Proteins were separated by SDS-PAGE using 4%–12% Bis-Tris Bolt gels (Thermo Fisher) and transferred onto 0.45-μm nitrocellulose membranes (Bio-Rad) using a semidry transfer apparatus (Trans-Blot Turbo Transfer System, Bio-Rad). The membranes were blocked for >30 min with Odyssey blocking buffer (PBS) (LI-COR Biosciences) and incubated in primary antibodies overnight at 4°C. Membranes were washed with PBST (phosphate-buffered saline with 0.01% Tween-20) and incubated with an antimouse secondary antibody conjugated to IRDye 800CW at a 1:15,000 dilution (LI-COR Biosciences 926-32212) and/or an antirabbit antibody conjugated to IRDye 680RD at a 1:15,000 dilution (LI-COR Biosciences 926-68071) for >1 h at room temperature. Immunoblots were imaged and quantified using the Odyssey system (LI-COR Biosciences).
Antibodies used in this study were mouse anti-V5 antibody (Thermo Fisher R960-25) used at a 1:2000 dilution; rabbit antihexokinase antibody (US Biological H2035) used at a 1:20,000 dilution; rabbit anti-Sis1 antibody (kind gift from E. Craig) used at a 1:5000 dilution, and a rabbit anti-H3 serine 10 phosphorylation antibody (Sigma 06-570) used at 1:1000 dilution.
The levels of each protein of interest were typically normalized to Hxk2, except for in Figure 6F. For the quantification in Figure 6F, Ndc80 levels were not normalized to that of Hxk2 because Hxk2 levels declined after cycloheximide treatment for the ama1Δ strain. To reflect the experimental variations, we performed five independent biological replicates and graphed the mean and the standard errors of the mean for all five replicates.
Spot growth assay
Cells were grown on YPG (2% glycerol + YEP) plates overnight, resuspended in milliQ water, and then diluted to an OD600 of 0.1 or 0.2. Fivefold serial dilutions were performed. Cells were spotted onto YPD plates or YPD plates supplemented with 15 µg/mL benomyl (benomyl plates). The cells on the YPD plates were incubated for 1–2 d at 30°C, and those on benomyl plates were incubated for 2 d at 23°C.
Reproducibility
All of the experiments in this study were performed at least twice independently, and one representative result was shown for all Western blots. All of the RT-qPCR experiments were performed three times independently, with the mean and standard errors of the mean graphed.
Supplementary Material
Acknowledgments
We thank Michael Rape, David Drubin, Rebecca Heald, and all members of the Ünal and Brar laboratories for experimental suggestions and critiques of this manuscript. We thank Sue Biggins and Anna de Regt for the recombinant Aurora B proteins, as well as Rachel Cohen-Kupiec for technical support. This work was supported by funds from the Pew Charitable Trusts (00027344), Damon Runyon Cancer Research Foundation (35-15), and National Institutes of Health (DP2 AG055946-01) to E.Ü., and a National Science Foundation Graduate Research Fellowship (grant no. DGE-1106400) to J.C.
Author contributions: J.C. conceived the study; performed the data curation, formal analysis, investigation, methodology, validation, and visualization; and wrote the draft of the manuscript. A.L., E.N.P., and H.L. performed the investigation, formal analysis, validation, and visualization and edited the manuscript. L.A.K. performed the investigation, data curation, methodology, and formal analysis for the denaturing IP and edited the manuscript. R.E. provided key resources (purifying the Ndc80 complex) and edited the manuscript. R.M.H. performed the investigation for the truncation analysis of Ndc80 and edited the manuscript. J.K.K. performed the investigation and formal analysis for quantitative mass spectrometry. M.J. supervised the study and was responsible for the project administration. E.Ü. conceived the study; performed the formal analysis, methodology, and visualization; supervised the study; was responsible for project administration and funding acquisition; and wrote the draft of the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.333997.119.
References
- Ahuja JS, Sandhu R, Mainpal R, Lawson C, Henley H, Hunt PA, Yanowitz JL, Börner GV. 2017. Control of meiotic pairing and recombination by chromosomally tethered 26S proteasome. Science 355: 408–411. 10.1126/science.aaf4778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyoshi B, Nelson CR, Ranish JA, Biggins S. 2009. Analysis of Ipl1-mediated phosphorylation of the Ndc80 kinetochore protein in Saccharomyces cerevisiae. Genetics 183: 1591–1595. 10.1534/genetics.109.109041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyoshi B, Nelson CR, Duggan N, Ceto S, Ranish JA, Biggins S. 2013. The Mub1/Ubr2 ubiquitin ligase complex regulates the conserved Dsn1 kinetochore protein. PLoS Genet 9: e1003216 10.1371/journal.pgen.1003216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Memisoglu G, Haber J. 2017. Cas9-mediated gene editing in Saccharomyces cerevisiae. Protocol Exchange 10.1038/protex.2017.021a [DOI] [Google Scholar]
- Asakawa H, Hayashi A, Haraguchi T, Hiraoka Y. 2005. Dissociation of the Nuf2-Ndc80 complex releases centromeres from the spindle-pole body during meiotic prophase in fission yeast. Mol Biol Cell 16: 2325–2338. 10.1091/mbc.e04-11-0996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au WC, Crisp MJ, DeLuca SZ, Rando OJ, Basrai MA. 2008. Altered dosage and mislocalization of histone H3 and Cse4p lead to chromosome loss in Saccharomyces cerevisiae. Genetics 179: 263–275. 10.1534/genetics.108.088518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchowitz LE, Gajadhar AS, van Werven FJ, De Rosa AA, Samoylova ML, Brar GA, Xu Y, Xiao C, Futcher B, Weissman JS, et al. 2013. A developmentally regulated translational control pathway establishes the meiotic chromosome segregation pattern. Genes Dev 27: 2147–2163. 10.1101/gad.224253.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S. 2013. The composition, functions, and regulation of the budding yeast kinetochore. Genetics 194: 817–846. 10.1534/genetics.112.145276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzblau HG, Chan CS, Hochwagen A, Bell SP. 2012. Separation of DNA replication from the assembly of break-competent meiotic chromosomes. PLoS Genet 8: e1002643 10.1371/journal.pgen.1002643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Hochwagen A, Ee LS, Amon A. 2009. The multiple roles of cohesin in meiotic chromosome morphogenesis and pairing. Mol Biol Cell 20: 1030–1047. 10.1091/mbc.e08-06-0637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Alani E, Kleckner N. 1990. A pathway for generation and processing of double-strand breaks during meiotic recombination in S. cerevisiae. Cell 61: 1089–1101. 10.1016/0092-8674(90)90072-M [DOI] [PubMed] [Google Scholar]
- Carlile TM, Amon A. 2008. Meiosis I is established through division-specific translational control of a cyclin. Cell 133: 280–291. 10.1016/j.cell.2008.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheerambathur DK, Prevo B, Chow TL, Hattersley N, Wang S, Zhao Z, Kim T, Gerson- Gurwitz A, Oegema K, Green R, et al. 2019. The kinetochore-microtubule coupling machinery is repurposed in sensory nervous system morphogenesis. Dev Cell 48: 864–872.e7. 10.1016/j.devcel.2019.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Drubin DG, Barnes G. 2002. Simple centromere, complex kinetochore: linking spindle microtubules and centromeric DNA in budding yeast. J Cell Biol 157: 199–203. 10.1083/jcb.200201052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Riley DJ, Chen PL, Lee WH. 1997. HEC, a novel nuclear protein rich in leucine heptad repeats specifically involved in mitosis. Mol Cell Biol 17: 6049–6056. 10.1128/MCB.17.10.6049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Tresenrider A, Chia M, McSwiggen DT, Spedale G, Jorgensen V, Liao H, van Werven FJ, Unal E. 2017. Kinetochore inactivation by expression of a repressive mRNA. Elife 6: e27417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhao J, Jiao Z, Wang W, Wang D, Yu X, Shi Z, Ge N, Pan Q, Xia J, et al. 2018. SKA1 overexpression is associated with poor prognosis in hepatocellular carcinoma. BMC Cancer 18: 1240 10.1186/s12885-018-5119-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Bao X, Gan X, Luo S, Rao H. 2017. Multiple E3s promote the degradation of histone H3 variant Cse4. Sci Rep 7: 8565 10.1038/s41598-017-08923-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Otto GM, Powers EN, Keskin A, Mertins P, Carr SA, Jovanovic M, Brar GA. 2018. Pervasive, coordinated protein-level changes driven by transcript isoform switching during meiosis. Cell 172: 910–923.e16. 10.1016/j.cell.2018.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia M, Tresenrider A, Chen J, Spedale G, Jorgensen V, Unal E, van Werven FJ. 2017. Transcription of a 5′ extended mRNA isoform directs dynamic chromatin changes and interference of a downstream promoter. Elife 6: e27420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Roca AC, Muscat CC, Wignall SM. 2017. Caenorhabditis elegans oocytes detect meiotic errors in the absence of canonical end-on kinetochore attachments. J Cell Biol 216: 1243–1253. 10.1083/jcb.201608042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Regt AK, Asbury CL, Biggins S. 2018. Tension on kinetochore substrates is insufficient to prevent Aurora-triggered detachment. bioRxiv 10.1101/415992 [DOI]
- Dhatchinamoorthy K, Shivaraju M, Lange JJ, Rubinstein B, Unruh JR, Slaughter BD, Gerton JL. 2017. Structural plasticity of the living kinetochore. J Cell Biol 216: 3551–3570. 10.1083/jcb.201703152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhatchinamoorthy K, Unruh JR, Lange JJ, Levy M, Slaughter BD, Gerton JL. 2019. The stoichiometry of the outer kinetochore is modulated by microtubule-proximal regulatory factors. J Cell Biol 218: 2124–2135. 10.1083/jcb.201810070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond AE, Park JS, Inoue I, Tachikawa H, Neiman AM. 2009. The anaphase promoting complex targeting subunit Ama1 links meiotic exit to cytokinesis during sporulation in Saccharomyces cerevisiae. Mol Biol Cell 20: 134–145. 10.1091/mbc.e08-06-0615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont J, Oegema K, Desai A. 2010. A kinetochore-independent mechanism drives anaphase chromosome separation during acentrosomal meiosis. Nat Cell Biol 12: 894–901. 10.1038/ncb2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti C, Totta P, Fiore M, Mattiuzzo M, Schillaci T, Ricordy R, Di Leonardo A, Degrassi F. 2010. Expression of the kinetochore protein Hec1 during the cell cycle in normal and cancer cells and its regulation by the pRb pathway. Cell Cycle 9: 4174–4182. 10.4161/cc.9.20.13457 [DOI] [PubMed] [Google Scholar]
- Finley D, Ulrich HD, Sommer T, Kaiser P. 2012. The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 192: 319–360. 10.1534/genetics.112.140467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Cheeseman IM. 2013. CDK-dependent phosphorylation and nuclear exclusion coordinately control kinetochore assembly state. J Cell Biol 201: 23–32. 10.1083/jcb.201301006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- Giroux CN, Dresser ME, Tiano HF. 1989. Genetic control of chromosome synapsis in yeast meiosis. Genome 31: 88–94. 10.1139/g89-017 [DOI] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW. 1991. Cyclin is degraded by the ubiquitin pathway. Nature 349: 132–138. 10.1038/349132a0 [DOI] [PubMed] [Google Scholar]
- Hayama S, Daigo Y, Kato T, Ishikawa N, Yamabuki T, Miyamoto M, Ito T, Tsuchiya E, Kondo S, Nakamura Y. 2006. Activation of CDCA1-KNTC2, members of centromere protein complex, involved in pulmonary carcinogenesis. Cancer Res 66: 10339–10348. 10.1158/0008-5472.CAN-06-2137 [DOI] [PubMed] [Google Scholar]
- Herrero E, Thorpe PH. 2016. Synergistic control of kinetochore protein levels by Psh1 and Ubr2. PLoS Genet 12: e1005855 10.1371/journal.pgen.1005855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heun P, Erhardt S, Blower MD, Weiss S, Skora AD, Karpen GH. 2006. Mislocalization of the Drosophila centromere-specific histone CID promotes formation of functional ectopic kinetochores. Dev Cell 10: 303–315. 10.1016/j.devcel.2006.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochwagen A, Tham WH, Brar GA, Amon A. 2005. The FK506 binding protein Fpr3 counteracts protein phosphatase 1 to maintain meiotic recombination checkpoint activity. Cell 122: 861–873. 10.1016/j.cell.2005.07.010 [DOI] [PubMed] [Google Scholar]
- Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, Grushcow JM, Brame CJ, Caldwell JA, Hunt DF, et al. 2000. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell 102: 279–291. 10.1016/S0092-8674(00)00034-9 [DOI] [PubMed] [Google Scholar]
- Hwang LH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW. 1998. Budding yeast Cdc20: a target of the spindle checkpoint. Science 279: 1041–1044. 10.1126/science.279.5353.1041 [DOI] [PubMed] [Google Scholar]
- Hyland KM, Kingsbury J, Koshland D, Hieter P. 1999. Ctf19p: a novel kinetochore protein in Saccharomyces cerevisiae and a potential link between the kinetochore and mitotic spindle. J Cell Biol 145: 15–28. 10.1083/jcb.145.1.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA. 2010. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11: 579–592. 10.1038/nrm2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Hyman AA, Sorger PK. 1997. Regulating the yeast kinetochore by ubiquitin-dependent degradation and Skp1p-mediated phosphorylation. Cell 91: 491–500. 10.1016/S0092-8674(00)80435-3 [DOI] [PubMed] [Google Scholar]
- Keeney S, Giroux CN, Kleckner N. 1997. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88: 375–384. 10.1016/S0092-8674(00)81876-0 [DOI] [PubMed] [Google Scholar]
- Kim S, Meyer R, Chuong H, Dawson DS. 2013. Dual mechanisms prevent premature chromosome segregation during meiosis. Genes Dev 27: 2139–2146. 10.1101/gad.227454.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapholz S, Waddell CS, Esposito RE. 1985. The role of the SPO11 gene in meiotic recombination in yeast. Genetics 110: 187–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster MJ, Timmers HT. 2015. Regulation of anti-sense transcription by Mot1p and NC2 via removal of TATA-binding protein (TBP) from the 3′-end of genes. Nucleic Acids Res 43: 143–152. 10.1093/nar/gku1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegenburg F, Jakopec V, Poulsen EG, Nielsen SV, Roguev A, Krogan N, Gordon C, Fleig U, Hartmann-Petersen R. 2014. A chaperone-assisted degradation pathway targets kinetochore proteins to ensure genome stability. PLoS Genet 10: e1004140 10.1371/journal.pgen.1004140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laband K, Le Borgne R, Edwards F, Stefanutti M, Canman JC, Verbavatz JM, Dumont J. 2017. Chromosome segregation occurs by microtubule pushing in oocytes. Nat Commun 8: 1499 10.1038/s41467-017-01539-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Amon A. 2003. Role of Polo-like kinase CDC5 in programming meiosis I chromosome segregation. Science 300: 482–486. 10.1126/science.1081846 [DOI] [PubMed] [Google Scholar]
- Li R, Murray AW. 1991. Feedback control of mitosis in budding yeast. Cell 66: 519–531. 10.1016/0092-8674(81)90015-5 [DOI] [PubMed] [Google Scholar]
- Li J, Xuan JW, Khatamianfar V, Valiyeva F, Moussa M, Sadek A, Yang BB, Dong BJ, Huang YR, Gao WQ. 2014. SKA1 over-expression promotes centriole over-duplication, centrosome amplification and prostate tumourigenesis. J Pathol 234: 178–189. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961. [DOI] [PubMed] [Google Scholar]
- Maier P, Rathfelder N, Finkbeiner MG, Taxis C, Mazza M, Le Panse S, Haguenauer-Tsapis R, Knop M. 2007. Cytokinesis in yeast meiosis depends on the regulated removal of Ssp1p from the prospore membrane. EMBO J 26: 1843–1852. 10.1038/sj.emboj.7601621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RE, Kim S, Obeso D, Straight PD, Winey M, Dawson DS. 2013. Mps1 and Ipl1/Aurora B act sequentially to correctly orient chromosomes on the meiotic spindle of budding yeast. Science 339: 1071–1074. 10.1126/science.1232518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RE, Chuong HH, Hild M, Hansen CL, Kinter M, Dawson DS. 2015. Ipl1/Aurora-B is necessary for kinetochore restructuring in meiosis I in Saccharomyces cerevisiae. Mol Biol Cell 26: 2986–3000. 10.1091/mbc.E15-01-0032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K. 1997. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91: 35–45. 10.1016/S0092-8674(01)80007-6 [DOI] [PubMed] [Google Scholar]
- Miller MP, Ünal E, Brar GA, Amon A. 2012. Meiosis I chromosome segregation is established through regulation of microtubule-kinetochore interactions. Elife 1: e00117 10.7554/eLife.00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MP, Asbury CL, Biggins S. 2016. A TOG protein confers tension sensitivity to kinetochore-microtubule attachments. Cell 165: 1428–1439. 10.1016/j.cell.2016.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscat CC, Torre-Santiago KM, Tran MV, Powers JA, Wignall SM. 2015. Kinetochore-independent chromosome segregation driven by lateral microtubule bundles. Elife 4: e06462 10.7554/eLife.06462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelschlaegel T, Schwickart M, Matos J, Bogdanova A, Camasses A, Havlis J, Shevchenko A, Zachariae W. 2005. The yeast APC/C subunit Mnd2 prevents premature sister chromatid separation triggered by the meiosis-specific APC/C-Ama1. Cell 120: 773–788. 10.1016/j.cell.2005.01.032 [DOI] [PubMed] [Google Scholar]
- Ohkuni K, Takahashi Y, Fulp A, Lawrimore J, Au WC, Pasupala N, Levy-Myers R, Warren J, Strunnikov A, Baker RE, et al. 2016. SUMO-targeted ubiquitin ligase (STUbL) Slx5 regulates proteolysis of centromeric histone H3 variant Cse4 and prevents its mislocalization to euchromatin. Mol Biol Cell 27: 1500–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okaz E, Argüello-Miranda O, Bogdanova A, Vinod PK, Lipp JJ, Markova Z, Zagoriy I, Novak B, Zachariae W. 2012. Meiotic prophase requires proteolysis of M phase regulators mediated by the meiosis-specific APC/CAma1. Cell 151: 603–618. 10.1016/j.cell.2012.08.044 [DOI] [PubMed] [Google Scholar]
- Ottoz DS, Rudolf F, Stelling J. 2014. Inducible, tightly regulated and growth condition-independent transcription factor in Saccharomyces cerevisiae. Nucleic Acids Res 42: e130 10.1093/nar/gku616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. 2008. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5: 877–879. 10.1038/nmeth.1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano A, Guse A, Krascenicova I, Schnabel H, Schnabel R, Glotzer M. 2003. CSC-1: a subunit of the Aurora B kinase complex that binds to the survivin-like protein BIR-1 and the incenp-like protein ICP-1. J Cell Biol 161: 229–236. 10.1083/jcb.200207117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarangapani KK, Duro E, Deng Y, Alves Fde L, Ye Q, Opoku KN, Ceto S, Rappsilber J, Corbett KD, Biggins S, et al. 2014. Sister kinetochores are mechanically fused during meiosis I in yeast. Science 346: 248–251. 10.1126/science.1256729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer EM, Joshi PR, Jorgensen V, Yunus J, Berchowitz LE, Ünal E. 2019. Developmental regulation of an organelle tether coordinates mitochondrial remodeling in meiosis. J Cell Biol 218: 559–579. 10.1083/jcb.201807097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher JM, Golden A, Donovan PJ. 1998. AIR-2: an Aurora/Ipl1-related protein kinase associated with chromosomes and midbody microtubules is required for polar body extrusion and cytokinesis in Caenorhabditis elegans embryos. J Cell Biol 143: 1635–1646. 10.1083/jcb.143.6.1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Yang M, Lin Q, Zhang Z, Miao C, Zhu B. 2016. SKA1 regulates the metastasis and cisplatin resistance of non-small cell lung cancer. Oncol Rep 35: 2561–2568. 10.3892/or.2016.4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirk K, Jin H, Giddings TH Jr, Winey M, Yu HG. 2011. The Aurora kinase Ipl1 is necessary for spindle pole body cohesion during budding yeast meiosis. J Cell Sci 124: 2891–2896. 10.1242/jcs.086652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonn MA, McCarroll R, Murray AW. 2000. Requirement of the spindle checkpoint for proper chromosome segregation in budding yeast meiosis. Science 289: 300–303. 10.1126/science.289.5477.300 [DOI] [PubMed] [Google Scholar]
- Shrestha RL, Ahn GS, Staples MI, Sathyan KM, Karpova TS, Foltz DR, Basrai MA. 2017. Mislocalization of centromeric histone H3 variant CENP-A contributes to chromosomal instability (CIN) in human cells. Oncotarget 8: 46781–46800. 10.18632/oncotarget.18108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes EK, Uren A, Vaux D, Horvitz HR. 2000. The survivin-like C. elegans BIR-1 protein acts with the Aurora-like kinase AIR-2 to affect chromosomes and the spindle midzone. Mol Cell 6: 211–223. 10.1016/S1097-2765(00)00023-X [DOI] [PubMed] [Google Scholar]
- Spencer F, Gerring SL, Connelly C, Hieter P. 1990. Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics 124: 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC, Zhang DX, Lee SE, Xu YN, Kim NH. 2011. Ndc80 regulates meiotic spindle organization, chromosome alignment, and cell cycle progression in mouse oocytes. Microsc Microanal 17: 431–439. 10.1017/S1431927611000274 [DOI] [PubMed] [Google Scholar]
- Tsuchiya D, Gonzalez C, Lacefield S. 2011. The spindle checkpoint protein Mad2 regulates APC/C activity during prometaphase and metaphase of meiosis I in Saccharomyces cerevisiae. Mol Biol Cell 22: 2848–2861. 10.1091/mbc.e11-04-0378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A. 1997. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science 278: 460–463. 10.1126/science.278.5337.460 [DOI] [PubMed] [Google Scholar]
- Washburn MP, Wolters D, Yates JR III. 2001. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 19: 242-247. 10.1038/85686 [DOI] [PubMed] [Google Scholar]
- Wigge PA, Jensen ON, Holmes S, Souès S, Mann M, Kilmartin JV. 1998. Analysis of the Saccharomyces spindle pole by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. J Cell Biol 141: 967–977. 10.1083/jcb.141.4.967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. 2007. Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell 27: 3–16. 10.1016/j.molcel.2007.06.009 [DOI] [PubMed] [Google Scholar]
- Zhao G, Oztan A, Ye Y, Schwarz TL. 2019. Kinetochore proteins have a post-mitotic function in neurodevelopment. Dev Cell 48: 873–882.e4. 10.1016/j.devcel.2019.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.