

The reactivation of herpesviruses, most commonly varicella-zoster virus (VZV) and pseudorabies virus (PRV), may cause cranial nerve disorder and unbearable pain. Clinical studies have also reported that HSV-1 causes postherpetic neuralgia and chronic occipital neuralgia in humans. The current work meticulously studies the functional expression profile changes of VGSCs during the processes of HSV-1 latency establishment and reactivation using human dorsal root ganglion-derived neuronal HD10.6 cells as an in vitro model. Our results indicated that VGSC activity was eliminated upon infection but steadily recovered during latency establishment and that latent neurons exhibited even higher VGSC activity. This finding advances our knowledge of how ganglion neurons generate uncharacteristic electrical impulses due to abnormal VGSC functional expression influenced by the latent virus.
Keywords: herpesvirus, voltage-gated sodium channel, dorsal root ganglion, latency, reactivation
ABSTRACT
The molecular mechanisms of pain associated with alphaherpesvirus latency are not clear. We hypothesize that the voltage-gated sodium channels (VGSC) on the dorsal root ganglion (DRG) neurons controlling electrical impulses may have abnormal activity during latent viral infection and reactivation. We used herpes simplex virus 1 (HSV-1) to infect the human DRG-derived neuronal cell line HD10.6 in order to study the establishment and maintenance of viral latency, viral reactivation, and changes in the functional expression of VGSCs. Differentiated cells exhibited robust tetrodotoxin (TTX)-sensitive sodium currents, and acute infection significantly reduced the functional expression of VGSCs within 24 h and completely abolished VGSC activity within 3 days. A quiescent state of infection mimicking latency can be achieved in the presence of acyclovir (ACV) for 7 days followed by 5 days of ACV washout, and then the viruses can remain dormant for another 3 weeks. It was noted that during the establishment of HSV-1 latency, the loss of VGSC activity caused by HSV-1 infection could not be blocked by ACV treatment. However, neurons with continued ACV treatment for another 4 days showed a gradual recovery of VGSC functional expression. Furthermore, the latently infected neurons exhibited higher VGSC activity than controls. The overall regulation of VGSCs by HSV-1 during quiescent infection was proved by increased transcription and possible translation of Nav1.7. Together, these observations demonstrated a very complex pattern of electrophysiological changes during HSV infection of DRG neurons, which may have implications for understanding of the mechanisms of virus-mediated pain linked to latency and reactivation.
IMPORTANCE The reactivation of herpesviruses, most commonly varicella-zoster virus (VZV) and pseudorabies virus (PRV), may cause cranial nerve disorder and unbearable pain. Clinical studies have also reported that HSV-1 causes postherpetic neuralgia and chronic occipital neuralgia in humans. The current work meticulously studies the functional expression profile changes of VGSCs during the processes of HSV-1 latency establishment and reactivation using human dorsal root ganglion-derived neuronal HD10.6 cells as an in vitro model. Our results indicated that VGSC activity was eliminated upon infection but steadily recovered during latency establishment and that latent neurons exhibited even higher VGSC activity. This finding advances our knowledge of how ganglion neurons generate uncharacteristic electrical impulses due to abnormal VGSC functional expression influenced by the latent virus.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a neurotropic pathogen associated with excruciating orofacial sensations and other chronic neuropathic pains (1 – 10). It is also known that patients with HSV-1 complications may experience paresthesias, such as allodynia, dysesthesia, or tingling, but the underlying molecular mechanisms are unclear (7, 11, 12). After the acute infection, the virus may establish a lifelong latency in the neurons of the trigeminal ganglion (TG) as well as in other sensory and autonomic ganglia, including the dorsal root and geniculate ganglia, through innervating sensory nerves (13). Reactivation may occur in response to various stress signals and trauma (14). The resulting pain and other complications have a detrimental impact on patients’ lives, as well as imposing economic and health burdens.
A variety of ion channels and receptors are expressed in ganglion neurons, such as transient receptor potential (TRP) channels, voltage-gated sodium channels (VGSCs), acid-sensing ion channels, ATP-sensitive receptors, potassium channels, voltage-gated calcium channels, and glutamate receptors (15). Active infection with the neurotropic pathogen HSV-1 could cause nerve injury, evidenced by the loss of neuronal processes or sometimes by neuronal cell death (16 – 18). However, nerve injury in the dorsal root ganglion (DRG) neurons could cause increased neuronal excitability by lowering the action potential threshold or increasing the firing frequency (19, 20). The transmission of pain information is highly dependent on the generation of action potential. VGSCs play a role in the generation and propagation of action potentials needed for the transmission and processing of pain signals (15). Changes in the molecular and/or functional expression of VGSCs during HSV-1 infections can potentially regulate sensory pain transmission by altering electrical excitability and the generation of electrical signals in pain-transmitting neurons (21). In addition, reduced electrical excitability has been shown to increase viral replication in HSV-1-infected neurons (22). These observations suggested that HSV-1 infection reduces neuronal excitability during early infection in order to enhance viral replication and spreading.
VGSCs consist of a multimeric complex containing a large pore-forming α subunit and several auxiliary subunits. In mammals, the main pore-forming subunit is generated by the product of 10 genes (23). Of these subunits, Nav1.1, -1.6, -1.7, -1.8, and -1.9 were distributed in DRG and TG (23), and Nav1.7, -1.8, and -1.9 were found to participate in mediating chronic pain in DRG (24). Disruption of Na+ channel expression has been associated with altered excitability in pain-transmitting neurons (25). Furthermore, inhibition of Na+ channels can reverse mechanical allodynia and thermal hyperalgesia in a model of herpesvirus-induced postherpetic pain (26).
Our hypothesis is that quiescent HSV-1 infections can modulate the functional expression of Na+ channels and alter neuronal excitability, thus participating in the establishment of latency and the regulation of pain. Changes in VGSC activity may play a role in the sensory abnormalities described in patients suffering chronic pain associated with herpesvirus infection (9, 10).
RESULTS
Differentiated HD10.6 cells supported the establishment of quiescent HSV-1 infection, and increased viral replication could be observed by TSA treatment.
To test the ability of HD10.6 cells to support quiescent infection with green fluorescent protein (GFP)-labeled HSV-1 strain McKrae (McKrae-GFP), differentiated cells were infected at a multiplicity of infection (MOI) of 1 in the absence or presence of acyclovir (ACV), followed by qualitative fluorescence microscopy to monitor the progress of viral infection. GFP fluorescence helps us identify the infected cells at different time points of latency establishment (LE), latency maintenance (LM), or reactivation. It has been reported that the intensity of fluorescent reporter signals is correlated with recombinant HIV replication in infected cells (27). According to our experience with other GFP-fused HSV-1 strains, GFP expression also correlates with HSV-1 replication. Strong GFP intensity indicates increased viral replication, while weak GFP intensity indicates decreased viral replication. The results showed that without ACV, HD10.6 cells were well infected by McKrae-GFP at an MOI of 1 and replicated efficiently at 3 days postinfection (dpi) (compare Fig. 1A and B). In the presence of ACV at 3 dpi, fluorescence microscopy indicated that cells were infected with decreased replication (Fig. 1C). The green fluorescence continued to decrease at 7 dpi (Fig. 1D) and was barely detected at 12 dpi after 5 days of ACV washout (Fig. 1E). This quiescent state of infection, found in approximately all of the infected cells, can be maintained for another 2 weeks without evidence of viral replication (data not shown). These quiescently infected cells, nonetheless, displayed increased green fluorescence and increased numbers of GFP-positive cells after the addition of the histone deacetylase (HDAC) inhibitor trichostatin A (TSA) for 2 days (Fig. 1F). These findings suggested that differentiated HD10.6 cells were able to support HSV-1 infection in a dormant state mimicking latency and maintained the ability to replicate, a scenario similar to reactivation.
FIG 1.

Establishing a quiescent state of HSV-1 infection with ACV and triggering replication with TSA. Differentiated HD10.6 cells were infected with HSV-1, followed by fluorescence microscopy examining the progress of infection. (A) No-infection control. (B) Lytic infection. The virus replicated, and green fluorescence can be clearly seen at 3 dpi. (C) Infection with ACV at 3 dpi. Cells with green fluorescence can be observed with weaker intensity than that seen in lytic infection. (D) Infection with ACV at 7 dpi. Green fluorescent cells can barely be detected, and their number has decreased. (E) Infection at 12 dpi with 5 days of ACV washout. Fluorescence microscopy can hardly detect the green fluorescent cells, suggesting that a quiescent infection mimicking latency has been established and maintained. (F) Infection at 12 dpi with 3 days of ACV washout, followed by 2 days of TSA treatment. Fluorescence microscopy records more cells with stronger green fluorescence, suggesting that the dormant state of infection has been disrupted with increased viral gene expression and replication, a scenario similar to the reactivation observed in animal models.
TSA increased viral gene expression and replication but not virus release.
The quiescent state of infection and the effects of TSA on the virus were further analyzed by quantitative reverse transcription-PCR (qRT-PCR) in order to investigate changes in viral gene expression. The results showed that HSV-1 thymidine kinase (TK) and GFP expression under the control of the cytomegalovirus (CMV) promoter were increased 3.47-fold and 124-fold by TSA, respectively, following reactivation (Fig. 2A and B). The phosphoinositide 3-kinase (PI3K) inhibitor LY294002, which was used previously to induce HSV-1 reactivation (28 – 30), did not appear to increase viral gene expression, probably due to the fact that in order to maintain neuronal excitability, neurotrophins such as nerve growth factor (NGF) and glial cell-derived neurotrophic factor (GDNF) were not removed (Fig. 2A and B). The release of infectious viral particles was tested by collecting the media of infected HD10.6 cultures, followed by plaque assays. Neither the TSA nor the LY294002 treatment was able to trigger the release of infectious viral particles (data not shown). The same experiment was performed using infected HD10.6 cell lysates, and the results indicated that TSA treatment produced a virus titer approximately 32 PFU/ml, while latent cells and cells under LY294002 treatment generated no plaques (Fig. 2C). These results indicated that the HDAC inhibitor TSA promoted viral gene expression and replication of the latent HD10.6 cells, generating a small quantity of cell-associated virions. This interruption of viral release was not due to the specific viral strain, since TSA could not trigger viral release using strain 17-Syn+ (data not shown). TSA was able to induce HSV-1 reactivation and the release of infectious virus in other neuronal cells or neuron-like cells (31, 32). Therefore, it is likely that this lack of viral release is due to a cell-specific mechanism.
FIG 2.
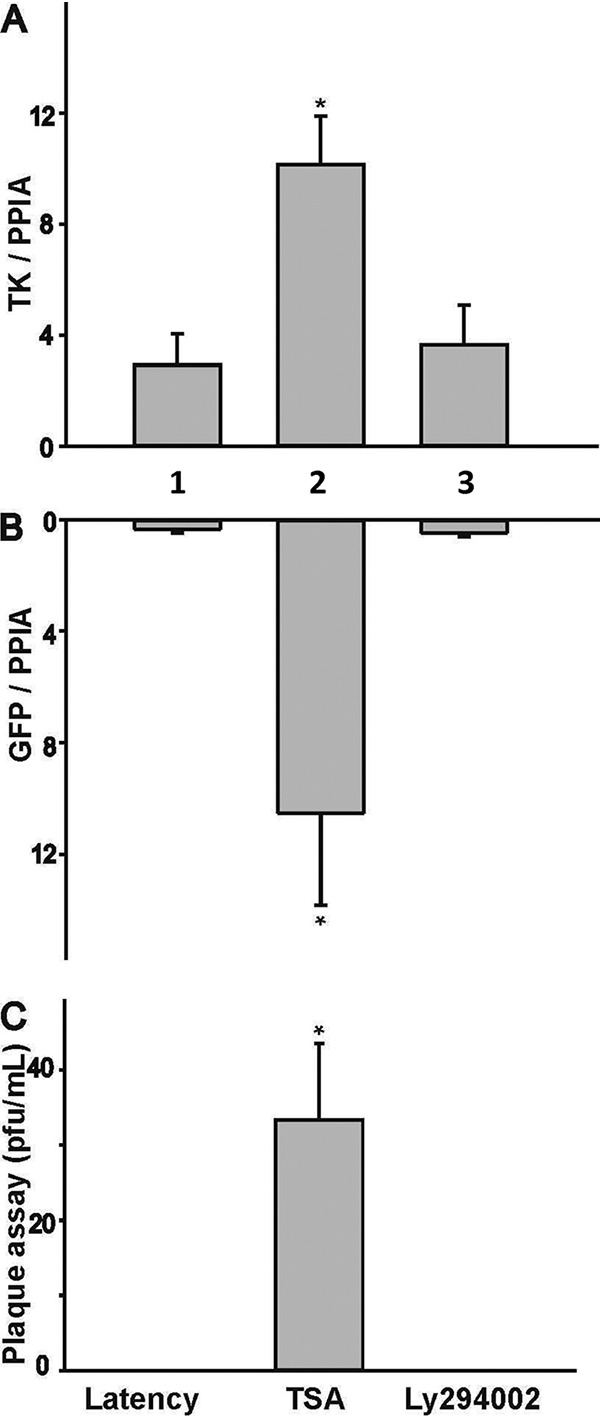
TSA induced viral gene expression and replication from the latent reservoir of HD10.6 cells. Viral latency and reactivation were assessed by qRT-PCR and plaque assays, respectively. (A) HSV-1 TK was analyzed by qRT-PCR. Shown are results for latent neurons that were either left untreated (no TSA or LY294002) (bar 1) or treated with TSA (bar 2) or LY294002 (bar 3). (B) GFP was measured by qRT-PCR. Latent neurons were either left untreated or treated as described for panel A. (C) Plaque assays were performed using lysates from infected HD10.6 cells. Shown are results for untreated lysates from latent neurons that were either left untreated (left) or treated with TSA (center) or LY294002 (right). An asterisk indicates a significant statistical difference with a P value of <0.05 for comparison with the latent group.
HSV-1 gene expression decreased during latency establishment and maintenance but increased when the virus was reactivated.
The protocol of latency establishment, maintenance, and reactivation is summarized in Fig. 3A. For LE, infected cultures were treated with 100 μM ACV for 7 days to initiate a quiescent HSV-1 infection. For LM, ACV was then removed, and the dormant state of infection continued for 5 days. Reactivation was attempted by adding 1 μM TSA for 2 days after 3 days of ACV washout. The transcription of the ICP0, TK, and latency-associated transcript (LAT) viral genes in the absence and presence of ACV at different time points (3 dpi, 7 dpi, and 12 dpi) was analyzed by qRT-PCR. It was shown that compared to levels in lytic infection, levels of ICP0 and TK gene expression decreased 50% and 75%, respectively, at 3 dpi in the presence of ACV (Fig. 3B). LAT, on the other hand, was accumulated to a level comparable to that during lytic infection at 3 dpi without viral replication (Fig. 3B). Levels of all three viral transcripts, nonetheless, declined at 7 dpi and 12 dpi, but TSA reversed the diminishing trend with significant increases (Fig. 3B). Collectively, these results suggested that differentiated HD10.6 cells supported the establishment of a quiescent HSV-1 infection, thus mimicking the maintenance of latency. TSA treatment overturned the dormant state, increased viral gene expression, and promoted replication. LAT did not accumulate when the virus established latency in vitro but was relatively abundant temporarily at 3 dpi without viral replication.
FIG 3.

Transcription profiles of three HSV-1 genes during the course of latency establishment and reactivation. (A) Schematic of a human DRG cell line-derived neuronal system used to investigate HSV-1 latency establishment, latency maintenance, and reactivation, as well as the related time point for sodium current recording. (B) The transcription of three viral genes (ICP0, TK, and LAT) was assessed by qRT-PCR at different time points (3 dpi, 7 dpi, and 12 dpi) in the absence of viral replication and was compared to those for lytic infection at 3 dpi and reactivation by TSA at 14 dpi. Asterisks indicate statistically significant differences (P < 0.05) from the corresponding gene in the 3-dpi no-ACV group (*) or the 12-dpi latency group (**). NS, not statistically significant.
Na+ channel activity was abolished by HSV-1 infection but gradually recovered during latency establishment.
To test the impact of viral infection on sodium channel activity, differentiated cells were infected with McKrae-GFP, followed by electrophysiological recordings of Na+ currents from cells expressing green fluorescence as evidence of infection. VGSC activity was measured as current density, which was calculated by dividing the maximal current amplitude by the cell capacitance (Fig. 4A). Differentiated control cells exhibited robust sodium currents, and lytic infection at 3 dpi eliminated the Na+ currents (Fig. 4B, bar 3), similar to what we observed previously in ND7/23 cells (16). The effects of infection in the presence of ACV on the Na+ currents were assessed at 3 dpi and 7 dpi. It can be seen that Na+ currents were not affected by ACV in controls (Fig. 4B, bar 2) but were reduced at 3 dpi with ACV (Fig. 4B, bar 4). This is different from the previous report that ACV treatment can maintain VGSC activity after HSV-1 infection (33). The sodium current densities were recovered, without significant differences from those for the control, during LE at 7 dpi (Fig. 4B, bar 5). The loss of VGSC activity was not due to cell death, since viability assays showed that no difference was recorded among the groups (Fig. 4C). These results, similar to what was observed previously with varicella-zoster virus (34), suggested that the VGSC activity lost upon HSV-1 infection was rescued during LE.
FIG 4.
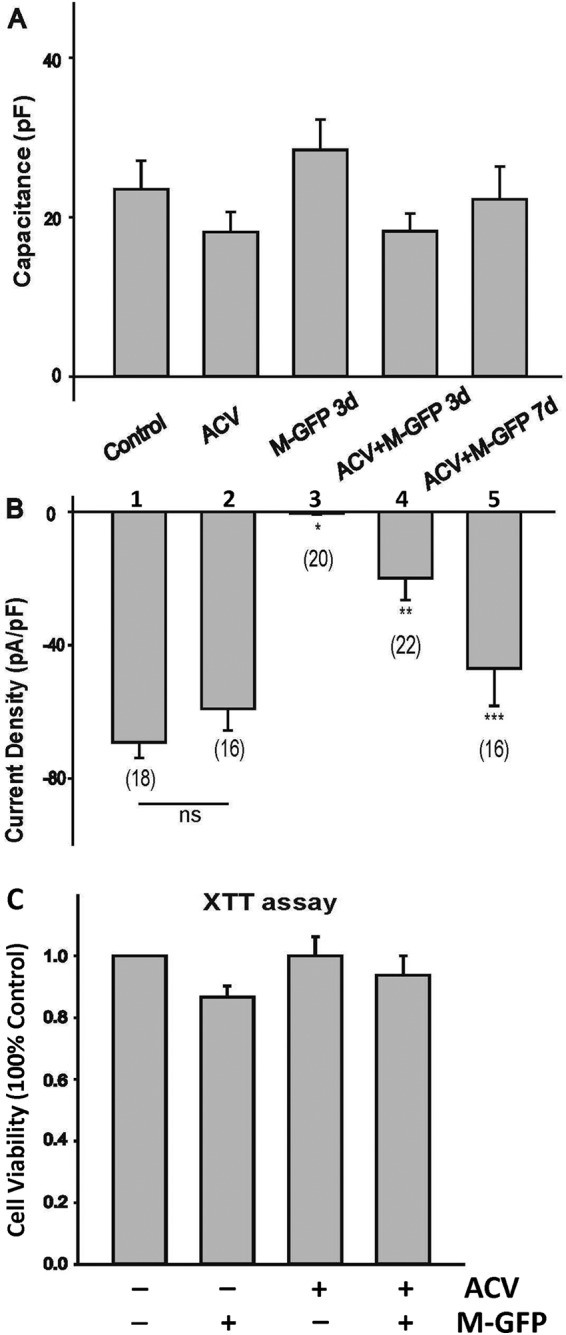
Sodium current recording during HSV-1 latency establishment in HD10.6 cells. (A) Comparison of capacitances of HD10.6 cells under differentiation conditions. Control, no ACV treatment; ACV, ACV treatment; M-GFP 3d, lytic infection with GFP-labeled HSV-1 McKrae for 3 days; ACV+M-GFP 3d, latency establishment for 3 days; ACV+M-GFP 7d, latency establishment for 7 days. (B) Mean Na+ current densities generated in HD10.6 cells under different conditions. The voltage-gated Na+ current density was calculated by dividing the peak current amplitude by the cell capacitance. Asterisks indicate significant differences (P < 0.05) from the control group (*), the ACV treatment group (**), or the group infected with McKrae-GFP in the presence of ACV at 3 dpi (***). Numbers in parentheses indicate the number of cells recorded for each condition. ns, not statistically significant. (C) Cell viability was determined by the XTT assay. Four different groups were tested: control, M-GFP, ACV, and ACV+M-GFP. No statistically significant difference was detected among groups, although a minor reduction in viability at 3 dpi without ACV was observed.
HD10.6 cells exhibited robust TTX-sensitive Na+ currents.
To assess the nature of HD10.6 cell VGSCs, the Na+ current was recorded after tetrodotoxin (TTX) treatment. It appeared from analyses of the Na+ current trace (Fig. 5A) and the current-voltage (I–V) relationship (Fig. 5B) that TTX at 250 nM significantly reduced the Na+ current. Quantitative studies showed that approximately 88% of the Na+ current was eliminated by TTX (Fig. 5C), in agreement with the previous finding that HD10.6 cells and DRG neurons utilized primarily TTX-sensitive VGSCs for their physiological activity (35, 36). The expression of two TTX-sensitive channels, Nav1.3 and Nav1.7, was analyzed by qRT-PCR, and the results indicated that Nav1.7 expression, but not Nav1.3 expression, was steadily increased during LE (Fig. 5D). Western blot analyses showed that Nav1.7, but not Nav1.3, was expressed in differentiated HD10.6 cells (Fig. 5E). Together, these studies suggested that VGSC activity lost due to viral infection was recovered during LE and was likely derived from TTX-sensitive Nav1.7.
FIG 5.

Identification of voltage-gated Na+ channel subtypes in HD10.6 cells during HSV-1 infection. (A) The current traces of the ACV group generated by voltage steps to 0 mV from a holding potential of –100 mV were eliminated following TTX treatment. The peak currents were obtained by voltage steps to +20 mV after treatment with 250 nM TTX. (B) The current-voltage (I–V) relationship for the peak Na+ current amplitude in differentiated HD10.6 cells before and after TTX treatment was generated by a series of voltage steps from a holding potential of −100 mV. (C) Capacitance and Na+ current densities were compared between cells with or without TTX treatment. The asterisk (*) indicates a statistically significant difference (P < 0.05) from the ACV control. Numbers in parentheses indicate the number of cells recorded for each condition. ns, no statistically significant difference. (D) Nav1.3 and Nav1.7 expression was analyzed by qRT-PCR during latency establishment. Asterisks indicate significant differences (P < 0.05) from results for McKrae-GFP infection in the absence (*) or presence (**) of ACV at 3 dpi. (E) Nav1.3 and Nav1.7 protein expression was detected by Western blot analyses.
Nav1.7 expression was increased in latently infected neurons.
We continued to study Nav1.7 on the basis of the observation that HD10.6 cells displayed TTX-sensitive VGSC activity. The expression profile of the sodium channels in the latent neurons was analyzed by Western blotting using antibodies against the TTX-sensitive channel subunit Nav1.7. The results showed that the Nav1.7 protein was expressed in control cells but that its expression was higher in latent neurons (Fig. 6A, lanes 1 and 2). TSA slightly decreased protein expression from that in control cells (Fig. 6A, lane 3) but caused a significant reduction in cells with the latent reservoir (Fig. 6A, lane 4). Additional studies by qRT-PCR revealed that the Nav1.7 transcript of latent cells displayed a 4.25-fold increase over others (Fig. 6B, bar 2). These results suggested that Nav1.7 protein expression was elevated in latent neurons, probably due to increased transcription influenced by the latent virus. Nav1.7 expression on individual neurons was further assessed by immunofluorescence microscopy, which showed that Nav1.7 expression was as high in latent neurons as in uninfected neurons, if not higher (Fig. 6C). The signal, nonetheless, became weaker in neurons with viral reactivation triggered by TSA (Fig. 6D), suggesting that viral replication may have effects that reduce Nav1.7 expression. Although there appeared to be a change in Nav1.7 expression following the establishment of latency and reactivation, the VGSCs are multimeric proteins, and it is not clear whether other axillary subunits also contribute to the increased molecular and functional expression of VGSCs.
FIG 6.
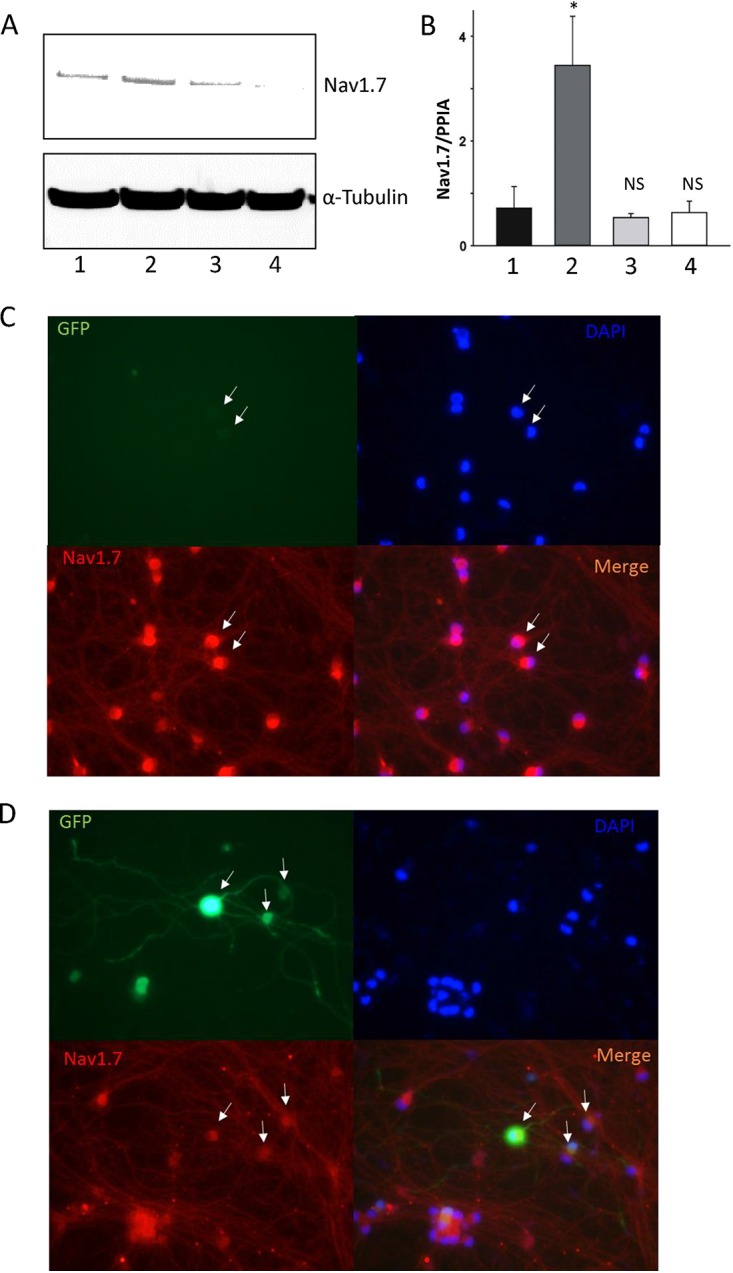
Expression of Nav1.7 in HD10.6 cells. (A and B) The expression of Nav1.7 protein and mRNA was assessed by Western blotting (A) and qRT-PCR (B), respectively. (A) Lane 1, control; lane 2, latent cells; lane 3, control plus TSA; lane 4, latent cells plus TSA. (B) The asterisk (*) indicates a statistically significant difference (P < 0.05) from the control; NS, no significant difference from the control. (C and D) Nav1.7 expression was further evaluated in individual cells by immunofluorescence microscopy. (C) Latent cells; (D) latent cells plus TSA. White arrows indicate the latent or reactivated cells evidenced by GFP.
Latent neurons exhibited very high VGSC activity.
Electrophysiological studies were performed to analyze the sodium current density. The results indicated that the capacitance of cells under treatment did not change and that the Na+ current density from latent neurons was approximately 20% higher than that from control cells (compare Fig. 7A and B). TSA treatment, nevertheless, decreased the current density by 50% regardless of the status of latency (Fig. 7B). Current density plot analyses of the VGSC activities of the individual neurons demonstrated that control cells (Fig. 7C) displayed a wide range of current densities, from very high (>80 pA/pF) (40% of cells) to high (between 60 and 80 pA/pF) (40% of cells) and moderate (between 40 and 60 pA/pF) (20% of cells) densities. Latently infected neurons, in contrast, presented a unique pattern, with approximately 90% of the neurons exhibiting very high current densities (Fig. 7D). With the TSA treatment, charts for both control (Fig. 7E) and infected (Fig. 7F) groups revealed the current density shifting toward the left, with 45 to 50% of neurons exhibiting low (<40 pA/pF) density values, suggesting a trend of decreased VGSC number and activity. Collectively, these results indicated that latent neurons appeared to have higher VGSC activity, presumably resulting from the increased Nav1.7 protein expression due to the elevated transcription of Nav1.7 influenced by latency. This VGSC increase was nullified upon reactivation by TSA. It is noteworthy, though, that TSA treatment per se did not considerably affect the level of protein expression but significantly decreased VGSC activity.
FIG 7.

Evaluation of VGSC activity during latency and after induced viral replication. Na+ currents of the whole cell were recorded to measure the activity. (A) Cell capacitance was measured and compared to that for controls. (B) Na+ current density was calculated based on the peak current and capacitance. Asterisks indicate statistically significant differences (P < 0.05) from the control (*) or the latent (**) group. Numbers in parentheses indicate the number of cells recorded for each condition. (C to F) Current density plots were analyzed to compare the VGSC activities of neurons in each group: control neurons (C), latent neurons (D), control neurons plus TSA (E), and latent neurons plus TSA (F). Note that for latent neurons, <0.02% of the cells had bright GFP, and these were regarded as failing to establish latency; none of our current recordings are from those bright GFP cells.
DISCUSSION
The glossary of viral latency and reactivation has been an interesting topic for virologists, especially those who study alphaherpesviruses (37). It is generally believed that HSV-1, for example, employs two alternative infection plans: lytic and latent infections. Episodes are launched by initial acute lytic infection, marked by active viral replication in epithelial cells, followed by latency established inside the sensory neurons of the ganglia, such as TG or DRG, innervating mucosae of the primary infection site (38). The operational definition of latency was suggested to be that a viral genome remained within a cell population over time, maintaining a dormant state without detectable lytic viral transcripts, proteins, or infectious particles, yet retaining the capacity to replicate and reactivate (38 – 44). The best examples recapitulating these features of HSV latency are the in vivo rodent models (45 – 47). Besides the animal models, a number of cell culture models generated in neuroblastoma cell lines, a differentiated embryonal carcinoma cell line, induced pluripotent stem cells, neuronal stem cells, and embryonic stem cells have been developed (43, 47 – 52). Although they barely satisfied all characteristics of latency, these in vitro models offered a number of benefits in studying HSV latency, particularly in understanding the molecular mechanisms.
The current cell culture model derived from human DRG was first used to study nociceptive properties (35). Recent progress made through infection with HSV-1 in the presence of ACV demonstrated a unique platform revealing many properties mimicking HSV-1 latency, such as a silent phase of infection and low-frequency reactivation (42). Our present results using a different virus strain, McKrae, largely agreed with this previous finding while adding novel observations (42). First, a quiescent infection was achieved without detection of the infectious virus inside or outside the cells, and TSA treatment appeared to switch the latent reservoir into a replication mode, imitating the viral reactivation observed in vivo. Second, the TSA action resulted in increased viral transcription and production of viral progeny within the cells, which was not observed previously. Third, and last, the transcription of HSV-1 ICP0 and TK progressively diminished during the latency establishment stage but increased significantly during TSA-mediated reactivation. A slightly different pattern was observed for HSV-1 LAT, which temporarily accumulated at 3 dpi, followed by a steady decline thereafter. It should be noted that the PCR oligonucleotide primers were designed to target the stable LAT intron (53), so it is not impossible that this accumulation resulted from delayed degradation. However, we cannot rule out the possibility that HSV-1 LAT plays a role in latency initiation, which warrants future studies.
Our results indicated that VGSC activity in HD10.6 cells was quickly eliminated by lytic HSV-1 infection, a finding consistent with the preclinical investigation showing that peripheral nerve injury decreased pain-associated voltage-gated channels in injured DRG (15). Additional studies indicated that sodium currents gradually recuperated during the establishment of latency, in line with a previous report suggesting an increase in sodium current density associated with another alphaherpesvirus, varicella-zoster virus (34). Furthermore, latent neurons demonstrated a unique functional expression profile in which 90% of the cells exhibited increased sodium currents with a very high density of VGSCs. This observation suggested that HSV-1 employed a strategy of first facilitating replication during lytic infection by bringing down VGSC activity in order to decrease neuronal excitability and then sustaining the quiescent state by promoting the functional expression of VGSCs, thus increasing excitability. In other words, increased neuronal excitability seemed to impede viral replication and participate in the maintenance of latency. This is further supported by a previous report (22) and our observation that neuronal depolarization by potassium chloride significantly inhibits viral gene expression and replication (unpublished data). On the other hand, the sensation of neuropathic pain may be part of an innate immune mechanism to suppress viral replication and prevent viral reactivation. Additional investigations to elucidate the underlying molecular mechanisms are under way.
The observation of Na+ current recovery in neurons following the establishment of latency prompted us to study which sodium channel subtypes may contribute to the rescue. Our rationale is based on the literature and our own data. First, the HD10.6 cells were originally generated as small-diameter sensory DRG neurons (35), and the VGSC activities of the small-diameter neurons were expected primarily from Nav1.7 (TTX sensitive), Nav1.8 (TTX resistant), and Nav1.9 (TTX resistant) (15). These three were the most abundantly expressed sodium channels in the peripheral nervous system (PNS) and DRG neurons, with important functions (54). Next, during DRG nerve injury, significant changes in expression were expected for Nav1.3, Nav1.7, Nav1.8, and Nav1.9 (15). A number of studies revealed that increased Nav1.3 and Nav1.8 expression in DRG neurons was related to neuropathic pain induced by peripheral nerve injury (55 – 57). Last, our TTX experiment showed that TTX-sensitive sodium channels are the main contributors to the functional current in differentiated HD10.6 cells (Fig. 5). Therefore, we ruled out Nav1.8 and Nav1.9, since they are TTX resistant. Among the TTX-sensitive channels, the role of Nav1.1 in neuropathic pain is elusive (15). As for Nav1.2, the evidence suggested that it is unlikely to participate in the development of neuropathic pain as well (58 – 60). Thus, we decided to focus on Nav1.3 and Nav1.7 expression during viral infection, since Nav1.3 was accumulated in the neuromas of patients with painful neuropathy (61), and Nav1.7 was further reported to be predominately expressed in human DRG neurons and significantly increased in several chronic pain disorders (36). The first subunit examined was Nav1.3, since it was reported to be upregulated in DRG neuronal injury, and the protein was accumulated in patients suffering painful neuropathy (15, 61 – 64). However, we did not detect any changes in Nav1.3 transcription, and there was no Nav1.3 protein expression in differentiated HD10.6 cells (Fig. 5). Our investigation on Nav1.7 indicated that its transcription was active and that the total protein levels were slightly higher during latency but reduced upon TSA treatment, which causes viral reactivation. Immunofluorescence microscopy revealed that latent neurons under the influence of TSA displayed reduced expression of Nav1.7, suggesting that the neurons with latent HSV-1 exhibited Nav1.7 induction and that reactivation impeded this increase. Subsequent electrophysiological studies investigating VGSC activity demonstrated that sodium currents, though higher in latent neurons and lower after reactivation, revealed a decrease in the TSA control (Fig. 6). Given the fact that the total Nav1.7 protein was not significantly decreased by TSA, it is likely that TSA modulated cell signaling and obstructed the trafficking of Nav1.7 to the cell surface. TSA decreased neuronal excitability and induced viral replication, which indicated that decreased neuronal excitability may facilitate viral reactivation.
Nav1.7 has been shown to be physiologically associated with the function of other VGSCs to amplify generator potentials and boost subthreshold stimuli, thus causing VGSC activation and efficient recovery from inactivation, as well as generating high-frequency action potentials (65). Blockade of the channel indicated that Nav1.7 participated in the key TTX-sensitive Na+ current in small-diameter DRG neurons (66). Nav1.7 activity has been linked to posttranslational modifications, such as phosphorylation (67). Signaling transduction studies suggested that phosphorylation on an intracellular loop by extracellular signal-related kinase 1/2 (ERK1/2) influenced Nav1.7 activity, thus increasing its efficacy during neuronal firing (67, 68). Other reports indicated that Fyn tyrosine kinase can phosphorylate Nav1.7, thus influencing channel expression and gating (69). It is not known how HSV-1 latency can influence VGSC activity. Understanding the molecular mechanisms involved in the regulation of Nav1.7 activity may have important implications for HSV-1 latency and reactivation.
Our current study demonstrated that differentiated HD10.6 cells displayed robust VGSC activity and steady recovery of Na+ currents after elimination by HSV-1 infection. Differentiated HD10.6 cells supported the establishment of a quiescent state of infection mimicking latency and maintaining the capacity to replicate, a scenario similar to the reactivation observed in vivo. During the maintenance stage of dormant infection, the neurons harboring the latent reservoir of the virus appeared to have very high functional expression of VGSCs, and reactivation decreased VGSC activity. This abnormal pattern of the Na+ channel expression profile following lytic HSV-1 infection, latency, and reactivation may disturb the ability of sensory neurons to transmit pain information. HSV-1 infection often alters pain sensory transmission, with both diminished and enhanced pain sensations (7, 12, 70). Functionally, elevated sensory neurotransmission and increased excitability due to higher expression of voltage-activated Na+ currents during latency could potentially enhance pain signaling but benefit the host by suppressing viral replication. This unique change in ion channel expression at different stages of viral infection, because of the host responses to virus assault, remains to be investigated and may have implications for the development of therapeutic protocols to treat herpesvirus-mediated pain, trigeminal neuralgia, and postherpetic neuralgia.
MATERIALS AND METHODS
Cells and virus.
The human DRG neuronal cell line HD10.6 was used as an in vitro model as described previously (41, 42) and was a gift from Celgene Corporation (San Diego, CA). The HD10.6 cells were cultured and differentiated as described previously by Thellman et al. (42). Briefly, undifferentiated HD10.6 cells were cultured on fibronectin (catalog no. FC010; Millipore)-coated flasks and were grown in advanced Dulbecco’s modified Eagle medium (DMEM)–nutrient mixture F-12 (catalog no. 12634-010; Fisher Scientific) supplemented with 1× GlutaMAX, 1× B-27 supplement, 10 ng/ml prostaglandin E1, 0.5 ng/ml basic fibroblast growth factor (β-FGF), and 50 μg/ml G418 solution. For differentiation, HD10.6 cells were seeded on plates or coverslips (for whole-cell recordings) precoated with 2% Matrigel (catalog no. 356234; Corning) and 10 μg/ml poly-d-lysine for 1 h, at a density of 5 × 104/well in 24-well plates (or coverslips) or 6 × 105/well in 6-well plates in differentiation medium (DM). Differentiation of the HD10.6 cells was evoked by NeuralQ basal medium (catalog no. GSM-9420; GlobalStem) supplemented with 1× GlutaMAX, 1× B-27 Plus neuronal supplement (catalog no. A3582801; Thermo Fisher), 50 ng/ml 2.5S nerve growth factor (NGF), 25 ng/ml each of ciliary neurotrophic factor (CNTF), glial cell-derived neurotrophic factor (GDNF), and neurotrophin-3 (NT-3), 1 μg/ml tetracycline, and 25 μM forskolin. Half of the medium was changed every 3 or 4 days, while the whole medium was changed for the ACV treatment or the ACV washout.
African green monkey kidney Vero cells were purchased from the ATCC (catalog no. CCL-81). The culture conditions for these cells have been described previously (32, 71). Only cells passaged <15 times were used in this work. None of the cell lines used in this work had been misidentified, according to the International Cell Line Authentication Committee (ICLAC), and cell line authentication was performed by the providers using short-tandem repeat (STR) analysis. HSV-1 strain McKrae with GFP expression (M-GFP) under the control of a cytomegalovirus (CMV) promoter (72) was a gift from Gus Kousoulas and was used throughout the current study.
Infection, latency establishment, and reactivation.
For lytic HSV-1 infection, differentiated (≥7 days) HD10.6 cells were infected with the virus for 1 h at a multiplicity of infection (MOI) of 1. To establish HSV-1 latency, cells were pretreated with 100 μM acycloguanosine (also known as acyclovir; catalog no. A4669; Sigma-Aldrich) overnight, followed by infection. Unbound viral particles were washed out after 1 h; DM supplemented with 100 μM ACV was applied for 3 days and was then replaced with fresh DM with 100 μM ACV for another 4 days to allow for the establishment of latent HSV-1 infection. ACV was then removed by a vigorous wash with DM to allow for the maintenance of latent HSV-1 infection. The reactivation of HSV-1 from latency was tested by adding 1 μM TSA (catalog no. T1952; Sigma-Aldrich) or 20 μM LY294002 (catalog no. 440202; Sigma-Aldrich) for 2 days.
Fluorescence microscopy.
Morphometric analysis was performed, and the GFP fluorescence of cultured HD10.6 cells was determined, with a Nikon Eclipse Ti microscope with a 20× inverted objective and a Photometrics CoolSNAP EZ cooled camera. The intensity of the GFP fluorescence change was used to monitor the change in HSV-1 activity in infected HD10.6 cells (16, 73).
Quantitative RT-PCR.
Viral gene expression was quantified as described previously (16, 32, 53). RNA was collected using the iScript sample preparation reagent (catalog no. 170-8898; Bio-Rad), followed by reverse transcription using iScript RT Supermix (catalog no. 17008841; Bio-Rad) to produce the cDNA template. qPCRs were performed using SsoAdvanced Universal SYBR green supermix (catalog no. 1725271; Bio-Rad) on triplicate samples with primers specific for the selected viral or human genes listed in Table 1. The qPCRs were carried out initially at 95°C for 30 s, followed by 39 cycles at 95°C for 10 s, the respective annealing temperature (Table 1) for 30 s, and 65°C for 5 s. For each gene, mRNA expression was normalized by peptidylprolyl isomerase A (PPIA) expression (32, 71, 74).
TABLE 1.
Genes, primers, and annealing temperatures for qPCR
| Target genea | NCBI RSb | Primer sequencec | Annealing temp (°C) |
|---|---|---|---|
| Nav1.3 (h) | NM_006922.4 | AGACAGCGAAAGCAGGAGAG (f) | 58 |
| ACCCACCAAGGAAACCACAC (r) | |||
| Nav1.7 (h) | NM_002977.3 | TATGACCATGAATAACCCGC (f) | 58 |
| TCAGGTTTCCCATGAACAGC (r) | |||
| PPIA (h) | NM_005612.5 | AGCATACGGGTCCTGGCATCT (f) | 65 |
| CATGCTTGCCATCCAACCACTCA (r) | |||
| GFP | ACTTCAAGATCCGCCACAACA (f) | 58 | |
| TGATCGCGCTTCTCGTTGG (r) | |||
| ICP0 (v) | GACGGGCAATCAGCGGTTC (f) | 60 | |
| GTAGTCTGCGTCGTCCAGGT (r) | |||
| TK (v) | TACCCGAGCCGATGACTTAC (f) | 62 | |
| AAGGCATGCCCATTGTTATC (r) | |||
| LAT (v) | CGGCGACATCCTCCCCCTAAGC (f) | 60 | |
| GACAGACGAACGAAACGTTCCG (r) |
h, human; v, viral.
RS, reference single nucleotide polymorphism identification number.
f, forward primer; r, reverse primer.
Plaque assay.
Plaque assays were performed as described previously (16, 32, 71). Lysates of infected HD10.6 cells were first mixed with 500 μl medium and then subjected to three freeze-thaw cycles so that the virus was released into the medium. The cell lysates and the supernatants from infected cultures were serially diluted before being added to Vero cell monolayers in 24-well plates seeded at 8 × 104 cells/well. After 1 hpi, the medium was removed and was replaced by a fresh culture medium supplemented with 5% fetal bovine serum (FBS). After 2 days, the cells were fixed with ice-cold methanol and were stained with crystal violet before plaques were counted. Data were collected from triplicates.
Cell viability assay.
HD10.6 cells (1.5 × 104) were seeded in quadruplicate for each group on 96-well plates. Cell viability was determined with an XTT cell viability assay kit (catalog no. 30007; Biotium, Hayward, CA) according to the manufacturer’s instructions. Briefly, 25 μl Activation Reagent was mixed with 5 ml XTT solution, and 50 μl of the activated XTT solution was added to each well. After 3 h of incubation, the absorbances of the samples were measured at a wavelength of 450 nm, and the background absorbance at 610 nm was measured, using a Multiskan FC microplate reader (Fisher Scientific). Cell viability was calculated in quadruplicate and was normalized to control values.
Electrophysiology.
During recordings, HD10.6 cells were visualized with a Nikon Eclipse Ti inverted microscope (Nikon Instruments, Melville, NY) equipped with Hoffman optics and epifluorescence filters, which can be used to identify infected HD10.6 cells by the expression of GFP. Voltage-activated sodium currents from individual cells were recorded using the whole-cell patch-clamp technique. Recordings were performed at room temperature (22 to 24°C). The pipette electrode solution consisted of 120 mM CsCl, 2 mM MgCl2, 10 mM HEPES-KOH, 10 mM EGTA, 1 mM ATP, and 0.1 mM GTP, pH 7.4, adjusted with CsOH. The extracellular solution used to measure Na+ currents contained 145 mM NaCl, 5.3 mM KCl, 0.54 mM CaCl2, 5.7 mM MgCl2, 13 mM HEPES, and 5 mM glucose, pH 7.4, adjusted with KOH. Na+ currents were generated by applying a 20-ms depolarizing step to various potentials from a holding potential of −100 mV. A MultiClamp 700A amplifier was used to apply voltage commands, and pCLAMP software (Axon Instruments, Foster City, CA) was used to perform data acquisition and analysis. MultiClamp 700B Commander was used to compensate for pipette offset, whole-cell capacitance, and series resistance (≤10 MΩ) automatically. Sampling rates were between 5 and 10 kHz. For quantitative analysis, cell size was normalized as described previously (75, 76). Voltage-gated Na+ current density was calculated by dividing the maximal current amplitude, generated by a voltage step around +10 mV from a holding potential of −100 mV, by the cell capacitance. TTX was purchased from Tocris (catalog no. 1069).
Western blot analysis.
Immunoblot analyses of the voltage-gated Na+ channel subunits were conducted as described previously (16), using rabbit anti-Nav1.3 (dilution, 1:1,000; catalog no. ASC-004) and anti-Nav1.7 (1:1,000; catalog no. ASC-008) antibodies (both from Alomone Labs, Jerusalem, Israel). Proteins were first separated on precast 8% SDS-PAGE gels (catalog no. NW0080; Thermo Fisher) and then transferred to nitrocellulose membranes using the Invitrogen iBlot system. After incubation with a horseradish peroxidase-conjugated anti-rabbit secondary antibody (catalog no. 211-035-109; Jackson ImmunoResearch Labs, West Grove, PA) for 1 h at room temperature, a chemiluminescent substrate (SuperSignal West Pico; catalog no. 34080; Thermo Fisher) was applied, and the signals were captured using a ChemiDoc XRS+ documentation system (Bio-Rad, Hercules, CA). For controls, membranes were stripped and reprobed with a tubulin-specific antibody (1:2,000; catalog no. 05-829; Millipore), followed by incubation with a peroxidase-conjugated anti-mouse secondary antibody (catalog no. 115-035-146; Jackson ImmunoResearch Labs, West Grove, PA) and chemiluminescent detection.
Immunofluorescence assays.
The immunofluorescence assay was performed as described previously by Figliozzi et al. (32). Briefly, infection of HD10.6 cells seeded on 24-well plates precoated with Matrigel and poly-d-lysine was performed before fixation of cell cultures. Cells were washed with phosphate-buffered saline (PBS) twice, for 5 min each time, before 4% paraformaldehyde (PFA) was applied to fix the cells. After 15 min of incubation at room temperature, cells were washed with PBS three times, for 5 min each time. The cells were subjected to 0.25% Triton X-100 for 5 min and were then washed with PBS. Then the cells were incubated with blocking buffer (1× PBS, 1% bovine serum albumin [BSA], 0.3 M glycine, 0.1% Tween 20) supplemented with 5% goat serum for 1 h at room temperature. After overnight incubation with a Nav1.7 antibody (catalog no. ASC-008; Alomone Labs, Jerusalem, Israel) diluted 1:250 in antibody dilution buffer (1× PBS, 1% BSA, 0.1% Tween 20) at 4°C, the cells were washed with PBS three times, for 5 min each time. A preadsorbed, Alexa Fluor 594-conjugated goat anti-rabbit IgG(H+L) secondary antibody (diluted 1:500 in antibody dilution buffer; catalog no. ab150088; Abcam) was used to incubate the cells for 2 h at room temperature in the dark. After three washes with PBS, cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/ml in PBS) for 10 min in the dark, followed by imaging. Microscopy was performed with an Olympus IX2-RFACA motorized fluorescence microscope with a 40× inverted objective, and images were colorized using Olympus cellSens Dimension imaging software.
Data analysis.
Statistical analyses were performed with Student’s unpaired t test and one-way analysis of variance (ANOVA) followed by post hoc analysis using Tukey’s honestly significant difference test (Statistica Software, Tulsa, OK). All data values are presented as means ± standard errors of the means (SEM), and a P value of ≤0.05 was regarded as significant.
ACKNOWLEDGMENTS
We are grateful to Celgene Corporation (San Diego, CA) for the gift of HD10.6 cells. We thank Catherine Pearce and Nicholas Baird of the University of Colorado School of Medicine for scientific support and discussions. We thank Gus Kousoulas of LSU Baton Rouge for the virus. We appreciate the editorial support of Donna Satterlee of the University of Maryland Eastern Shore.
This project was supported in part by the University of Maryland Eastern Shore School of Pharmacy and NINDS/NIH grant R01NS081109 (to S.V.H.). The content of this work does not necessarily represent the official views of the NIH and is solely the responsibility of the authors. NINDS had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
We declare no conflict of interest.
S.V.H. started the project with the initial hypotheses and funding acquisition. M.M.-C. and S.V.H. designed the experimental approaches, which were further modified by Q.Z. All cells and viruses were cultured, differentiated, and maintained by Q.Z. Infection, gene expression analyses, and plaque assays were performed by Q.Z. and S.V.H. Electrophysiology was performed by Q.Z. with assistance from M.M.-C. Immunofluorescence assays were performed by Q.Z. and S.V.H. The results were interpreted and discussed by Q.Z., M.M.-C., and S.V.H. The manuscript was written by S.V.H., with help from Q.Z. and M.M.-C.
REFERENCES
- 1.Nagel MA, Choe A, Traktinskiy I, Gilden D. 2015. Burning mouth syndrome due to herpes simplex virus type 1. BMJ Case Rep 2015:bcr2015209488. doi: 10.1136/bcr-2015-209488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martins I, Cabral L, Pinto A, Wilson SP, Lima D, Tavares I. 2011. Reversal of inflammatory pain by HSV-1-mediated overexpression of enkephalin in the caudal ventrolateral medulla. Eur J Pain 15:1008–1014. doi: 10.1016/j.ejpain.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Martins I, Costa-Araujo S, Fadel J, Wilson SP, Lima D, Tavares I. 2010. Reversal of neuropathic pain by HSV-1-mediated decrease of noradrenaline in a pain facilitatory area of the brain. Pain 151:137–145. doi: 10.1016/j.pain.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 4.Walwyn WM, Matsuka Y, Arai D, Bloom DC, Lam H, Tran C, Spigelman I, Maidment NT. 2006. HSV-1-mediated NGF delivery delays nociceptive deficits in a genetic model of diabetic neuropathy. Exp Neurol 198:260–270. doi: 10.1016/j.expneurol.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Sasaki A, Takasaki I, Andoh T, Shiraki K, Takeshima H, Takahata H, Kuraishi Y. 2008. Nociceptin-receptor deficiency prevents postherpetic pain without effects on acute herpetic pain in mice. Neuroreport 19:83–86. doi: 10.1097/WNR.0b013e3282f35839. [DOI] [PubMed] [Google Scholar]
- 6.Takasaki I, Andoh T, Nitta M, Takahata H, Nemoto H, Shiraki K, Nojima H, Kuraishi Y. 2000. Pharmacological and immunohistochemical characterization of a mouse model of acute herpetic pain. Jpn J Pharmacol 83:319–326. doi: 10.1254/jjp.83.319. [DOI] [PubMed] [Google Scholar]
- 7.Takasaki I, Andoh T, Shiraki K, Kuraishi Y. 2000. Allodynia and hyperalgesia induced by herpes simplex virus type-1 infection in mice. Pain 86:95–101. doi: 10.1016/s0304-3959(00)00240-2. [DOI] [PubMed] [Google Scholar]
- 8.Theil D, Derfuss T, Paripovic I, Herberger S, Meinl E, Schueler O, Strupp M, Arbusow V, Brandt T. 2003. Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. Am J Pathol 163:2179–2184. doi: 10.1016/S0002-9440(10)63575-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzales GR. 1992. Postherpes simplex type 1 neuralgia simulating postherpetic neuralgia. J Pain Symptom Manage 7:320–323. doi: 10.1016/0885-3924(92)90065-p. [DOI] [PubMed] [Google Scholar]
- 10.Valyi-Nagy T, Rathore JS, Rakic AM, Rathore RS, Jain P, Slavin KV. 2017. Herpes simplex virus type 1 human cervical dorsal root ganglionitis. Case Rep Neurol 9:188–194. doi: 10.1159/000479146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klockner U, Lee JH, Cribbs LL, Daud A, Hescheler J, Pereverzev A, Perez-Reyes E, Schneider T. 1999. Comparison of the Ca2+ currents induced by expression of three cloned α1 subunits, α1G, α1H and α1I, of low-voltage-activated T-type Ca2+ channels. Eur J Neurosci 11:4171–4178. doi: 10.1046/j.1460-9568.1999.00849.x. [DOI] [PubMed] [Google Scholar]
- 12.Andoh T, Shiraki K, Kurokawa M, Kuraishi Y. 1995. Paresthesia induced by cutaneous infection with herpes simplex virus in rats. Neurosci Lett 190:101–104. doi: 10.1016/0304-3940(95)11512-u. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell BM, Bloom DC, Cohrs RJ, Gilden DH, Kennedy PG. 2003. Herpes simplex virus-1 and varicella-zoster virus latency in ganglia. J Neurovirol 9:194–204. doi: 10.1080/13550280390194000. [DOI] [PubMed] [Google Scholar]
- 14.Hsia SC, Bedadala GR, Balish MD. 2011. Effects of thyroid hormone on HSV-1 gene regulation: implications in the control of viral latency and reactivation. Cell Biosci 1:24. doi: 10.1186/2045-3701-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W, Gu J, Li YQ, Tao YX. 2011. Are voltage-gated sodium channels on the dorsal root ganglion involved in the development of neuropathic pain? Mol Pain 7:16. doi: 10.1186/1744-8069-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Q, Hsia SC, Martin-Caraballo M. 2017. Regulation of T-type Ca2+ channel expression by herpes simplex virus-1 infection in sensory-like ND7 cells. J Neurovirol 23:657–670. doi: 10.1007/s13365-017-0545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conrady CD, Drevets DA, Carr DJ. 2010. Herpes simplex type I (HSV-1) infection of the nervous system: is an immune response a good thing? J Neuroimmunol 220:1–9. doi: 10.1016/j.jneuroim.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson RL, Sawtell NM. 2001. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J Virol 75:6660–6675. doi: 10.1128/JVI.75.14.6660-6675.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song XJ, Vizcarra C, Xu DS, Rupert RL, Wong ZN. 2003. Hyperalgesia and neural excitability following injuries to central and peripheral branches of axons and somata of dorsal root ganglion neurons. J Neurophysiol 89:2185–2193. doi: 10.1152/jn.00802.2002. [DOI] [PubMed] [Google Scholar]
- 20.Xie RG, Chu WG, Hu SJ, Luo C. 2018. Characterization of different types of excitability in large somatosensory neurons and its plastic changes in pathological pain states. Int J Mol Sci 19:E161. doi: 10.3390/ijms19010161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oakes SG, Petry RW, Ziegler RJ, Pozos RS. 1981. Electrophysiological changes of HSV-1-infected dorsal root ganglia neurons in culture. J Neuropathol Exp Neurol 40:380–389. doi: 10.1097/00005072-198107000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Zhang CX, Ofiyai H, He M, Bu X, Wen Y, Jia W. 2005. Neuronal activity regulates viral replication of herpes simplex virus type 1 in the nervous system. J Neurovirol 11:256–264. doi: 10.1080/13550280590952781. [DOI] [PubMed] [Google Scholar]
- 23.Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. 2000. Nomenclature of voltage-gated sodium channels. Neuron 28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 24.Strickland IT, Martindale JC, Woodhams PL, Reeve AJ, Chessell IP, McQueen DS. 2008. Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur J Pain 12:564–572. doi: 10.1016/j.ejpain.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Wood JN, Boorman JP, Okuse K, Baker MD. 2004. Voltage-gated sodium channels and pain pathways. J Neurobiol 61:55–71. doi: 10.1002/neu.20094. [DOI] [PubMed] [Google Scholar]
- 26.Garry EM, Delaney A, Anderson HA, Sirinathsinghji EC, Clapp RH, Martin WJ, Kinchington PR, Krah DL, Abbadie C, Fleetwood-Walker SM. 2005. Varicella zoster virus induces neuropathic changes in rat dorsal root ganglia and behavioral reflex sensitisation that is attenuated by gabapentin or sodium channel blocking drugs. Pain 118:97–111. doi: 10.1016/j.pain.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Terahara K, Yamamoto T, Mitsuki YY, Shibusawa K, Ishige M, Mizukoshi F, Kobayashi K, Tsunetsugu-Yokota Y. 2011. Fluorescent reporter signals, EGFP, and DsRed, encoded in HIV-1 facilitate the detection of productively infected cells and cell-associated viral replication levels. Front Microbiol 2:280. doi: 10.3389/fmicb.2011.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. 2010. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. 2012. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog 8:e1002540. doi: 10.1371/journal.ppat.1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M. 2015. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18:649–658. doi: 10.1016/j.chom.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Danaher RJ, Jacob RJ, Steiner MR, Allen WR, Hill JM, Miller CS. 2005. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript-independent manner in neuronal cells. J Neurovirol 11:306–317. doi: 10.1080/13550280590952817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Figliozzi RW, Chen F, Balish M, Ajavon A, Hsia SV. 2014. Thyroid hormone-dependent epigenetic suppression of herpes simplex virus-1 gene expression and viral replication in differentiated neuroendocrine cells. J Neurol Sci 346:164–173. doi: 10.1016/j.jns.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storey N, Latchman D, Bevan S. 2002. Selective internalization of sodium channels in rat dorsal root ganglion neurons infected with herpes simplex virus-1. J Cell Biol 158:1251–1262. doi: 10.1083/jcb.200204010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kennedy PG, Montague P, Scott F, Grinfeld E, Ashrafi GH, Breuer J, Rowan EG. 2013. Varicella-zoster viruses associated with post-herpetic neuralgia induce sodium current density increases in the ND7-23 Nav-1.8 neuroblastoma cell line. PLoS One 8:e51570. doi: 10.1371/journal.pone.0051570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raymon HK, Thode S, Zhou J, Friedman GC, Pardinas JR, Barrere C, Johnson RM, Sah DW. 1999. Immortalized human dorsal root ganglion cells differentiate into neurons with nociceptive properties. J Neurosci 19:5420–5428. doi: 10.1523/JNEUROSCI.19-13-05420.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang W, Berta T, Kim YH, Lee S, Lee SY, Ji RR. 2018. Expression and role of voltage-gated sodium channels in human dorsal root ganglion neurons with special focus on Nav1.7, species differences, and regulation by paclitaxel. Neurosci Bull 34:4–12. doi: 10.1007/s12264-017-0132-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sawtell NM, Thompson RL. 2016. Herpes simplex virus and the lexicon of latency and reactivation: a call for defining terms and building an integrated collective framework. F1000Res 5:2038. doi: 10.12688/f1000research.8886.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bloom DC. 2016. Alphaherpesvirus latency: a dynamic state of transcription and reactivation. Adv Virus Res 94:53–80. doi: 10.1016/bs.aivir.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Pires de Mello CP, Bloom DC, Paixão IC. 2016. Herpes simplex virus type-1: replication, latency, reactivation and its antiviral targets. Antivir Ther 21:277–286. doi: 10.3851/IMP3018. [DOI] [PubMed] [Google Scholar]
- 40.Pesola JM, Zhu J, Knipe DM, Coen DM. 2005. Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. J Virol 79:14516–14525. doi: 10.1128/JVI.79.23.14516-14525.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thellman NM, Triezenberg SJ. 2017. Herpes simplex virus establishment, maintenance, and reactivation: in vitro modeling of latency. Pathogens 6:E28. doi: 10.3390/pathogens6030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thellman NM, Botting C, Madaj Z, Triezenberg SJ. 2017. An immortalized human dorsal root ganglion cell line provides a novel context to study herpes simplex virus 1 latency and reactivation. J Virol 91:e00080-17. doi: 10.1128/JVI.00080-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pourchet A, Modrek AS, Placantonakis DG, Mohr I, Wilson AC. 2017. Modeling HSV-1 latency in human embryonic stem cell-derived neurons. Pathogens 6:E24. doi: 10.3390/pathogens6020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang S, Ljubimov AV, Jin L, Pfeffer K, Kronenberg M, Ghiasi H. 2018. Herpes simplex virus 1 latency and the kinetics of reactivation are regulated by a complex network of interactions between the herpesvirus entry mediator, its ligands (gD, BTLA, LIGHT, and CD160), and the latency-associated transcript. J Virol 92:e01451-18. doi: 10.1128/JVI.01451-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K, Mahalingam G, Imai Y, Pesnicak L, Margolis TP, Margolis TT, Straus SE, Cohen JI. 2009. Cell type specific accumulation of the major latency-associated transcript (LAT) of herpes simplex virus type 2 in LAT transgenic mice. Virology 386:79–87. doi: 10.1016/j.virol.2008.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson RL, Preston CM, Sawtell NM. 2009. De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog 5:e1000352. doi: 10.1371/journal.ppat.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson RL, Sawtell NM. 2011. The herpes simplex virus type 1 latency associated transcript locus is required for the maintenance of reactivation competent latent infections. J Neurovirol 17:552–558. doi: 10.1007/s13365-011-0071-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spivack JG, Fraser NW. 1987. Detection of herpes simplex virus type 1 transcripts during latent infection in mice. J Virol 61:3841–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 235:1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- 50.Kubat NJ, Amelio AL, Giordani NV, Bloom DC. 2004. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J Virol 78:12508–12518. doi: 10.1128/JVI.78.22.12508-12518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilcox CL, Johnson EM Jr. 1988. Characterization of nerve growth factor-dependent herpes simplex virus latency in neurons in vitro. J Virol 62:393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, Margolis TP. 2011. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J Virol 85:6669–6677. doi: 10.1128/JVI.00204-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bedadala GR, Pinnoji RC, Palem JR, Hsia SC. 2010. Thyroid hormone controls the gene expression of HSV-1 LAT and ICP0 in neuronal cells. Cell Res 20:587–598. doi: 10.1038/cr.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu FH, Catterall WA. 2003. Overview of the voltage-gated sodium channel family. Genome Biol 4:207. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan AM, Samad OA, Dib-Hajj SD, Waxman SG. 2015. Virus-mediated knockdown of Nav1.3 in dorsal root ganglia of STZ-induced diabetic rats alleviates tactile allodynia. Mol Med 21:544–552. doi: 10.2119/molmed.2015.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He XH, Zang Y, Chen X, Pang RP, Xu JT, Zhou X, Wei XH, Li YY, Xin WJ, Qin ZH, Liu XG. 2010. TNF-α contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain 151:266–279. doi: 10.1016/j.pain.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 57.Thakor DK, Lin A, Matsuka Y, Meyer EM, Ruangsri S, Nishimura I, Spigelman I. 2009. Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol Pain 5:14. doi: 10.1186/1744-8069-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Black JA, Liu S, Tanaka M, Cummins TR, Waxman SG. 2004. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108:237–247. doi: 10.1016/j.pain.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 59.Kim CH, Oh Y, Chung JM, Chung K. 2001. The changes in expression of three subtypes of TTX sensitive sodium channels in sensory neurons after spinal nerve ligation. Brain Res Mol Brain Res 95:153–161. doi: 10.1016/s0169-328x(01)00226-1. [DOI] [PubMed] [Google Scholar]
- 60.Berta T, Poirot O, Pertin M, Ji RR, Kellenberger S, Decosterd I. 2008. Transcriptional and functional profiles of voltage-gated Na+ channels in injured and non-injured DRG neurons in the SNI model of neuropathic pain. Mol Cell Neurosci 37:196–208. doi: 10.1016/j.mcn.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 61.Black JA, Nikolajsen L, Kroner K, Jensen TS, Waxman SG. 2008. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann Neurol 64:644–653. doi: 10.1002/ana.21527. [DOI] [PubMed] [Google Scholar]
- 62.Fukuoka T, Kobayashi K, Yamanaka H, Obata K, Dai Y, Noguchi K. 2008. Comparative study of the distribution of the alpha-subunits of voltage-gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. J Comp Neurol 510:188–206. doi: 10.1002/cne.21786. [DOI] [PubMed] [Google Scholar]
- 63.Lindia JA, Kohler MG, Martin WJ, Abbadie C. 2005. Relationship between sodium channel NaV1.3 expression and neuropathic pain behavior in rats. Pain 117:145–153. doi: 10.1016/j.pain.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 64.Hains BC, Saab CY, Klein JP, Craner MJ, Waxman SG. 2004. Altered sodium channel expression in second-order spinal sensory neurons contributes to pain after peripheral nerve injury. J Neurosci 24:4832–4839. doi: 10.1523/JNEUROSCI.0300-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rush AM, Cummins TR, Waxman SG. 2007. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol 579:1–14. doi: 10.1113/jphysiol.2006.121483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alexandrou AJ, Brown AR, Chapman ML, Estacion M, Turner J, Mis MA, Wilbrey A, Payne EC, Gutteridge A, Cox PJ, Doyle R, Printzenhoff D, Lin Z, Marron BE, West C, Swain NA, Storer RI, Stupple PA, Castle NA, Hounshell JA, Rivara M, Randall A, Dib-Hajj SD, Krafte D, Waxman SG, Patel MK, Butt RP, Stevens EB. 2016. Subtype-selective small molecule inhibitors reveal a fundamental role for Nav1.7 in nociceptor electrogenesis, axonal conduction and presynaptic release. PLoS One 11:e0152405. doi: 10.1371/journal.pone.0152405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Asiedu MN, Han C, Dib-Hajj SD, Waxman SG, Price TJ, Dussor G. 2017. The AMPK activator A769662 blocks voltage-gated sodium channels: discovery of a novel pharmacophore with potential utility for analgesic development. PLoS One 12:e0169882. doi: 10.1371/journal.pone.0169882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stamboulian S, Choi JS, Ahn HS, Chang YW, Tyrrell L, Black JA, Waxman SG, Dib-Hajj SD. 2010. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel NaV1.7 and alters its gating properties. J Neurosci 30:1637–1647. doi: 10.1523/JNEUROSCI.4872-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Y, Zhu T, Yang H, Dib-Hajj SD, Waxman SG, Yu Y, Xu TL, Cheng X. 2018. Nav1.7 is phosphorylated by Fyn tyrosine kinase which modulates channel expression and gating in a cell type-dependent manner. Mol Pain 14:1744806918782229. doi: 10.1177/1744806918782229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krohel GB, Richardson JR, Farrell DF. 1976. Herpes simplex neuropathy. Neurology 26:596–597. doi: 10.1212/wnl.26.6.596. [DOI] [PubMed] [Google Scholar]
- 71.Chen F, Figliozzi RW, Bedadala G, Palem J, Hsia SV. 2016. Overexpression of thyroid hormone receptor β1 altered thyroid hormone-mediated regulation of herpes simplex virus-1 replication in differentiated cells. J Neurovirol 22:555–563. doi: 10.1007/s13365-016-0423-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.David AT, Saied A, Charles A, Subramanian R, Chouljenko VN, Kousoulas KG. 2012. A herpes simplex virus 1 (McKrae) mutant lacking the glycoprotein K gene is unable to infect via neuronal axons and egress from neuronal cell bodies. mBio 3:e00144-12. doi: 10.1128/mBio.00144-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Q, Hsia SC, Martin-Caraballo M. 2019. Regulation of T-type Ca2+ channel expression by interleukin-6 in sensory-like ND7/23 cells post herpes simplex virus (HSV-1) infection. J Neurochem 151:238–254. doi: 10.1111/jnc.14697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watson S, Mercier S, Bye C, Wilkinson J, Cunningham AL, Harman AN. 2007. Determination of suitable housekeeping genes for normalisation of quantitative real time PCR analysis of cells infected with human immunodeficiency virus and herpes viruses. Virol J 4:130. doi: 10.1186/1743-422X-4-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pachuau J, Martin-Caraballo M. 2007. Extrinsic regulation of T-type Ca2+ channel expression in chick nodose ganglion neurons. Dev Neurobiol 67:1915–1931. doi: 10.1002/dneu.20560. [DOI] [PubMed] [Google Scholar]
- 76.Pachuau J, Martin-Caraballo M. 2007. Expression pattern of T-type Ca2+ channels in embryonic chick nodose ganglion neurons. Dev Neurobiol 67:1901–1914. doi: 10.1002/dneu.20563. [DOI] [PubMed] [Google Scholar]