

To establish infection and complete the viral life cycle, human papillomavirus (HPV) needs to alter the transcriptional program of host cells. Until recently, studies were restricted to keratinocyte-derived cell lines immortalized by HPV due to the lack of experimental systems to efficiently infect primary keratinocytes. Need for selection and immortalization made it impossible to distinguish between alterations induced by HPV and secondary adaptation due to selection and immortalization. With our recent establishment of an extracellular matrix (ECM)-to-cell transfer system allowing efficient infection of primary keratinocytes, we were able to identify transcriptional changes attributable to HPV16 infection. Most perturbed genes fall into the class of S-phase genes, which are regulated by pocket proteins. Indeed, infection with viruses lacking E7 abrogated most transcriptional changes. It is important to note that many transcriptional alterations thought to be important for the HPV life cycle are actually late events that may reflect immortalization and, possibly, disease progression.
KEYWORDS: E7, HPV16, RB, infection model, organotypic raft culture, p53, pocket protein, primary foreskin keratinocytes, transcriptome
ABSTRACT
It is established that the host cell transcriptomes of natural lesions, organotypic rafts, and human papillomavirus (HPV)-immortalized keratinocytes are altered in the presence of HPV genomes. However, the establishment of HPV-harboring cell lines requires selection and immortalization, which makes it impossible to distinguish between alterations directly induced by HPV or indirectly by the need for immortalization or selection. To address direct effects of HPV infection on the host cell transcriptome, we have used our recently established infection model that allows efficient infection of primary keratinocytes with HPV16 virions. We observed only a small set of genes to be deregulated at the transcriptional level at 7 days postinfection (dpi), most of which fall into the category regulated by pocket proteins pRb, p107, and p130. Furthermore, cell cycle genes were not deregulated in cells infected with a virus lacking E7 despite the presence of episomal genome and viral transcripts. These findings imply that the majority of transcriptional changes are due to the E7 protein impairing pocket protein function. Additional pathways, such as the Fanconi anemia-BRCA pathway, became perturbed only after long-term culturing of infected cells. When grown as organotypic raft cultures, keratinocytes infected with wild-type but not E7 mutant virus had perturbed transcriptional regulation of pathways previously identified in natural lesions and in rafts derived from immortalized keratinocytes. We conclude that the HPV infection model provides a valuable tool to distinguish immediate transcriptional alterations from those induced by persistent infection and the need for selection and immortalization.
IMPORTANCE To establish infection and complete the viral life cycle, human papillomavirus (HPV) needs to alter the transcriptional program of host cells. Until recently, studies were restricted to keratinocyte-derived cell lines immortalized by HPV due to the lack of experimental systems to efficiently infect primary keratinocytes. Need for selection and immortalization made it impossible to distinguish between alterations induced by HPV and secondary adaptation due to selection and immortalization. With our recent establishment of an extracellular matrix (ECM)-to-cell transfer system allowing efficient infection of primary keratinocytes, we were able to identify transcriptional changes attributable to HPV16 infection. Most perturbed genes fall into the class of S-phase genes, which are regulated by pocket proteins. Indeed, infection with viruses lacking E7 abrogated most transcriptional changes. It is important to note that many transcriptional alterations thought to be important for the HPV life cycle are actually late events that may reflect immortalization and, possibly, disease progression.
INTRODUCTION
Infection by high-risk human papillomaviruses (HPVs) such as HPV16 is linked to epithelial tumors (1). Cervical and oropharyngeal squamous cell carcinomas are the most prevalent tumors associated with HPV infection. HPV is ubiquitous in the human population and infections are common. However, only a small fraction of HPV infections results in clinical disease, typically benign self-limited outgrowth of the skin and mucosa, and an even smaller percentage results in cancer. In cancers driven by HPV infection, the viral genome is often integrated, resulting in loss of viral functions linked to viral genome replication and transcriptional regulation and thus an aborted life cycle.
The productive life cycle of HPV depends on the terminal differentiation process of the stratified epithelia (1). Viruses gain access to the basal cell compartment of the tissue through (micro)lesions. They preferentially bind to the basement membrane from which they are endocytosed by keratinocytes (2). After infectious entry, the viral genome is amplified and infection established. During latency and genome maintenance, copy number remains unchanged and the viral transcription is tightly regulated. When infected cells enter the terminal differentiation program, viral promoters are activated, the viral genome is amplified, capsid proteins are expressed, and progeny virions are assembled and released (3, 4). In addition to viral replication factors E1 and E2, which initiate replication and function as a helicase (E1), the cellular replication machinery is required for genome amplification. Normally, keratinocytes entering the terminal differentiation program withdraw from the cell cycle. However, in the presence of E7, one of the viral oncoproteins, keratinocytes are retained in the cell cycle, allowing for expression of S-phase genes such as cellular replication factors (5). Another viral oncoprotein, E6, binds to p53 to prevent cell cycle arrest due to unscheduled DNA replication (6, 7).
It is well established that the presence of HPV genomes in keratinocytes deregulates host cell gene expression, mostly due to expression of the two virally encoded oncoproteins E6 and E7 (8). While E6 interacts with and induces the degradation of p53, thus abrogating p53 transcriptional regulatory activity, E7 protein binds to and induces the degradation of pocket proteins such as pRb, p107, and p130 (9 – 11). This results in the release of members of the E2F transcription factor family and the expression of S-phase genes (12). E7-mediated degradation of p107 and p130 has also been linked to release of DREAM complex-mediated gene repression (13, 14). Additionally, many reports have demonstrated interactions of E7 protein with other host cell factors involved in multiple regulatory pathways with downstream transcriptional targets (for review see reference 5 and 15).
Global transcriptome analyses have been applied to HPV-harboring patient tissue samples comparing tissues of differing severity (16 – 20), HPV-immortalized established cell lines (21 – 24), and cell lines individually expressing viral oncoproteins (25 – 27). The analysis of patient tissue samples has identified genes that are deregulated in the stratified epithelium and allowed the identification of putative markers of tumor progression (16). The analysis of HPV-immortalized cells, whether harboring the complete viral genome or individual viral genes, has the caveat that the establishment of these cell lines requires immortalization and outgrowth of primary keratinocytes. Therefore, it is difficult to distinguish between virally induced alterations and those that are due to the selection, immortalization, and long-term culturing of cells. The study of the transcriptome of cells immediately after viral infection without the need for immortalization and selection was hampered by the lack of an efficient infection-based cell culture model using primary keratinocytes. For unknown reasons, primary keratinocytes did not readily endocytose HPV virions, which instead remained bound to the cell surface (28).
We have recently reported a new infection model that allows efficient infection of primary keratinocytes via the basolateral surface with HPV16 virions produced in the 293TT packaging cell line (29). This model system not only greatly facilitates the study of immediate early events of the viral life cycle following infectious delivery of the viral genome but is also amenable to the genetic dissection of viral factors important for establishing and maintaining an episomal viral genome absent of any need for immortalization. Herein, we compare changes to the host cell transcriptome after HPV16 infection without the need for immortalization and compare the alterations to those observed in human foreskin-derived primary keratinocytes (HFK) infected with a mutant lacking E7 protein and HPV16-immortalized HFK.
RESULTS
HPV16 infection results in limited changes to the host transcriptome compared to that of HPV16-immortalized cells.
To examine HPV16-induced changes in the cell host transcriptome, primary human foreskin-derived keratinocytes (HFK) were infected with HPV16 virions for 7 days using the extracellular matrix (ECM)-to-cell transfer system recently described. To facilitate infection, the cell culture medium was supplemented with the Rho kinase (ROCK) inhibitor Y-27632 and grown in the presence of mouse fibroblasts as feeder cells (30, 31). We recently used RNAscope and reverse transcription-quantitative PCR to correlate the efficiency of infection with transcript levels (Fig. 1A and Table 1). The controls were mock-infected and HPV16-immortalized HFK grown under the same conditions for at least 7 days. HPV16-immortalized cell lines were established after the transfection of the viral genome into primary HFK, selection, and outgrowth of an immortalized population as previously described. RNAs were isolated and subjected to next-generation RNA-sequencing (RNA-seq). RNA-seq was carried out on RNAs obtained from triplicate biological populations of HFK grown in monolayer culture under nondifferentiating conditions. Based on E7 transcript levels (difference in cycle thresholds [ΔCT] [E7 – cyclophilin A] values of 1.6, 3.4, and 3.3) in samples subjected to RNA-seq, we estimate that at least 50% of cells had been infected at the time of harvest (compare to Table 1).
FIG 1.

HPV16-induced changes to the host cell transcriptome at 7 dpi. (A) Representative RNAscope images of HPV16-infected HFK at 7 dpi. (B) Transcripts per million (TPM) values of viral E6 and E1̂E4 transcript reads in samples from HPV16-infected and -immortalized cells grown as a monolayer in the presence of Y-27632. (C) Numbers of genes whose expression was significantly modified in HPV16-infected and -immortalized cells. (D) Heat map illustrating the cluster of genes whose expression was significantly modified in HPV16-immortalized cells analyzed as sample groups from controls and HPV16-immortalized and HPV16-infected cells grown in the presence of Y-27632. (E) Venn diagram showing the number of altered host genes common to HPV16-infected and -immortalized cells grown in the presence of Y-27632. (F) Principal-component analysis (PCA) diagram of RNA sequencing results from HPV16-infected and -immortalized cells grown as a monolayer with and without Y-27632 based on TPM values in the bulk RNA-seq data set. Similarities between data sets are correlated to the distances in the projection of the space defined by the principal components.
TABLE 1.
Efficiency of infection of HFK with HPV16 at 7 dpi
CT of viral E7 minus CT of housekeeping gene (CyPA) transcripts.
Percentage of E6/E7-positive cells in RNAscope.
As described previously, viral transcript levels in HPV16-infected HFK were consistently lower than in HPV16-immortalized keratinocytes, with variations due to the efficiency of infection (Fig. 1A). Transcript levels of almost 6,000 genes were altered in HPV16-immortalized HFK compared to that in HFK controls grown under identical conditions, of which 3,196 and 2,658 were up- and downregulated, respectively. In contrast, the expression of only 113 genes was deregulated in HFK at 7 days postinfection (dpi) with HPV16, 98 of which were significantly upregulated (Fig. 1B and C). Ninety-five genes were common between HPV16-infected and -immortalized keratinocytes (Fig. 1D). Principal-component analysis (PCA) demonstrated that HPV16-infected HFK cluster closely with mock-infected but not with HPV16-immortalized HFK (Fig. 1E). The horizontal spread among individual samples of groups reflects differences in the donors used in the biological replicates. To control for effects of ROCK inhibitor treatment on the host transcriptome, we also subjected RNA obtained from HPV16-immortalized and control HFK grown in the absence of the inhibitor Y-27632 to RNA-seq. While the deregulated genes clustered differently from those in HFK grown in the presence of this inhibitor, pointing to significant alterations induced by Y-27632, both control and HPV16-immortalized HFK shifted in the same direction. Therefore, the inhibitor’s alterations to the transcriptome were independent of HPV16. Also, clustering was not observed with HPV16-infected HFK grown in the presence of this inhibitor and thus could not explain the stark differences observed between HPV16-infected and -immortalized HFK.
To exclude the possibility that the limited number of alterations induced by HPV16 infection compared to that with HPV16 immortalization is solely due to low viral transcript levels, we performed RNA-seq analyses of HPV16-infected HFK at later times postinfection. These cells were grown in the absence of the ROCK inhibitor Y-27632. Even though the number of transcriptionally deregulated genes increased with increasing time postinfection (358 and 238 up- and downregulated genes, respectively, at up to 63 dpi), these transcriptional changes never reached levels observed in HPV16-immortalized keratinocytes grown under identical conditions (1,239 and 1,200) despite similar viral transcript levels (Fig. 2A and B). Transcript levels of most of the genes identified as significantly deregulated at later times after infection were also altered at earlier time points without initially reaching significance (Fig. 2C). One hundred eighty-five genes were common among HPV16-infected and -immortalized HFK (Fig. 2D); 149 of which are upregulated. PCA demonstrated increasing separation of mock- and HPV16-infected HFK with prolonged times after infection (Fig. 2E).
FIG 2.

Extended culturing of HPV16-infected cells increases the number of deregulated genes. (A) Numbers of down- or upregulated genes in WT and E7-TTL mutant HPV16-infected and HPV16-immortalized HFK (p2/3, 27 to 33 dpi; p3/4, 36 to 55 dpi; p4/5, 47 to 63 dpi). All cells were infected and cultured in the absence of Y-27632. (B) TPM values of viral transcript reads in samples from HPV16-infected and HPV16-immortalized cells used for analysis. (C) Heat map analysis of genes altered in HPV16-infected cells. The subgroup of 113 genes identified in Fig. 1 were selected for the analysis and analyzed as sample groups from controls and WT and E7-TTL HPV16-infected cells at different times postinfection. (D) Venn diagram showing the overlap between differentially expressed genes in HPV16-immortalized and -infected cells (p4/5). (E) PCA analysis of RNA sequencing data from control and WT and E7-TTL HPV16-infected cells based on TPM values in the bulk RNA-seq data set.
Ingenuity pathway analysis (IPA) predicted an impairment of pRb and p53 regulatory functions in infected cells at 7 dpi, suggesting that most changes are driven by E6 and E7 expression. Indeed, the E2F family transcription factors were identified as positive upstream regulators for most of the deregulated genes. In line with this, cell cycle control of chromosomal replication was identified as the top canonical pathway in HPV16-infected HFK, including but not limited to genes such as MCM3, -4, -5, and -7, POLD1, and POLE (Fig. 3A). Downstream effect analysis identified “cell cycle,” “cellular assembly and organization,” and “DNA replication, recombination, and repair” as the main molecular and cellular functions altered by HPV16 infection (Fig. 3B). Surprisingly, only one deregulated gene among the 113 identified as a classical p53 transcriptional target according to a recent meta-analysis (32) (Table 2), suggesting that E6 protein may not be expressed at sufficient levels to functionally repress p53 at 7 dpi. IPA of the 185 genes overlapping between long-term-infected and immortalized groups again identified pRb and the E2F family of transcription factors as top upstream regulators and “cell cycle control of chromosomal replication” as the top canonical function, and downstream effect analysis predicted the same cellular functions to be affected as at 7 dpi (Fig. 4A). Expression of 13 p53 transcriptional targets was found to be altered (Table 2). Perturbation of the Fanconi anemia-BRCA pathway was observed upon long-term culturing of HPV16-infected HFK (Fig. 4B and C), suggesting it is an adaptation to continuous presence of HPV16. Taken together, these data suggest that HPV16 infection perturbs the HFK transcriptome only modestly compared to that of HPV16-immortalized HFK. These findings imply that the majority of changes observed in HPV16-immortalized HFK are likely due to selection, the need for immortalization, and/or long-term culturing.
FIG 3.

Members of the Fanconi anemia-BRCA pathway become activated upon long-term culturing of HPV16-infected HFK. (A) Downstream effect analysis of genes altered in HPV16-infected HFK at 63 dpi. The top 5 molecular and cellular functions affected by HPV16 are shown. (B) Fold changes of altered genes of the Fanconi anemia-BRCA pathway in HPV16-infected versus control cells at 63 dpi. (C) Genes of the Fanconi anemia-BRCA pathway highlighted in pink are transcriptionally altered in HPV16-infected cells at 63 dpi.
TABLE 2.
Genes identified as the p53, pocket protein, and cell cycle gene network targets according to the TargetGeneReg database (32)
| Sample typea | Total no. of genes | p53 targets |
Cell cycle genes |
DREAM targets |
MMB-FOXM1 targets |
RB-E2F targets |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | n | % | n | % | ||
| Monolayer | |||||||||||
| WT 7 dpi Y+ | 113 | 1 | 0.9 | 66 | 58.4 | 63 | 55.8 | 27 | 23.9 | 32 | 28.3 |
| WT 63 dpi Y− | 597 | 13 | 2.2 | 123 | 20.6 | 105 | 17.6 | 26 | 4.4 | 70 | 11.7 |
| Immort Y− | 2,439 | 37 | 1.5 | 480 | 19.7 | 407 | 16.7 | 139 | 5.7 | 227 | 9.3 |
| Immort Y+ | 5,854 | 109 | 1.9 | 718 | 12.3 | 546 | 9.3 | 172 | 2.9 | 302 | 5.2 |
| E7-TTL 27 dpi Y+ | 17 | 0 | 0 | 2 | 11.8 | 0 | 0 | 0 | 0 | 0 | 0 |
| Raft | |||||||||||
| WT | 1,837 | 52 | 2.8 | 379 | 20.6 | 322 | 17.5 | 94 | 5.1 | 207 | 11.3 |
| E7-TTL | 35 | 1 | 2.9 | 2 | 5.7 | 2 | 5.7 | 0 | 0.0 | 1 | 2.9 |
Y+, in the presence of Y-27632; Y−, in the absence of Y-27632; WT, cells infected wild-type HPV16; E7-TTL, cells infected with HPV-16 E7-TTL mutant; Immort, immortalized cells.
FIG 4.

IPA analysis of genes falling into the cell cycle of chromosomal replication category. (A) Genes highlighted in pink are transcriptionally altered in HPV16-infected cells at 7 dpi. (B) Downstream effect analysis of genes altered in HPV16-infected HFK at 7 dpi. The top 5 molecular and cellular functions affected by HPV16 are shown.
HPV E7 is primarily responsible for deregulating the host transcriptome following infection.
Our analyses so far suggested that E7 protein-driven impairment of pocket protein function is responsible for most transcriptional alterations. To test for this more directly, we also analyzed the transcriptome of HFK infected with a previously described mutant HPV16 virus harboring a termination translation linker (TTL) in the E7 open reading frame (HPV16-E7-TTL). Interestingly, only 17 genes were deregulated in HFK at 27 dpi with HPV16-E7-TTL (Fig. 2A and C). PCA demonstrated close clustering of HPV16-E7-TTL with mock-infected HFK (Fig. 2E). Taken together, our findings suggest that the modest alterations observed in HPV16-infected HFK can be attributed to E7-mediated functional inactivation of pocket proteins.
Host cell transcriptome analysis of HFK grown as organotypic rafts.
In our RNA-seq analyses of HPV16-infected HFK grown as monolayer, we noticed the absence of a number of pathways found to be deregulated in HPV16-immortalized keratinocytes in previous studies. However, some of the absent pathways have also been demonstrated to play important roles in late stages of the viral life cycle, such as upregulation of DNA repair and downregulation of the STAT/JAK signaling pathways. We have previously demonstrated that HPV16-infected HFK support the complete viral life cycle when subjected to growth as organotypic raft cultures. Therefore, perturbation of these pathways by HPV16 was either not required for the HPV16 life cycle or only induced during growth as stratified epithelia and required for late stages of the viral life cycle. Thus, we examined by RNA-seq analysis organotypic raft cultures derived from HPV16-infected HFK. We also included rafts derived from HFK infected with HPV16-E7-TTL mutant virus to determine the role of E7 under conditions of differentiation. As expected, growth as organotypic rafts significantly increased viral transcript levels in wild-type (WT) HPV16-infected cells but not HPV16-E7-TTL-infected cells (Fig. 5A and D). Surprisingly, viral genome levels were similar in rafts derived from WT HPV16- and HPV16-E7-TTL-infected HFK. Therefore, we employed highly sensitive fluorescent in situ hybridization to visualize the viral genome in raft-derived tissue sections. Viral genome was detected in essentially all cells infected with WT HPV16 with the expected distribution: stronger signal in differentiated layers of the epithelium indicative of differentiation-induced genome amplification. However, the basal cell compartment had higher levels of viral genome, and the genome was lost in differentiated keratinocytes in HPV16-E7-TTL-harboring rafts (Fig. 5C). Transcriptome analyses identified 838 and 999 genes up- and downregulated, respectively, in expression in wild-type HPV16 compared to that in uninfected control rafts (Fig. 5E). Again, pathways involved in cell cycle and DNA metabolism were identified as main cellular and molecular functions (Fig. 6A). In addition to S-phase genes, we observed an upregulation of genes involved in DNA repair (Fig. 6B and C). Activation of S-phase (PCNA, RPA2, and MCM7) and DNA repair (RPA2 and Rad51) markers was confirmed by immunofluorescent staining in rafts from WT but not E7-TTL mutant HPV16-infected HFK (Fig. 7). Genes regulated by the JAK/STAT1 signaling axis were downregulated in WT HPV16 rafts (Fig. 8), which is in line with the literature (33, 34). We also observed subtle reductions in STAT1 and STAT2 but not JAK transcript levels, which did not reach significance (data are available in the GEO database under accession number GSE137965). Despite the presence of high levels of viral genome in the basal cell compartment, the expression of only 34 genes was deregulated in rafts derived from HPV16-E7-TTL-infected HFK, 14 of which were also found in WT HPV16-harboring rafts, again suggesting that most changes are driven by E7 protein (Fig. 5E). The deregulated genes do not fall into any specific pathway. The lack of activation of S-phase and DNA repair markers in HPV16-E7-TTL-containing rafts was also confirmed by immunofluorescence (Fig. 7).
FIG 5.

HPV16-induced changes in the transcriptome of cells grown in organotypic rafts. Viral transcript levels in undifferentiated (mono) and differentiated cells infected with WT and E7-TTL mutant viruses. RNA and DNA were collected from monolayer cells at 7 dpi and from 20-day-old rafts. Viral E6 transcript (A) and DNA (B) are expressed as ΔCT relative to the household gene controls cyclophilin A (A) and β-actin (B), respectively. (C) Detection of viral DNA in infected rafts by highly sensitive fluorescent in situ hybridization. (D) TPM values for viral transcripts in samples used for sequencing. (E) Numbers of genes whose expression was significantly modified in WT and E7-TTL HPV16-infected cells grown in organotypic raft cultures.
FIG 6.

The Fanconi anemia-BRCA pathway is activated in HPV16-infected organotypic rafts. (A) Downstream effect analysis of genes altered in HPV16-infected rafts. The top 5 molecular and cellular functions affected by HPV16 are shown. (B) Genes of the Fanconi anemia-BRCA pathway highlighted in pink are transcriptionally altered in HPV16-infected rafts. (C) Fold changes of altered genes of the Fanconi anemia-BRCA pathway in HPV16-infected versus control rafts based on next-generation sequencing (NGS) analysis.
FIG 7.
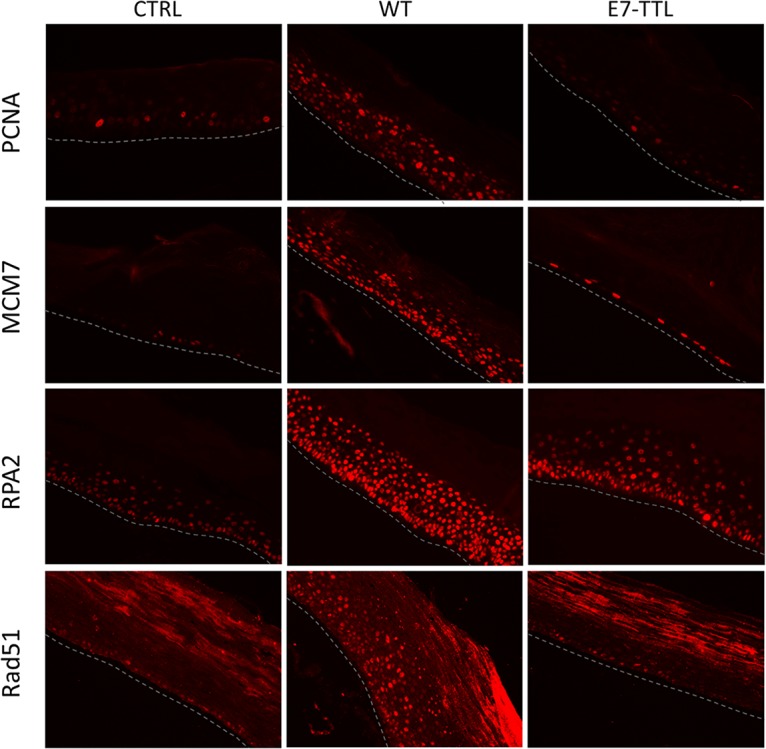
Immunofluorescent staining of organotypic rafts. Organotypic rafts derived from control and WT and E7-TTL HPV16-infected cells were paraffin embedded, sectioned, and processed for staining of S-phase markers (PCNA and MCM7) as well as markers of the DNA damage response (RPA2 and Rad51).
FIG 8.

Genes regulated by the JAK/STAT signaling axis are downregulated in HPV16-infected organotypic rafts.
In WT HPV16-harboring rafts, 52 p53 transcriptional targets were deregulated in their expression (Table 2), in line with the complete HPV-induced loss of p53 protein from these rafts (Fig. 9) (35). Surprisingly, only one p53 transcriptional target was found in the pool of genes identified in HPV16-E7-TTL-harboring rafts, even though we have previously shown that cells infected by HPV16-E7-TTL express E6, and the presence of E6 protein was confirmed in E7-TTL infected rafts (Fig. 9B). In line with this, p53 was detectable in E7-TTL-harboring rafts at levels similar to those in control rafts (Fig. 9A).
FIG 9.

p53, E6 protein, and viral genome in organotypic rafts. (A) Organotypic rafts derived from control (ctrl) and from WT and E7-TTL HPV16-infected HFK were paraffin embedded, sectioned, and processed for detection of p53 and viral DNA (vDNA) by immunofluorescent and fluorescent in situ hybridization, respectively. Adjacent sections were used for in situ hybridization and immunofluorescent staining. (B) Furthermore, E6 protein was detected using the OncoE6 cervical test. Note detection of p53 despite the presence of E6 protein and viral genome E7-TTL-harboring rafts.
DISCUSSION
The aim of our study was to examine how HPV16 influences the host cell transcriptome after infection of primary keratinocytes in the absence of selection, outgrowth, and immortalization. It was previously reported that HPV-immortalized human keratinocytes from various origins exhibit large perturbations of their transcriptome, with thousands of genes affected. We have confirmed this observation herein. In contrast, using our newly developed HPV16 infection model, which allows efficient infection of primary HFK, we found a very limited set of genes deregulated at 7 dpi, most of which were upregulated. This implies that primary infection of keratinocytes perturbs the host cell only minimally at the transcriptional level. Mostly S-phase genes are upregulated, likely giving HPV16-harboring cells a growth advantage in order to expand lesions and increase their life span.
The majority of the observed changes in HPV16-infected cells can be linked to the main function of the E7 protein: binding and degradation of pocket proteins, which results in activation of E2F transcription factors to drive transcription of S-phase genes. Indeed, 65 of the 113 genes identified have been categorized as cell cycle genes regulated by pRb/E2F and the DREAM and/or the MMB-FOXM1 complex (32). p107 and p130 are subunits of the DREAM and MMB-FOXM1 complexes, suggesting that all three pocket proteins are functionally impaired in HPV16-infected HFK. In overexpression systems and/or HPV16-immortalized keratinocytes, E7 has also been demonstrated to down- or upregulate pathways not under the control of pocket proteins and E2F family members. For example, E7 downregulates the constitutively active interferon response (36, 37) and transforming growth factor β (TGF-β) signaling (38) and has also been shown to activate pathways associated with the DNA damage response (39). In HFK infected for 7 days with HPV16, these pathways were not affected at the transcriptional level, suggesting that their perturbation is not required for establishment of infection. The Fanconi anemia-BRCA pathway becomes activated when infected cells are cultured and survive beyond their normal life expectancy. This DNA repair pathway is constitutively active in HPV-immortalized keratinocytes and is essential for viral DNA replication. Recent studies suggest that relocation of members of this pathway to viral replication foci and impairment with deubiquitination of FancD2 may actually contribute to genomic instability observed in HPV-harboring cells (40). The slow onset of perturbations in this pathway after infection might allow mechanistic studies of the temporal deregulation of this pathway. In contrast to the Fanconi anemia-BRCA pathway, which is activated during persistent infection, interferon and TGF-β signaling pathways are not perturbed, even in cells cultured up to 63 dpi. These results imply that their perturbation in HPV16-immortalized keratinocytes may be due to outgrowth and selection of HPV-harboring cells and/or the immortalization process rather than immediate alterations induced by virally encoded proteins. However, the additional changes to the host transcriptome may reflect biologically meaningful “adaptive changes” that are required for long-term persistent infections, a hallmark of high-risk HPVs and a major risk factor for cancer formation. In case these pathways become deregulated after even longer passaging of infected keratinocytes, the infection model may provide a means to investigate progression of HPV-induced lesions in vitro.
We extended our studies to the analysis of organotypic raft cultures derived from HPV16-infected cells to determine whether the stark differences to immortalized HFK in undifferentiated monolayer cell culture are also observed in differentiated tissues. Comparing our data with published results suggests that many of the same pathways are altered in rafts grown from immortalized and infected HFK, including the induction of S-phase genes and pathways associated with the DNA damage response as well as downregulation of the JAK/STAT signaling axis. Also, interferon (IFN) and TGF-β signaling pathways are downregulated. This is in line with multiple previous reports which demonstrated the importance of these pathways for completion of the viral life cycle. However, the gene expression profile is not completely congruent due to the fact that the published studies used keratinocytes derived from different tissues, mostly cervical, and microarray analysis rather than RNA-seq.
Our analysis of cells infected with the E7-TTL mutant HPV16 revealed that the E7 oncoprotein orchestrates the vast majority of changes in the host cell transcriptome, both in undifferentiated and differentiated cells. HFK infected with the E7-TTL mutant virus no longer altered the expression of genes regulated by the E2F family despite the presence of an episomal viral genome and viral transcripts. Indeed, only 17 genes showed changes in their expression in HPV16-E7-TTL-infected HFK grown in monolayer. No specific pathways were identifiable, and only 7 of these genes were also perturbed in wild-type HPV16-infected cells. Similarly, transcriptome analysis of organotypic rafts derived from cells infected with a mutant virus lacking E7 abolished most changes to the host cell transcriptome. Not unexpectedly, lack of E7 protein abolished the retention of differentiated cells in the cell cycle, viral genome amplification, and retention of viral genomes in differentiated layers of raft tissue despite elevated levels of viral genome in the basal and suprabasal cell compartments.
According to a meta-analysis recently undertaken by Fischer et al. (32), only one of the affected genes identified in monolayer culture classified as classical p53 transcriptional target at 7 dpi, despite the fact that we have previously shown E6 protein to be expressed in these HPV-infected cells. However, it has previously been demonstrated that HPV16-harboring cells and even cells overexpressing E6 protein retain low levels of p53 (41). In contrast, some 50 p53 targets were identified in WT HPV16 rafts, which is in line with our finding that rafts derived from these cells are completely deficient of p53. We assume that HPV-harboring keratinocytes, which are continuously passaged, have different characteristics from cells found in the basal cell compartment of an intact epithelium, which may include the ability to degrade p53 in an E6-dependent manner. Given that tight junctions are continuously being disrupted during passaging, we assume that the cultured cells are more representative of wound-healing keratinocytes. To investigate if these differences indeed exist on the transcriptional level, laser capture microdissection has to be employed to compare the transcriptomes of the basal cells excised from an intact epithelium with those from cultured undifferentiated keratinocytes. Unfortunately, because the viral genome was not maintained in cells infected with an E6-deficient HPV16, we were not able to extend our studies to a setting that lacked E6 protein exclusively. So far, we have no mechanistic explanation why complete p53 degradation is not evident in E7-TTL-harboring rafts; however, the data suggest that unidentified E7-induced alterations to the keratinocytes may be required. Alternatively, E6 expression levels may be too low for efficient degradation of p53.
MATERIALS AND METHODS
Cell lines.
Human embryonic kidney 293TT cells used for HPV16 virus particle generation were obtained from John Schiller (NIH, Bethesda, MD). They were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), nonessential amino acids, antibiotics, and GlutaMAX. Spontaneously immortalized human keratinocyte HaCaT cells were purchased from the American Type Culture Collection (ATCC) and grown in low-glucose DMEM containing 5% FBS and antibiotics. Primary epithelial keratinocytes were purchased from ATCC (PCS-200-010) and maintained in E medium containing mouse epidermal growth factor (EGF) and mitomycin-treated mouse 3T3 J2 fibroblasts as previously described (42). Where indicated, we maintained and infected primary keratinocytes in the presence of the Rho kinase inhibitor (ROCK) Y-27632 (SCM075; Millipore), which was reported to increase their life span (30, 31). However, the ROCK inhibitor was excluded prior and during experiments involving long-term culturing of infected primary cells. Stable, immortalized human keratinocyte cell lines containing HPV16 episomes were generated after transfection of HPV16 genome and outgrowth of colonies as previously described (43) and were provided by Jason Bodily (LSUHSC, Shreveport, LA). Before harvesting the cells for RNA or DNA extraction, fibroblast feeders were removed by short trypsin treatment, followed by two washes in phosphate-buffered saline (PBS).
Ethics statement.
Foreskin keratinocytes were collected from discarded tissue following routine circumcisions from anonymous donors attending University Health, Shreveport, LA. Because the samples were deidentified, would otherwise have been discarded, and were not collected specifically for our studies, the LSUHSC-S institutional review board (IRB) ruled that they fell under the NIH’s definition of “exempt” from human subjects research, including informed consent (Institutional IRB approval number STUDY00000187).
Generation of HPV16 quasivirions.
The pSheLL16 L1/L2 packaging plasmid was a kind gift from John Schiller (NIH, Bethesda, MD). The plasmid pEGFP-N1 containing the entire floxed HPV16 genome (pEGFP-N1-HPV16) and the pBCre plasmid have been described previously (44). Quasivirions were generated using 293TT cells following the improved protocol of Buck and Thompson (45) with minor modifications. Briefly, 293TT cells were first cotransfected with the pSheLL16 L1/L2 and pEGFP-N1-HPV16 plasmids and, 24 h later, were transfected with the pBCre plasmid. An additional 2 days later, cells were harvested and viral particles were purified as described previously (45, 46). Because activity of the Cre recombinase generates two circular plasmids of packable size (pEGFP-N1 and HPV16 genome), isolated viral particles comprise a mixture of pseudovirions (pEGFP-N1 plasmid) and quasivirions (HPV16 genome). Viral genome equivalents (vge) were determined by real-time quantitative PCR (RT-qPCR) of encapsidated DNA isolated using the NucleoSpin Blood QuickPure kit (740569.250; Macherey-Nagel). The E7-TTL mutant has been described previously (43).
Infection using extracellular matrix-to-cell transfer.
HaCaT cells were seeded in 60-mm cell culture dishes and grown for 24 to 48 h until they reached confluence to allow secretion of ECM. Cells were incubated in Dulbecco’s PBS supplemented with 0.5 mM EDTA for up to 2 h in order to remove the cells. This removes essentially all cells. However, to prevent outgrowth of any residual HaCaT cells, the dishes were treated with 8 μg/ml mitomycin for 4 h prior to EDTA treatment and removal of HaCaT cells. OptiPrep-purified viral particles (>5 × 107 vge/dish) diluted in 2 ml E medium were added to the ECM for at least 2 h at 37°C. At this time, 5 × 105 low-passage-number primary keratinocytes were added. Two hours later, approximately 1 × 105 mitomycin-treated fibroblast feeder cells were added to the dish. When different size culture dishes (ranging from 12-well plates to 100-mm dishes) were used, cell and vge numbers were scaled proportionally to the surface area. Infection was continued for up to 7 days or until cells reached confluence. Under these conditions, at least 50% of HFK are infected according to RNAscope at 7 dpi. However, the rate of infectivity varies significantly from donor to donor.
Detection of HPV16 RNA by RNAscope.
Viral RNA detection (RNAscope) in infected cells was performed by utilizing the Multiplex Fluorescent v2 kit (catalog number [cat. no.] 323100; Advanced Cell Diagnostics [ACD]) and a probe targeting HPV16/18 E6 and E7 (cat. no. 311121; ACD) according to the manufacturer’s protocol. Briefly, infected and control cells cultured on coverslips were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature at 7 dpi. Immediately after washing with PBS, samples were dehydrated with 50%, 70%, and 100% ethyl alcohol gradients for 5 min each at room temperature. Cells were then rehydrated with 70% and 50% ethyl alcohol gradients for 2 min each and finally incubated twice with PBS for 10 min. Next, slides were incubated with pretreatment reagents (cat. no. 322381; ACD): hydrogen peroxide and protease III at room temperature for 10 min each. After a PBS wash, samples were incubated with prewarmed target probes for 2 h at 40°C. In addition to the probe targeting HPV16/18 E6 and E7, the positive control for cellular RNA detection RNAscope 2.5 Duplex Positive Control Probe(Mm) PPIB-C1/POLR2A-C2 (cat no. 321651) provided by the supplier was used. Signal amplification and detection reagents (cat. no. 323110; ACD) were applied sequentially and incubated at 40°C in AMP 1, AMP 2, AMP 3, and HRP1 reagents for 30, 30, 15, and 15 min, respectively. Before adding each reagent, samples were washed twice with washing buffer (cat. no. 310091; ACD). For fluorescence detection, Alexa Fluor 488 tyramide (cat. no. T20912; Invitrogen) in RNAscope Multiplex TSA buffer (cat. no. 322809; ACD) was added for 30 min at 40°C. After extensive final washing, cells were mounted in Gold Antifade containing 4′,6-diamidino-2-phenylindole (DAPI) (P3693; Life Technologies) for imaging.
Organotypic raft cultures.
Organotypic raft cultures generated from HFK infected for 5 to 7 days with HPV16 quasivirions were grown as described (42, 47). Briefly, 1 million keratinocytes were seeded onto the surface of the collagen gel containing fibroblast feeders. Following attachment, the gel layer with keratinocytes was lifted and placed onto a stainless steel grid in a culture dish. Culture medium was added to the dish so that the keratinocyte/collagen plug was exposed to the air from above and to the medium from below. The medium was changed every other day, maintaining the air-liquid interface. Rafts were grown for 21 days, and samples were collected for RNA/DNA analysis. For immunofluorescent staining and fluorescent in situ hybridization (FISH), raft tissues were fixed for 30 min in 4% paraformaldehyde at 4°C, washed three times in cold 1× PBS and once in cold 70% ethanol, and stored in fresh cold 70% ethanol at 4°C prior to processing and paraffin embedding. Rafts generated from uninfected HFK seeded on ECM were used as a control.
DNA isolation and real-time qPCR.
DNA was isolated using the NucleoSpin Blood QuickPure kit (740569.250; Macherey-Nagel) according to the manufacturer’s instructions. One hundred nanograms of DNA was quantified by RT-qPCR using the IQ SYBR green Supermix (Bio-Rad) and a CFX96 Fast Real-Time system (Bio-Rad). PCRs were carried out in triplicates, and genome levels were normalized to actin. Primers were as follows: E7, 5′-ATGAGCAATTAAATGACAGCTCAGAG-3′ and 5′-CACACTTGCAACAAAAGGTTACAAT-3′; β-actin, 5′-GGCATCCTCACCCTGAAGTA-3′ and 5′-CAGAGGCGTACAGGGATAGC-3′. The Bio-Rad CFX Manager 3.1 and Bio-Rad CFX Maestro 1.1 software were used to analyze the data.
RNA isolation, cDNA synthesis, real-time qPCR.
Total RNA from HFK was extracted using the RNeasy Plus Mini RNA Isolation kit (74236; Qiagen). RNA from raft tissues was extracted using the Qiagen QIAcube via the miRNeasy aqueous-phase protocol. Isolated RNA samples were treated with DNase I (M0303L; NEB) prior to reverse transcription; 0.5 to 1 μg of total RNA was reverse transcribed into cDNA using an ImProm-II Reverse Transcriptase kit (Promega). Equal amounts of cDNA were quantified by RT-qPCR using the IQ SYBR green Supermix (Bio-Rad) and a CFX96 Fast Real-Time system (Bio-Rad). PCRs were carried out in triplicates, and transcript levels were normalized to cyclophilin A. Mock reverse-transcribed samples were included as a negative control. Primers were as follows: E6, 5′-CTGCAATGTTTCAGGACCC-3′ and 5′-TCACGTCGCAGTAACTGTTG-3′; cyclophilin A (CyPA), 5′-CTTGGGCCGCGTCTCC-3′ and 5′-GCAGGAACCCTTATAACCAAATCC-3′. The Bio-Rad CFX Manager 3.1 and Bio-Rad CFX Maestro 1.1 software were used to analyze the data.
RNA sequencing.
RNA-seq was carried out in triplicates for populations of cells grown in a monolayer culture and in quintuplicates for raft three-dimensional (3D) cultures. Total RNA was extracted as described above. RNA quality was assessed on an Agilent 2200 TapeStation. All samples showed an RNA integrity number (RIN) greater than 7.0. Strand-specific mRNA-seq libraries for the Illumina platform were prepared using the NEBNextUltra directional library kit (NEB) and the TruSeq stranded mRNA kit (Illumina). Barcoded libraries were sequenced on a NextSeq 550 (Illumina) at 2 × 75 bp (paired-end sequencing), obtaining >25 million reads per sample. Reads were aligned to the HPV16 (NC_001526.3) and human genome GRCh38p10 using STAR_2.4.2a and counted using RSEM 1.2.31. Differentially expressed genes were identified using EBSeq with a false-discovery rate (FDR) of <0.1 and >1 transcripts per million (TPM) in at least 1 of 6 analyzed samples. Partek Flow was used for hierarchical clustering, heat map construction, and principle-component analysis. Ingenuity Pathway Analysis (IPA) was used for ontological classification and identification of the biological pathways affected by HPV16.
Immunofluorescent staining.
Paraffin wax-embedded sections were dewaxed in series of xylene and alcohol washes, followed by antigen retrieval using a pressure cooker for 10 min in citrate buffer with 0.05% Tween. Slides were permeabilized with 0.5% Triton X-100 for 45 min and blocked with 5% goat serum for 1 h. Primary antibodies, including anti-PCNA (sc-7907; Santa Cruz Biotechnology), anti-MCM7 (ab52489; Abcam), anti-RPA2/32 (ab76420; Abcam), and anti-Rad51 (sc-53428; Santa Cruz Biotechnology), were added for overnight incubation at 4°C. After extensive washing with PBS, sections were incubated for 1 h with Alexa Fluor-tagged secondary antibodies (A21236, A21245, A11030, and A11035; Molecular Probes). After extensive final washing with PBS, cells were mounted in Gold Antifade containing DAPI (P3693; Life Technologies). All immunofluorescence (IF) images were captured by using a Leica CTR6000 fluorescence microscope or by confocal microscopy with a 63× lens objective using a Leica TCS SP5 spectral confocal microscope and processed with Adobe Photoshop software.
Detection of HPV16 genome.
Viral DNA detection (DNAscope) was performed by utilizing a Multiplex Fluorescent v2 kit (cat. no. 323100; ACD) and the probe targeting HPV16/18 E6/7 (cat no. 311121; ACD). We modified the RNAscope procedure as described by Deleage et al. (48) by adding an RNase tissue pretreatment step followed by a short denaturation step. Briefly, paraffin-embedded sections of raft tissues were heated at 60°C for 1 h, deparaffinized in xylene for 10 min, followed by dehydration in 100% ethanol for 5 min before air drying. Next, slides were incubated with RNAscope hydrogen peroxide for 10 min at room temperature to block endogenous peroxidases. Heat-induced epitope retrieval was performed by boiling sections in RNAscope Target Retrieval reagent for 15 min, and the sections were immediately washed in Milli-Q water and then dehydrated in 100% ethanol for 5 min before air drying. Tissue sections were then incubated with RNAscope Protease Plus for 30 min at 40°C. Slides were rinsed in Milli-Q water and incubated with RNase A (25 μg/ml, cat no. EN0531; Thermo Scientific) for 30 min at 37°C. The RNase tissue pretreatment step was followed by a short denaturation step in which we incubated the slides at 60°C with warmed probes for 15 min and then transferred them to 40°C for hybridization overnight. Amplification and detection were performed according to the manufacturer’s protocol, using RNAscope 0.5× wash buffer for all washing steps.
OncoE6 cervical test.
The presence of E6 protein in infected rafts was detected using a kit from ArborVita according to the manufacturer’s protocol. Briefly, the cell lysate was incubated with alkaline phosphatase (AP)-conjugated high-affinity E6 HPV16/18 monoclonal antibodies. Next, a nitrocellulose test strip with two capture lines consisting of immobilized monoclonal antibodies (MAbs) to HPV16/18 E6 was placed into the lysate/MAb-AP mix. The solution was allowed to migrate through the strip by capillary action. E6-MAb-AP present in the sample forms a ternary complex with the immobilized antibodies on the strip. The complex was visualized as a purple line in the respective location on the strip by the addition of an enzyme substrate solution provided in the kit.
Data availability.
RNA-seq data files have been submitted to Gene Expression Omnibus under the accession number GSE137965.
ACKNOWLEDGMENTS
We thank Jason Bodily and members of his group for RNA samples from HPV16-immortalized HFK.
This work was supported by the NIH through grants to M.S. (R01 CA211576 and P30 GM110703) and R.S.S. (R01 DE025565) as well as with continuous generous financial support by the LSUHSC-S Feist-Weiller Cancer Center.
REFERENCES
- 1.Zur-Hausen H, de Villiers EM. 1994. Human papillomaviruses. Annu Rev Microbiol 48:427–447. doi: 10.1146/annurev.mi.48.100194.002235. [DOI] [PubMed] [Google Scholar]
- 2.Roberts JN, Buck CB, Thompson CD, Kines R, Bernardo M, Choyke PL, Lowy DR, Schiller JT. 2007. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat Med 13:857–861. doi: 10.1038/nm1598. [DOI] [PubMed] [Google Scholar]
- 3.Doorbar J. 2005. The papillomavirus life cycle. J Clin Virol 32 Suppl 1:S7–S15. doi: 10.1016/j.jcv.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Hummel M, Hudson JB, Laimins LA. 1992. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J Virol 66:6070–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roman A, Munger K. 2013. The papillomavirus E7 proteins. Virology 445:138–168. doi: 10.1016/j.virol.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 7.Werness BA, Levine AJ, Howley PM. 1990. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 248:76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 8.Hebner CM, Laimins LA. 2006. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol 16:83–97. doi: 10.1002/rmv.488. [DOI] [PubMed] [Google Scholar]
- 9.Boyer SN, Wazer DE, Band V. 1996. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res 56:4620–4624. [PubMed] [Google Scholar]
- 10.Dyson N, Howley PM, Munger K, Harlow E. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. 2001. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol 75:7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLaughlin-Drubin ME, Münger K. 2009. The human papillomavirus E7 oncoprotein. Virology 384:335–344. doi: 10.1016/j.virol.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nor Rashid N, Yusof R, Watson RJ. 2011. Disruption of repressive p130-DREAM complexes by human papillomavirus 16 E6/E7 oncoproteins is required for cell-cycle progression in cervical cancer cells. J Gen Virol 92:2620–2627. doi: 10.1099/vir.0.035352-0. [DOI] [PubMed] [Google Scholar]
- 14.Nor Rashid N, Yusof R, Watson RJ. 2013. Disruption of pocket protein dream complexes by E7 proteins of different types of human papillomaviruses. Acta Virol 57:447–451. doi: 10.4149/av_2013_04_447. [DOI] [PubMed] [Google Scholar]
- 15.Songock WK, Kim SM, Bodily JM. 2017. The human papillomavirus E7 oncoprotein as a regulator of transcription. Virus Res 231:56–75. doi: 10.1016/j.virusres.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.den Boon JA, Pyeon D, Wang SS, Horswill M, Schiffman M, Sherman M, Zuna RE, Wang Z, Hewitt SM, Pearson R, Schott M, Chung L, He Q, Lambert P, Walker J, Newton MA, Wentzensen N, Ahlquist P. 2015. Molecular transitions from papillomavirus infection to cervical precancer and cancer: role of stromal estrogen receptor signaling. Proc Natl Acad Sci U S A 112:E3255–E3264. doi: 10.1073/pnas.1509322112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, Woodworth CD, Connor JP, Haugen TH, Smith EM, Kelsey KT, Turek LP, Ahlquist P. 2007. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res 67:4605–4619. doi: 10.1158/0008-5472.CAN-06-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong YF, Cheung TH, Tsao GS, Lo KW, Yim SF, Wang VW, Heung MM, Chan SC, Chan LK, Ho TW, Wong KW, Li C, Guo Y, Chung TK, Smith DI. 2006. Genome-wide gene expression profiling of cervical cancer in Hong Kong women by oligonucleotide microarray. Int J Cancer 118:2461–2469. doi: 10.1002/ijc.21660. [DOI] [PubMed] [Google Scholar]
- 19.Xu J, Liu H, Yang Y, Wang X, Liu P, Li Y, Meyers C, Banerjee NS, Wang HK, Cam M, Lu W, Chow LT, Xie X, Zhu J, Zheng ZM. 2019. Genome-wide profiling of cervical RNA-binding proteins identifies human papillomavirus regulation of RNASEH2A expression by viral E7 and E2F1. mBio 10:e02687-18. doi: 10.1128/mBio.02687-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhai Y, Kuick R, Nan B, Ota I, Weiss SJ, Trimble CL, Fearon ER, Cho KR. 2007. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res 67:10163–10172. doi: 10.1158/0008-5472.CAN-07-2056. [DOI] [PubMed] [Google Scholar]
- 21.Chang YE, Laimins LA. 2000. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J Virol 74:4174–4182. doi: 10.1128/jvi.74.9.4174-4182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karstensen B, Poppelreuther S, Bonin M, Walter M, Iftner T, Stubenrauch F. 2006. Gene expression profiles reveal an upregulation of E2F and downregulation of interferon targets by HPV18 but no changes between keratinocytes with integrated or episomal viral genomes. Virology 353:200–209. doi: 10.1016/j.virol.2006.05.030. [DOI] [PubMed] [Google Scholar]
- 23.Klymenko T, Gu Q, Herbert I, Stevenson A, Iliev V, Watkins G, Pollock C, Bhatia R, Cuschieri K, Herzyk P, Gatherer D, Graham SV. 2017. RNA-Seq analysis of differentiated keratinocytes reveals a massive response to late events during human papillomavirus 16 infection, including loss of epithelial barrier function. J Virol 91:e01001-17. doi: 10.1128/JVI.01001-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruutu M, Peitsaro P, Johansson B, Syrjanen S. 2002. Transcriptional profiling of a human papillomavirus 33-positive squamous epithelial cell line which acquired a selective growth advantage after viral integration. Int J Cancer 100:318–326. doi: 10.1002/ijc.10455. [DOI] [PubMed] [Google Scholar]
- 25.Cicchini L, Blumhagen RZ, Westrich JA, Myers ME, Warren CJ, Siska C, Raben D, Kechris KJ, Pyeon D. 2017. High-risk human papillomavirus E7 alters host DNA methylome and represses HLA-E expression in human keratinocytes. Sci Rep 7:3633. doi: 10.1038/s41598-017-03295-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duffy CL, Phillips SL, Klingelhutz AJ. 2003. Microarray analysis identifies differentiation-associated genes regulated by human papillomavirus type 16 E6. Virology 314:196–205. doi: 10.1016/s0042-6822(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 27.Garner-Hamrick PA, Fostel JM, Chien WM, Banerjee NS, Chow LT, Broker TR, Fisher C. 2004. Global effects of human papillomavirus type 18 E6/E7 in an organotypic keratinocyte culture system. J Virol 78:9041–9050. doi: 10.1128/JVI.78.17.9041-9050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Day PM, Lowy DR, Schiller JT. 2008. Heparan sulfate-independent cell binding and infection with furin pre-cleaved papillomavirus capsids. J Virol 82:12565–12568. doi: 10.1128/JVI.01631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bienkowska-Haba M, Luszczek W, Myers JE, Keiffer TR, DiGiuseppe S, Polk P, Bodily JM, Scott RS, Sapp M. 2018. A new cell culture model to genetically dissect the complete human papillomavirus life cycle. PLoS Pathog 14:e1006846. doi: 10.1371/journal.ppat.1006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chapman S, Liu X, Meyers C, Schlegel R, McBride AA. 2010. Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J Clin Invest 120:2619–2626. doi: 10.1172/JCI42297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chapman S, McDermott DH, Shen K, Jang MK, McBride AA. 2014. The effect of Rho kinase inhibition on long-term keratinocyte proliferation is rapid and conditional. Stem Cell Res Ther 5:60. doi: 10.1186/scrt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fischer M, Grossmann P, Padi M, DeCaprio JA. 2016. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res 44:6070–6086. doi: 10.1093/nar/gkw523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chatterjee S, Kang SD, Alam S, Salzberg AC, Milici J, van der Burg SH, Freeman W, Meyers C. 2019. Tissue-specific gene expression during productive HPV16 infection of cervical, foreskin and tonsil epithelium. J Virol 93:e00915-19. doi: 10.1128/jvi.00915-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong S, Mehta KP, Laimins LA. 2011. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J Virol 85:9486–9494. doi: 10.1128/JVI.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kho EY, Wang HK, Banerjee NS, Broker TR, Chow LT. 2013. HPV-18 E6 mutants reveal p53 modulation of viral DNA amplification in organotypic cultures. Proc Natl Acad Sci U S A 110:7542–7549. doi: 10.1073/pnas.1304855110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nees M, Geoghegan JM, Hyman T, Frank S, Miller L, Woodworth CD. 2001. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J Virol 75:4283–4296. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perea SE, Massimi P, Banks L. 2000. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor-1. Int J Mol Med 5:661–666. doi: 10.3892/ijmm.5.6.661. [DOI] [PubMed] [Google Scholar]
- 38.Lee DK, Kim BC, Kim IY, Cho EA, Satterwhite DJ, Kim SJ. 2002. The human papilloma virus E7 oncoprotein inhibits transforming growth factor-beta signaling by blocking binding of the Smad complex to its target sequence. J Biol Chem 277:38557–38564. doi: 10.1074/jbc.M206786200. [DOI] [PubMed] [Google Scholar]
- 39.Spardy N, Duensing A, Charles D, Haines N, Nakahara T, Lambert PF, Duensing S. 2007. The human papillomavirus type 16 E7 oncoprotein activates the Fanconi anemia (FA) pathway and causes accelerated chromosomal instability in FA cells. J Virol 81:13265–13270. doi: 10.1128/JVI.01121-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khanal S, Galloway DA. 2019. High-risk human papillomavirus oncogenes disrupt the Fanconi anemia DNA repair pathway by impairing localization and de-ubiquitination of FancD2. PLoS Pathog 15:e1007442. doi: 10.1371/journal.ppat.1007442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mesplède T, Gagnon D, Bergeron-Labrecque F, Azar I, Sénéchal H, Coutlée F, Archambault J. 2012. p53 degradation activity, expression, and subcellular localization of E6 proteins from 29 human papillomavirus genotypes. J Virol 86:94–107. doi: 10.1128/JVI.00751-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyers C, Laimins LA. 1994. In vitro systems for the study and propagation of human papillomaviruses. Curr Top Microbiol Immunol 186:199–215. doi: 10.1007/978-3-642-78487-3_11. [DOI] [PubMed] [Google Scholar]
- 43.Bodily JM, Mehta KP, Cruz L, Meyers C, Laimins LA. 2011. The E7 open reading frame acts in cis and in trans to mediate differentiation-dependent activities in the human papillomavirus type 16 life cycle. J Virol 85:8852–8862. doi: 10.1128/JVI.00664-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JH, Yi SM, Anderson ME, Berger KL, Welsh MJ, Klingelhutz AJ, Ozbun MA. 2004. Propagation of infectious human papillomavirus type 16 by using an adenovirus and Cre/LoxP mechanism. Proc Natl Acad Sci U S A 101:2094–2099. doi: 10.1073/pnas.0308615100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buck CB, Thompson CD. 2007. Production of papillomavirus-based gene transfer vectors. Curr Protoc Cell Biol Chapter 26:Unit 26.1. doi: 10.1002/0471143030.cb2601s37. [DOI] [PubMed] [Google Scholar]
- 46.Buck CB, Thompson CD, Pang YYS, Lowy DR, Schiller JT. 2005. Maturation of papillomavirus capsids. J Virol 79:2839–2846. doi: 10.1128/JVI.79.5.2839-2846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anacker D, Moody C. 2012. Generation of organotypic raft cultures from primary human keratinocytes. J Vis Exp 2012:3668. doi: 10.3791/3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deleage C, Wietgrefe SW, Del Prete G, Morcock DR, Hao XP, Piatak M Jr, Bess J, Anderson JL, Perkey KE, Reilly C, McCune JM, Haase AT, Lifson JD, Schacker TW, Estes JD. 2016. Defining HIV and SIV reservoirs in lymphoid tissues. Pathog Immun 1:68–106. doi: 10.20411/pai.v1i1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
RNA-seq data files have been submitted to Gene Expression Omnibus under the accession number GSE137965.