

Our work presents the first report on stress granule formation during hantavirus infection. We show that hantavirus infection actively inhibits stress granule formation, thereby escaping the detrimental effects on global translation imposed by host stress signaling. Our results highlight a previously uncharacterized aspect of hantavirus-host interactions with possible implications for how hantaviruses are able to cause persistent infection in natural hosts and for pathogenesis.
KEYWORDS: PKR, hantavirus, stress granule
ABSTRACT
Virus infection frequently triggers host cell stress signaling resulting in translational arrest; as a consequence, many viruses employ means to modulate the host stress response. Hantaviruses are negative-sense, single-stranded RNA viruses known to inhibit host innate immune responses and apoptosis, but their impact on host cell stress signaling remains largely unknown. In this study, we investigated activation of host cell stress responses during hantavirus infection. We show that hantavirus infection causes transient formation of stress granules (SGs) but does so in only a limited proportion of infected cells. Our data indicate some cell type-specific and hantavirus species-specific variability in SG prevalence and show SG formation to be dependent on the activation of protein kinase R (PKR). Hantavirus infection inhibited PKR-dependent SG formation, which could account for the transient nature and low prevalence of SG formation observed during hantavirus infection. In addition, we report only limited colocalization of hantaviral proteins or RNA with SGs and show evidence indicating hantavirus-mediated inhibition of PKR-like endoplasmic reticulum (ER) kinase (PERK).
IMPORTANCE Our work presents the first report on stress granule formation during hantavirus infection. We show that hantavirus infection actively inhibits stress granule formation, thereby escaping the detrimental effects on global translation imposed by host stress signaling. Our results highlight a previously uncharacterized aspect of hantavirus-host interactions with possible implications for how hantaviruses are able to cause persistent infection in natural hosts and for pathogenesis.
INTRODUCTION
Orthohantaviruses (hereinafter referred to as hantaviruses) are zoonotic pathogens that belong to the family Hantaviridae and harbor a trisegmented negative-sense single-stranded RNA genome. Hantaviruses are carried primarily by rodents, moles, shrews, and bats, but fishes and reptiles have also been recently found to carry hantaviruses (1). Hantaviruses are estimated to annually cause over 100,000 cases of human disease, including hemorrhagic fever with renal syndrome (HFRS) in Eurasia and hantavirus pulmonary syndrome (HPS) in the Americas, with mortality rates ranging from less than 1% to up to 40% depending on virus species (2). Key features of hantavirus pathogenesis include high levels of proinflammatory cytokine production, increased endothelial cell permeability, and inefficient viral clearance due to resistance to apoptosis (3, 4). The vascular endothelium is considered the primary target of hantavirus infection, but the molecular mechanisms underlying the observed clinical manifestations remain poorly defined.
Viral infection imposes various types of stress upon a host cell and often results in rapid establishment of an antiviral response. Besides the designated sensors of the innate immune system, viral infection frequently triggers host stress sensors which respond to changes in cellular homeostasis through phosphorylation of eukaryotic translation initiation factor 2 alpha subunit (eIF2α). These eIF2α kinases include general control nonderepressible 2 (GCN2), heme-regulated eIF2α kinase (HRI), protein kinase R (PKR), and PKR-like endoplasmic reticulum (ER) kinase (PERK), which respond to starvation, oxidative stress, double-stranded RNA (dsRNA) and ER stress, respectively (5). Phosphorylation of eIF2α results in global inhibition of translation and causes the formation of stress granules (SGs), which are dynamic, cytoplasmic, membraneless structures containing translationally silenced mRNA, 40S ribosomes, translation initiation factors, and various RNA-binding proteins (6). Besides functioning as depositories for translation initiation complexes during translational arrest, SGs are linked to the innate immune response by recruiting many antiviral proteins and acting as a platform for their activation (7, 8). Due to this antiviral role and to viral dependence on host translational machinery, the appearance of SGs is generally detrimental to virus infection. Accordingly, many viruses have evolved means to either counteract SG formation or divert SG components into novel roles that are beneficial for the virus (9). Some examples include inhibition of PKR by influenza A virus NS1 (10), cleavage of the SG protein G3BP1 by the poliovirus 3 C proteinase (11), and sequestration of G3BP1 by Semliki Forest virus nsP3 (12). The hantavirus nucleocapsid protein has been shown to inhibit PKR (13), but SG formation during hantavirus infection has not been investigated previously.
In this study, we sought to determine whether SGs form during hantavirus infection and to analyze the mechanism and kinetics of their formation. We show that hantavirus infection results in transient PKR-dependent SG formation. Furthermore, we show that hantaviruses specifically inhibit PKR- and PERK-mediated SG formation.
RESULTS
Puumala and Andes hantaviruses cause transient formation of SGs.
To assess whether hantavirus infection induces the formation of SGs, we infected human umbilical vein endothelial cells (HUVECs) with Puumala virus (PUUV), Andes virus (ANDV), and Hantaan virus (HTNV) and inspected the infected cells for formation of SGs by immunofluorescence (Fig. 1). PUUV and ANDV infections induced the accumulation of the SG marker G3BP1 into SG-like foci at 18 h, 24 h, and 48 h after infection but not at earlier or later time points (Fig. 1A and B). Further analysis using additional SG markers and treatment with SG inhibitor cycloheximide confirmed these foci to be bona fide SGs (Fig. 2A and B). Quantitative analysis showed that the most abundant SG formation was present at 24 h postinfection, with 5% to 10% of infected cells harboring SGs (Fig. 1B). Rare instances of colocalization of hantaviral proteins with SGs were observed but only in PUUV-infected HUVECs and in only approximately 4% of all cells harboring SGs (Fig. 1C and D). Curiously, no SGs were detected in HTNV-infected HUVECs, indicating a possible HTNV-specific mechanism in mitigating host cell stress responses. As the susceptibilities for SG formation can differ between different cell types, we next analyzed SG formation in infected A549 cells (Fig. 2C). Hantavirus infection induced SG formation in A549 cells but did so with a delayed time kinetic in comparison to HUVECs. In contrast to HUVECs, HTNV infection in A549 cells induced the formation of SGs, albeit to a lesser extent than PUUV infection and ANDV infection. We also investigated whether there is a connection between hantaviral pathogenesis and SG induction by analyzing SG formation induced by the nonpathogenic Prospect Hill and Tula hantaviruses. Both viruses induced SG formation in HUVECs with temporal kinetics similar to that seen with PUUV and ANDV, indicating that SG induction is not a feature of pathogenic hantaviruses only (Fig. 2D). These results show that hantavirus infection results in the formation of bona fide SGs but does so only at certain time points and in only a limited proportion of infected cells at any given time.
FIG 1.

PUUV and ANDV infection causes transient formation of SGs in HUVECs. (A) SG formation in hantavirus-infected HUVECs. HUVECs infected with different hantaviruses were fixed 24 h after the infection. Uninfected cells were used as a control. The cells were stained with antibodies against hantavirus proteins (red) and G3BP (green) and with DAPI (4′,6-diamidino-2-phenylindole) (blue). Arrows show SGs. Imaging was performed with confocal microscopy (60×). Scale bars = 10 μm. (B) Time kinetics of SG formation in hantavirus-infected HUVECs. HUVECs infected with different hantaviruses were fixed 12, 18, 24, 48, or 72 h after the infection. Uninfected cells were used as a control. The cells were stained for viral proteins and G3BP. During the investigation using fluorescence microscopy, several pictures of a total of >150 cells were taken at random positions and the number of infected cells showing SGs was determined. n = ≥3. (C) Colocalization of viral proteins with SGs in PUUV-infected cells. HUVECs were fixed 24 h after the infection with PUUV and stained with antibodies against hantavirus proteins (red) and G3BP (green) and with DAPI (blue). Arrows show colocalization of viral proteins with SGs. Imaging was performed with confocal microscopy (60×). Scale bars = 10 μm. (D) Number of infected cells showing colocalization of viral proteins with SGs. HUVECs infected with PUUV and ANDV were fixed 24 h after the infection. The cells were stained for viral proteins and G3BP. During the investigation using fluorescence microscopy, pictures of ≥100 cells with SGs were taken and the number of cells where viral proteins colocalized with SGs was determined. n = 3.
FIG 2.

Characterization of infection-induced SGs regarding SG-associated proteins, cell type, and virus pathogenicity. (A) Localization of different SG-associated proteins. HUVECs were infected with PUUV and fixed at 24 h after the infection. In addition, uninfected cells were treated with thapsigargin as described in Materials and Methods. The cells were stained with antibodies against hantavirus proteins (magenta), G3BP (red), and eIF4G, PABP, or HuR (green) and with DAPI (blue). The images show representative cells with stress granules. Imaging was performed with confocal microscopy (60×). Scale bars = 10 μm. (B) Treatment with cycloheximide dissolves infection-induced SGs. HUVECs infected with PUUV were incubated with 20 μg/ml of CHX for 1 h and fixed at 24 h after the infection. The cells were stained for viral proteins and G3BP and analyzed for SG formation under the microscope. During this investigation, several pictures of a total of >100 cells were taken at random positions and the number of infected cells showing SGs was determined. n = 4. (C) Time kinetics of SG formation in hantavirus-infected A549 cells. A549 cells infected with different hantaviruses were fixed 24, 48, or 72 h after the infection. Uninfected cells were used as a control. The cells were stained for viral proteins and G3BP. During the investigation using fluorescence microscopy, several pictures of a total of >150 cells were taken at random positions and the number of infected cells showing SGs was determined. n = 3. (D) Time kinetics of SG formation in HUVECs infected with the nonpathogenic hantaviruses Prospect Hill virus (PHV) and Tula orthohantavirus (TULV). HUVECs were infected with PHV and TULV and fixed 12, 18, 24, 48, or 72 h after the infection. The cells were stained for viral proteins and G3BP, and the number of cells harboring SGs was determined under the microscope. Several pictures were taken at random positions and showed a total of >100 cells. n = 3. **, P < 0.01.
SG formation is associated with translational shutdown of infected cells.
The appearance of stress granules is linked to global inhibition of translation, but some viruses utilize means to circumvent the SG-imposed translational block to allow continued viral translation. Examples of such measures include virus-encoded translational enhancers and noncanonical translation initiation strategies to enable efficient translation despite high levels of eIF2 phosphorylation (14, 15). To assess the impact of SG formation on hantavirus translation, we treated PUUV-infected and ANDV-infected HUVECs with puromycin and visualized global translation levels by immunofluorescence (Fig. 3). No traces of puromycin staining were observed in PUUV-infected and ANDV-infected cells harboring stress granules, indicating efficient shutdown of both host and viral translation. Interestingly, in comparison to poly(I·C)-induced SGs, the virus-induced SGs appear to have mediated a more pronounced inhibition of global translation as evidenced by the traces of puromycin staining that remained in cells harboring poly(I·C)-induced SGs. These results demonstrate that the appearance of SGs correlates with efficient inhibition of translation in hantavirus-infected cells.
FIG 3.
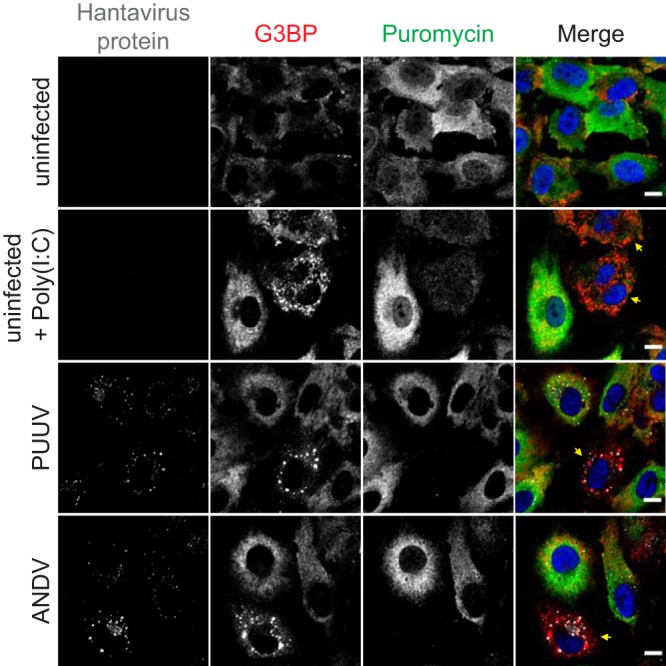
Infection-induced SG formation leads to inhibition of cellular translation. HUVECs infected with PUUV or ANDV were fixed 24 h after the infection. Uninfected untreated cells and uninfected cells treated with 1 μg poly(I·C) (see Materials and Methods) were used as controls. Before fixation, all samples were treated with 10 μg/ml puromycin for 5 min. The cells were stained with antibodies against hantavirus protein (magenta), G3BP (red), or puromycin (green) and DAPI (blue). Arrows show cells with SGs. Imaging was performed with confocal microscopy (60×). Scale bars = 10 μm.
Hantavirus-mediated SG induction is PKR dependent.
To investigate the mechanism of hantavirus-induced SG formation, we treated PUUV-infected and ANDV-infected HUVECs with known inhibitors of the cellular stress kinases and analyzed their impact on the proportion of SG-positive infected cells (Fig. 4A). No significant reduction in SG prevalence was observed with the GCN2 inhibitor indirubin-3’monoxime or the PERK inhibitor GSK2606414. However, a significant and dose-dependent decrease in SG prevalence was observed with PKR inhibitor C16, indicating a role for PKR in hantavirus-induced SG formation. As further confirmation, we used small interfering RNA (siRNA) to knock down PKR expression in HUVECs followed by ANDV infection and analysis of SG prevalence by immunofluorescence (Fig. 4B and C). The reduction in the level of SG-harboring cells was similar to that seen with C16, confirming the involvement of PKR in hantavirus-induced SG formation. In line with the expected antiviral role of PKR, a slight but significant increase in N protein expression and an increase in ANDV titer were observed in infected PKR knockdown cells compared to untreated ones (Fig. 4D and E). These results show that hantavirus-induced SG formation is PKR dependent and likely does not involve stress mediated by PERK or GCN2.
FIG 4.

Inhibition and knockdown of PKR reduce infection-induced SG formation. (A) Treatment with the PKR inhibitor C16 reduces infection-induced SG formation. HUVECs infected with PUUV or ANDV were treated with neat or 5× PKR inhibitor C16, PERK inhibitor GSK2606414, or GCN2 inhibitor indirubin-3′-monoxime (I-3′-m) (see Materials and Methods) and fixed at 24 h after the infection. The cells were stained for viral proteins and G3BP. During the investigation using fluorescence microscopy, several pictures of a total of >100 cells were taken at random positions and the number of infected cells showing SGs was determined. Ctrl, control. n ≥ 3. (B) Transfection with PKR siRNA decreases PKR expression. HUVECs were transfected with PKR siRNA or control siRNA as described in Materials and Methods and infected with ANDV after 24 h. Untreated ANDV-infected cells and ANDV-infected cells treated with C16 were used as controls. Cell lysates were collected at 24 h after the infection. In addition, cell lysates of untransfected or PKR siRNA transfected uninfected cells treated with poly(I·C) were collected. The lysates were then analyzed for the expression of PKR and N protein. β-actin was used as a loading control. (C) PKR knockdown reduces infection-induced SG formation in ANDV-infected HUVECs. HUVECs on coverslips were treated as described for panel B, fixed, and stained for viral proteins and G3BP and analyzed for SG formation under the microscope. During this investigation, several pictures of a total of >100 cells were taken at random positions and the number of infected cells showing SGs was determined. n ≥ 3. (D) PKR knockdown increases the production of N protein in ANDV-infected HUVECs. HUVECs were treated as described for panel B. N protein levels were normalized to cellular levels of β-actin. The data are presented as the fold change in relation to the N protein levels of untreated cells. n ≥ 3. (E) HUVECs were treated with control siRNA or PKR siRNA and infected with ANDV after 24 h. The supernatant was collected at 4 days postinfection (p.i.), and the virus titer was determined by endpoint dilution assay. Results shown as 50% tissue culture infectious doses (TCID50) per milliliter. n = 3. *, P < 0.05; **, P < 0.01.
Hantaviruses inhibit PKR- and PERK-mediated SG induction.
The hantavirus N protein has been previously reported to inhibit activation of PKR (13). Having established that hantavirus infection induces PKR-mediated SG formation, we next investigated whether hantavirus infection also inhibits SG formation. Such an inhibitory mechanism could explain why no SGs are observed at later stages of infection and why so few infected cells express SGs at any given time. To induce activation of the host cell stress kinases, PUUV-infected, ANDV-infected, HTNV-infected, and mock-infected HUVECs were treated at 72 h postinfection with poly(I·C), thapsigargin, CCT020312, rapamycin, or sodium arsenite, followed by immunofluorescence analysis and quantification of SG-harboring cells (Fig. 5A). All the inducers triggered SG formation in uninfected cells except CCT020312, which could be due to a lack of general unfolded protein response induction by the selective PERK activator (16). Responses to the inducers were consistent between cells infected with different hantaviruses, expressing little virus-specific variation. Significant increases in SG prevalence in infected cells compared to uninfected ones were observed in response to CCT020312, rapamycin, and sodium arsenite treatments, indicating that no inhibition of PERK, GCN2, or HRI was mediated by the hantavirus infections. However, hantavirus infection conferred resistance to poly(I·C) and to the ER stressor and PERK activator thapsigargin, resulting in significantly reduced SG prevalence in response to poly(I·C) or thapsigargin treatment.
FIG 5.

Hantaviruses inhibit PKR-mediated SG formation. (A) Hantavirus infection reduces SG formation induced by poly(I·C) and thapsigargin. HUVECs were infected with different hantaviruses and treated with poly(I·C), thapsigargin, CCT020312 (CCT), rapamycin (Rapa), or sodium arsenite (SA) (see Materials and Methods). Uninfected cells were used as controls. All samples were collected at 72 h after the infection. The cells were stained with antibodies against viral proteins and G3BP and analyzed for SG formation under the microscope. During this investigation, several pictures of a total of >100 cells were taken at random positions, and the number of infected cells showing SGs was determined. n ≥ 3. (B) Phosphorylation of eIF2α kinases. Uninfected (-) and PUUV-infected (+) HUVECs were treated as described for panel A, and cell lysates were collected at 72 h after the infection. The lysates were then analyzed to determine the amount of phospho-PKR, phospho-PERK, or phospho-GCN2. N protein was used to show infection, and β-actin was used as a loading control. *, P < 0.05; **, P < 0.01.
To gain clarity with respect to the observed discrepancy between the two PERK activators, we next analyzed PUUV-infected and mock-infected HUVEC lysates collected at 72 h postinfection using Western blotting for the activation of PKR, PERK, and GCN2 in response to the chemical stress inducers. Phosphorylation of PKR was observed in all infected samples, including the untreated cells (Fig. 5B), which was curious as SGs were not observed at that time point (Fig. 1B). Also, similar p-PKR levels were detected in poly(I·C)-treated cells regardless of whether they were infected or not, indicating that inhibition of PKR-mediated SG formation might take place downstream of PKR activation. With the exception of CCT020312-treated uninfected cells, all samples showed only low levels of p-PERK. Interestingly, PUUV infection caused a clear reduction in PERK phosphorylation levels, indicating that hantavirus infection inhibits PERK activation. Increased phosphorylation of GCN2 was seen in response to rapamycin and also in response to CCT020312, providing a possible explanation for the differences in SG responses to thapsigargin and CCT020312. Taken together, these results support the hypothesis of hantaviral inhibitory mechanisms regulating host SG formation and show that both PKR-mediated stress signaling and PERK-mediated stress signaling are inhibited during hantavirus infection.
Hantavirus RNA does not localize to SGs.
The composition of SGs typically varies depending on many factors, including cell type, type of stress associated with the SG formation, and any modulations induced by a possible disease state or virus infection (17). Having demonstrated only limited colocalization of hantaviral proteins with SGs and having found no evidence of virus-induced exclusion of any of the more typical SG marker proteins from SGs (Fig. 1A and 2A), we next investigated whether hantaviral RNA could be detected in virus-induced SGs. To that end, digoxigenin (DIG)-labeled DNA probes against ANDV and PUUV small (S), medium (M), and large (L) segments were used to detect viral RNA localization in HUVECs at 12 h, 18 h, and 24 h after infection. No localization of either ANDV or PUUV RNA in SGs could be detected (Fig. 6A and B; data not shown for M and L segments). Even though it had not colocalized with stress granules, some of the hantaviral RNA was found in close proximity to SGs. This could reflect localization of viral RNA in cytoplasmic processing bodies (P bodies), which are frequently found in proximity to SGs and function as sites of mRNA degradation and translational repression (18, 19). P bodies also have a role in hantavirus cap-snatching, as the N protein localizes to P bodies to bind host mRNA 5′ caps and protect them from degradation before use as primers for the viral polymerase (20). We were, however, unable to detect colocalization of viral RNA with P bodies (Fig. 6C), indicating that the viral foci near SGs represent sites of viral translation or replication rather than sites of interaction with P bodies.
FIG 6.
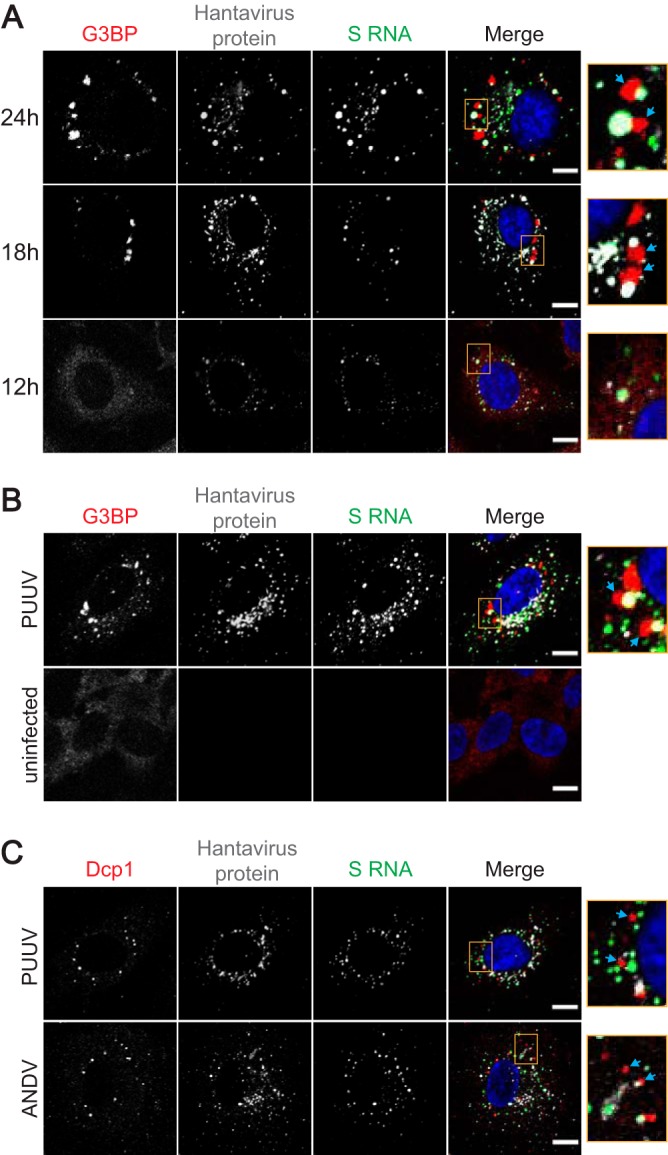
Viral RNA is excluded from SGs and P-bodies. (A) FISH analysis of the localization of ANDV S RNA and SGs at different time points. HUVECs were infected with ANDV and fixed at 12, 18, or 24 h after the infection. Hybridization and staining with antibodies against viral protein (magenta), G3BP (red), and digoxigenin (green) and with DAPI was performed as described in Materials and Methods. Arrows show SGs. (B) FISH analysis of the localization of PUUV S RNA and SGs at 24 h. HUVECs were infected with PUUV and fixed at 24 h after the infection. Hybridization and staining with antibodies against viral protein (magenta), G3BP (red), and digoxigenin (green) and with DAPI was performed as described in Materials and Methods. Arrows show SGs. Uninfected cells were used as a control. (C) FISH analysis of PUUV and ANDV S RNA and P-bodies at 24 h. HUVECs were infected with PUUV or ANDV and fixed at 24 h after the infection. Hybridization and staining with antibodies against viral protein (magenta), Dcp1 (red), and digoxigenin (green) and with DAPI was performed as described in Materials and Methods. Arrows show P-bodies. Imaging for all panels was performed with confocal microscopy (60×). Scale bars = 10 μm.
DISCUSSION
Cell stress responses are an integral part of the antiviral defense; consequently, many viruses have developed countermeasures to inhibit the formation and function of SGs. Here, we present the first report on hantavirus SG induction, showing that SGs can form during hantavirus infection but do so only transiently and in a low portion of infected cells. We identified PKR as the host cell stress kinase mainly involved in SG formation, and we show additional evidence supporting the idea of active inhibition of PKR and PERK stress signaling during hantavirus infection.
The observed PKR dependency of hantavirus-mediated SG formation is not surprising, considering the role of PKR as a key component of the innate immune response during virus infection. RNA viruses typically trigger activation of PKR either due to accumulation of dsRNA replication intermediates or through the activity of other noncanonical RNA ligands produced during the viral replication cycle (21). Negative-sense RNA viruses in general produce very little or negligible amounts of dsRNA; also, during hantavirus infection, dsRNA levels reportedly remain below detection levels (22, 23). This scarcity in available substrates for PKR, together with the N protein-mediated inhibition of PKR dimerization (13), may explain why SG formation occurs in only a few infected cells. However, the lack of dsRNA detection during hantavirus infection could also reflect the limitations of the dsRNA antibody, which fails to recognize dsRNA shorter than 40 bases (24). Analysis with a more sensitive dsRNA antibody has revealed dsRNA production during infection with many negative-sense RNA viruses (25), and the same could be the case for hantaviruses also. Curiously, the secondary dsRNA-like structures formed by hantavirus N mRNA have been shown to activate RIG-I (26). It is tempting to speculate that such secondary RNA structures could also trigger PKR activation. Moreover, single-stranded RNA (ssRNA) with even limited secondary structure has been shown previously to activate PKR (27), lending further plausibility to the notion of hantaviral mRNA acting as the source of PKR activation. An alternative explanation for the mechanism of hantavirus-induced PKR activation is that it occurs through the activity of PACT, a host cell PKR-binding protein capable of activating PKR in the absence of dsRNA (28). PACT is activated by various cellular stresses and is also involved in RIG-I activation (29 – 31). To test for the possible involvement of PACT, we treated PUUV-infected and ANDV-infected HUVECs with luteolin, a known inhibitor of the PKR/PACT interaction (32). No impact on SG formation was observed, indicating that PACT is likely not involved in hantavirus SG induction (data not shown).
SGs are dynamic by nature, having a propensity to form and dissociate in a matter of minutes in response to fluctuations in exposure to stress stimuli (33). Our observation of transient SG formation upon hantavirus infection suggests a time-dependent variation in the amount of stress inflicted on the host cell over the course of the infection, which likely represents the period of most abundant viral replication being reflected in the amount of ligand available for PKR activation. The transience of SG formation can also be due to temporal variation in the amount or activity of the PKR-inhibiting N protein, but this is unlikely since N is abundantly expressed throughout infection. Rather, the observed SG pattern likely reflects the PKR-inhibiting N efficiently suppressing SG formation at all times except during the periods of strongest dsRNA production, which can also explain the low SG prevalence even during peak periods of SG induction. Even though only a fraction of infected cells expresses SGs at any given time point, it is possible that all or most infected cells form SGs but do it so briefly that only a few are caught in steady-state analysis. Such oscillation of SGs has been shown to be a common feature of RNA virus infections and also of dsRNA-induced SGs in general (34). Establishing whether that is the case with hantaviruses would require additional studies using live-cell imaging techniques. In any case, the hantavirus-induced SGs are not likely to persist for long, since the observed total inhibition of host cell translation upon SG formation (Fig. 3) would result in cell death if prolonged.
Besides preventing the formation of SGs through inhibition of PKR, an active role of hantavirus in the clearance of stress granules cannot be ruled out. Such a mechanism has been reported for poliovirus, which causes early formation of SGs but later dissolves them through cleavage of G3BP (11). Our observation of very limited colocalization of hantaviral proteins with SGs suggests the absence of a direct interaction between hantaviral and SG proteins (Fig. 1), but an indirect impact on SG clearance remains a possibility. Stress granules are cleared by various means, most commonly through simple resumption of translation after the stress is resolved but also through chaperone-mediated disassembly and autophagy (35, 36). Interestingly, HTNV infection has been shown to induce expression of heat shock protein HSP70 (37), which is a major factor in chaperone-mediated disassembly of SGs (38, 39). Also, autophagy is modulated during hantavirus infection, resulting in induction of autophagy at early stages of infection and inhibition of autophagy flux at later stages (40, 41). Both these mechanisms could contribute to the observed SG dynamics and to the susceptibility of infected cells to external stress.
Our results also showed an interesting difference between hantavirus species in SG induction properties. Whereas PUUV and ANDV showed mostly similar levels of SG induction in both HUVECs and A549 cells, HTNV strikingly displayed no SG formation in HUVECs and clearly reduced SG formation in A549 cells compared to PUUV and ANDV (Fig. 1 and 2). This could indicate either more efficient PKR inhibition by the HTNV N or possibly species-specific differences in dsRNA production. Our own results (Fig. 5A) and previous studies (13) showed approximately equal levels of inhibition of PKR in response to poly(I·C) treatment with HTNV, ANDV, and PUUV, suggesting lower levels of production or better masking of PKR-activating dsRNA motifs during HTNV infection rather than stronger PKR inhibition. If the observed PKR activation was due to secondary structures formed by hantavirus RNA as speculated above, an intriguing possibility is that HTNV simply forms fewer RNA secondary structures capable of activating PKR than ANDV or PUUV. It is certainly possible, as HTNV belongs to a phylogenetic cluster different from that to which ANDV and PUUV belong and shares less than 70% N protein sequence identity with either of them (42). Interestingly, we did not observe SG formation in HUVECs infected with the closely related Seoul virus (data not shown), indicating that reduced SG formation could be a feature of the phylogenetic cluster containing HTNV, Seoul virus, and related viruses. Further studies using more sensitive dsRNA antibodies will be needed to clarify this issue.
Knockdown or chemical inhibition of PKR did not abolish SG formation completely in hantavirus-infected cells (Fig. 4), leaving open the possibility of a role for other stress sensors in addition to PKR in hantavirus SG induction. However, given the small impact of chemical inhibitors targeting the other eIF2α kinases on hantavirus SG induction (Fig. 4A), PKR likely is the strongest driver of SG formation during hantavirus infection. Moreover, we did not observe any increase in phosphorylated GCN2 or PERK levels in response to infection (Fig. 5B), indicating that these pathways are not efficiently activated. Hantavirus infection was instead found to decrease p-PERK levels in response to chemical induction by thapsigargin or CCT020312, indicating active inhibition of PERK activation during hantavirus infection. Hantavirus infection has been previously reported to induce ER stress (43), and inhibition of PERK could be a hantaviral measure to counteract ER stress signaling. PERK inhibition is clearly evident in reduced SG formation compared to that seen with uninfected cells after treatment with thapsigargin, though no inhibitory effect was seen in response to CCT020312 (Fig. 5A). This discrepancy could have been due to the different mechanisms of action of the inducers resulting in differences in the dynamics and extent of the stress response. A time kinetic experiment revealed SGs in approximately 20% of uninfected HUVECs treated with CCT020312 for 2 h, in contrast to no observable SGs present at 1 h or 4 h after treatment (data not shown). The stress already imposed on the host cell by the hantavirus infection might therefore make the host cell more susceptible to CCT020312-mediated SG induction despite viral inhibition of PERK phosphorylation. Further studies are required to confirm the impact of hantavirus infection on PERK phosphorylation and the possible consequences of this.
Besides PERK and PKR inhibition, hantavirus-infected cells remained sensitive to other stresses. Sodium arsenite treatment readily caused SG formation in infected cells, which displayed a significantly higher level of SG prevalence than uninfected cells (Fig. 5A). Rapamycin treatment had a similar impact, which is curious since rapamycin-mediated inhibition of mammalian target of rapamycin (mTOR) is known to induce autophagy and thus should induce SG clearance through granulophagy (36). Besides activation of GCN2, strong phosphorylation of PKR was seen in infected HUVECs in response to rapamycin treatment (Fig. 5B), suggesting that the observed increase in SG prevalence might have been due to rapamycin interfering with hantaviral inhibition of PKR. Accordingly, mTORC1 signaling has been shown to be important for ANDV replication (44). McNulty et al. showed that inhibition of mTORC1 specifically inhibited viral translation but had no detrimental effect on viral RNA synthesis and was even found to increase viral S RNA synthesis (44). Increased viral RNA synthesis coupled to reduced translation could result in less efficient encapsidation of newly produced viral RNA segments by the N protein and thus in more abundant ligand for PKR activation. Therefore, the observed increase in SG prevalence due to rapamycin treatment might have been a result of the combined effects of GCN2 activation and an amplified PKR response.
Finally, our results showed that hantavirus RNA does not colocalize with SGs (Fig. 6). This finding indicates that hantavirus-induced SGs are not “antiviral stress granules,” as such structures are characterized by the presence of viral RNA and antiviral proteins within SGs (7). Therefore, hantavirus-induced SGs are likely not involved in enhancement of the activation of the innate immune response. Analysis of the SG transcriptome has revealed that whereas only approximately 10% of total mRNA within a stressed cell localizes to SGs, mRNAs from basically all expressed genes are found in SGs albeit with widely variable efficiencies (45). Khong et al. reported mRNA length and translation efficiency as major factors influencing efficiency of mRNA inclusion in SGs and also noted the exclusion of ER-associated mRNA (45). The exclusion of hantaviral mRNA from SGs is probably due to the combined effects of the small size and ER association of the N and GPC mRNAs, respectively, and to the enhanced translation of virus mRNA mediated by the N protein (46).
In conclusion, we found that hantavirus infection induces PKR-dependent formation of stress granules, resulting in efficient inhibition of global translation. However, hantavirus infection also inhibits PKR-dependent SG formation, keeping SG formation transient and low in prevalence as well as limiting its impact on viral replication. The exclusion of viral RNA from SGs means that SGs probably do not have a direct role in the host innate immune response against hantavirus infection. Despite a lack of SG formation at later stages of infection, infected cells remain sensitive and even sensitized to external stressors, which could be a factor to consider in therapeutic intervention against hantaviruses.
MATERIALS AND METHODS
Cell culture.
Human umbilical vein endothelial cells (HUVECs) (Lonza, catalog no. C2517A) were grown in endothelial cell medium (ScienCell, catalog no. 1001) supplemented with 5% fetal bovine serum (FBS), 1% endothelial cell growth supplement (ScienCell, catalog no. 1052), and 1% penicillin/streptomycin solution (PeSt). Human lung endothelial A549 cells (ATCC CLL-185) were grown in a mixture containing minimal essential medium (MEM), 7.5% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Both cell types were grown at 37°C and 5% CO2.
Viruses and infection.
PUUV (strain Kazan-E6), ANDV (strain Chile-9717869), and HTNV (strain 76-118) were propagated on Vero E6 cells and titrated as described earlier (47). Hanks’ balanced salt solution (HBSS) supplemented with 2% fetal calf serum (FCS), 2% HEPES, and 1% PeSt (Thermo Fisher Scientific) was used for infections. At 1 h after infection, the virus suspension was removed, the cells were washed with phosphate-buffered saline (PBS), and fresh growth medium was added. Infections were done at a multiplicity of infection (MOI) of 2, and sample collection was performed at indicated time points.
Immunofluorescence microscopy.
Cells were washed with PBS and fixed for 30 min with 4% (vol/vol) paraformaldehyde–PBS. They were then permeabilized by treatment with PBS containing 0.5% (vol/vol) Triton X for 5 min and washed three times with PBS. Blocking was performed by treatment with 0.5% (wt/vol) bovine serum albumin (BSA)–PBS for 30 min. Samples were incubated with the following primary antibodies diluted in 0.5% (wt/vol) BSA–PBS for 1 h: polyclonal antibodies from convalescent patient sera (1:400), mouse anti-G3BP (abcam, ab56574) (1:500), rabbit anti-G3BP (abcam, ab181149) (1:300), mouse anti-eIF4G1 (Santa Cruz, sc-133155) (1:200), mouse anti-poly(A)-binding protein (Santa Cruz, sc-32318) (1:200), mouse anti-HuR (Santa Cruz, sc-5261) (1:200), and mouse anti-puromycin (Merck, MABE343) (1:500). After washing, the samples were incubated with the following respective secondary antibodies diluted 1:800 in 0.5% (wt/vol) BSA–PBS for 1 h: goat anti-human 594 (Life Technologies, 1316689), goat anti-human 647 (Invitrogen, 1852202), goat anti-mouse 488 (Life Technologies, 1752514), and goat anti-rabbit 594 (Cell Signaling, 88895). Imaging was done with Nikon resonant scanning confocal system A1R+ and image processing with NIS-Elements C version 4.50 software. The images shown are representative of results from at least three independent experiments. Manual quantification of cells was performed by counting 5 to 10 random microscope frames corresponding to 100 to 200 infected cells. Specifically, 100 to 200 cells per experiment were counted for at least three independent biological replicates and the percentage of cells harboring SGs was calculated. For the infected samples, only infected cells (positive for viral proteins) were included in the counting.
Western blotting.
Cells were washed with PBS and lysed with 100 μl cell lysis buffer (Biovision) supplemented with protease inhibitors (Complete; Roche) and phosphatase inhibitors (PhosStop; Sigma). Samples were incubated for 10 min at 96°C in NuPage sample buffer supplemented with 1.5% beta-mercaptoethanol and were then separated by SDS-PAGE (10% polyacrylamide gel). After the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane with a iBlot 2 dry blotting system, the membranes were blocked with 5% (wt/vol) milk powder–Tris-buffered saline (TBS)–0.1% Tween (TBS-T) for 1 h. The following primary antibodies were diluted in 5% (wt/vol) BSA–TBS-T and incubated at 4°C overnight: rabbit anti-PKR (Cell Signaling, catalog no. 3072S) (1:1,000), rabbit anti-phospho-PKR (abcam, ab32036) (1:1,000), rabbit anti-phospho-PERK (Thermo Fisher) (1:500), and rabbit anti-phospho-GCN2 (abcam, ab75836) (1:1,000). The following primary antibodies were diluted in 5% (wt/vol) milk powder–TBS-T and incubated for 1 h at room temperature: mouse anti-N protein (1C12) (48) (1:1,000) and mouse anti-β-actin (Cell Signaling) (1:10,000). The secondary, horseradish peroxidase (HRP)-conjugated goat antibodies were diluted 1:10,000 in 5% (wt/vol) milk powder–TBS-T and incubated for 1 h at room temperature. Images were obtained with GelCapture (DNR Bio-Imaging Systems). The images shown are representative of results from at least three independent experiments. Densitometric quantification of bands was done with ImageJ version 1.48 software.
siRNA transfection.
Transfection of PKR siRNA (Santa Cruz, sc-36263) or control scramble siRNA (Santa Cruz, sc-37007) was performed using siRNA transfection medium (Santa Cruz, sc-36868) and siRNA transfection reagent (Santa Cruz, sc-29528) according to the manufacturer’s protocol with minor adjustments. Cell and reagent amounts were adjusted for 24-well plates. Cells were grown in regular growth medium until they reached confluence of 60% to 80%. They were then transfected with 0.5 μl PKR siRNA or 0.5 μl control siRNA using 2 μl transfection reagent. Cells were washed once with PBS before addition of transfection medium containing siRNA and transfection reagent. Regular growth medium was added after 5.5 to 6 h, and the medium was changed to fresh medium after an additional 18 h. Cells were infected at 30 h after transfection. Samples were collected at 48 h after infection.
Treatments.
For the inhibition of different eIF2α kinases, mock-infected and virus-infected cells were treated with a 2 μM concentration of PKR inhibitor C16 (Sigma, I9785), a 30 nM concentration of PERK inhibitor GSK2606414 (Merck, 516535), or a 100 μM concentration of GCN2 inhibitor indirubin-3′-monoxime (abcam, ab141296) for 1 h. To induce stress granule formation, mock-infected and virus-infected cells were treated with 10 μM CCT020312 (Fisher Scientific, 32-487-95MG) for 1 h, 1 to 5 μM thapsigargin (Merck, T9033) for 5 h, 400 nM rapamycin (InvivoGen, catalog no. tlrl-rap) overnight, or 100 μM sodium arsenite (Sigma) for 1 h. Puromycin labeling was performed by treating cells with 10 μg/ml puromycin for 5 min. Cells were treated with 20 μg/ml cycloheximide (Merck, C7698) for 1 h. For each treatment, the medium was exchanged to fresh growth medium containing the given concentration of the desired compound. For the treatment with poly(I·C), cells were transfected with 1 to 2 μg poly(I·C) (Sigma) using Lipofectamine LTX with Plus reagent (Thermo Fisher, catalog no. 15338100) according to the manufacturer´s instructions and incubated overnight. All sample collections were done immediately after the treatments.
Endpoint dilution assay.
Vero E6 cells in 96-well plates were treated with supernatants from infected cells in a dilution series of 10−1 to 10−6 with eight wells per dilution. After 10 days, the Vero E6 cells were fixed with methanol. To detect infection, the cells were incubated with monkey anti-PUUV serum (diluted 1:100 in PBS with 0.1% Tween and 5% FCS) for 1 h at 37°C. The secondary HRP-conjugated goat anti-human antibody was diluted 1:1,000 in PBS with 0.1% Tween and 5% FCS and incubated for 1 h at 37°C. The cells were then incubated with 3,3′,5,5′-tetramethylbenzidine (TMB) in citrate buffer for ca. 2 min until blue color was observed. Wells staining positive for hantavirus infection were marked, and the number of 50% tissue culture infectious doses (TCID50) per milliliter was calculated using the Spearman-Karber method.
Fluorescent in situ hybridization (FISH).
Hot Start Taq 2× Master Mix (New England Biolabs, M04965) and DIG-11-dUTPs (Jena Bioscience, NU-803-DIGXS) were used to generate DIG-labeled virus-specific double-stranded DNA probes according to the manufacturer´s instructions. The primers were 5′-AAGCTGGAATGAGCACCC-3′ (forward primer) and 5′-GGTTTACGTATCCCATTGACTTCTTC-3′ (reverse primer) for the ANDV S segment, 5′-CTGATTGAGGGTCAGTGTTTC-3′ (forward) and 5′-GTACTGTGATTAGATTCAGGTACTATACC-3′ (reverse) for the ANDV M segment, 5′-ACACAAGCAATATATGATGGCTTATC-3′ (forward) and 5′-GGTGACACTTTAATAGAACCCATTAATAG-3′ (reverse) for the ANDV L segment, 5′-GTTGCCAGACAAAAACTCAAGG-3′ (forward) and 5′-GGTGTAAGTTCTTCAGCTTTCATG-3′ (reverse) for the PUUV S segment, 5′-GAAACACCATGCCAAATAGATCTTTC-3′ (forward) and 5′-ATGCTCTTTTCTGTAACTAGGTCTG-3′ (reverse) for the PUUV M segment, and 5′-ACTCAATCTTTATATGATGGTCTACGTG-3′ (forward) and 5′-GATACTGTTGTCTTCTTGGGTG-3′ (reverse) for the PUUV L segment. cDNAs generated from RNA isolated from virus stocks were used as templates. The generated probes were between 509 and 530 bp long. FISH analysis was performed as described previously (14), with minor adjustments: The cells were fixed, permeabilized, and blocked as described for the immunofluorescence analysis and then washed twice with 2× SSC buffer (Merck, S6639) (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). The DIG-labeled probes were boiled at 95°C for 5 min and diluted in hybridization buffer containing 0.5× formamide (Merck, 47671), 4× SSC, 1× Denhardt’s solution (Merck, D2532), 5% (wt/vol) dextran sulfate (Merck, D8906), 200 mM dithiothreitol (Merck, 43816), and 0.5 mg/ml denatured salmon sperm DNA (Merck, D7656) and were incubated on the cells at 44°C overnight. The cells were then washed twice with 2× SSC. Antibody staining for hantavirus protein, G3BP, and digoxigenin (abcam, ab420) (mouse; 1:500) was performed as described for the immunofluorescence analysis, except that all antibodies were diluted in 4× SSC and the cells were washed with 2× SSC.
Statistical analysis.
Error bars show standard deviations. t tests (Fig. 2B and 4E) were performed in Microsoft Excel, and one-way analysis of variance (ANOVA) (Fig. 4C and D and 5A) and two-way ANOVA (Fig. 4A) were performed in GraphPad Prism Version 8.0.1 (145) to calculate P values (*, P < 0.05; **, P < 0.01).
ACKNOWLEDGMENTS
This research was supported by grants from the Swedish Research Council (Project K2015-56X-22774-01-3 and Project 2018-02646), the Swedish Foundation for Strategic Research (Project SB12-0003), and Karolinska Institutet.
W.C. performed experiments, designed the research, and analyzed data. J.T. designed the research and performed experiments. J.K. designed the research. J.T., W.C., and J.K. wrote the manuscript.
REFERENCES
- 1.Shi M, Lin XD, Chen X, Tian JH, Chen LJ, Li K, Wang W, Eden JS, Shen JJ, Liu L, Holmes EC, Zhang YZ. 2018. The evolutionary history of vertebrate RNA viruses. Nature 556:197–202. doi: 10.1038/s41586-018-0012-7. [DOI] [PubMed] [Google Scholar]
- 2.Jonsson CB, Figueiredo LT, Vapalahti O. 2010. A global perspective on hantavirus ecology, epidemiology, and disease. Clin Microbiol Rev 23:412–441. doi: 10.1128/CMR.00062-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaheri A, Strandin T, Hepojoki J, Sironen T, Henttonen H, Makela S, Mustonen J. 2013. Uncovering the mysteries of hantavirus infections. Nat Rev Microbiol 11:539–550. doi: 10.1038/nrmicro3066. [DOI] [PubMed] [Google Scholar]
- 4.Klingström J, Smed-Sörensen A, Maleki KT, Solà-Riera C, Ahlm C, Björkström NK, Ljunggren HG. 2019. Innate and adaptive immune responses against human Puumala virus infection: immunopathogenesis and suggestions for novel treatment strategies for severe hantavirus-associated syndromes. J Intern Med 285:510–523. doi: 10.1111/joim.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. 2016. The integrated stress response. EMBO Rep 17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buchan JR, Parker R. 2009. Eukaryotic stress granules: the ins and outs of translation. Mol Cell 36:932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Onomoto K, Jogi M, Yoo JS, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, Matsumiya T, Namiki H, Yoneyama M, Fujita T. 2012. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. doi: 10.1371/journal.pone.0043031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reineke LC, Lloyd RE. 2015. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J Virol 89:2575–2589. doi: 10.1128/JVI.02791-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reineke LC, Lloyd RE. 2013. Diversion of stress granules and P-bodies during viral infection. Virology 436:255–267. doi: 10.1016/j.virol.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khaperskyy DA, Hatchette TF, McCormick C. 2012. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J 26:1629–1639. doi: 10.1096/fj.11-196915. [DOI] [PubMed] [Google Scholar]
- 11.White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson Hedestam GB, McInerney GM. 2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol Biol Cell 23:4701–4712. doi: 10.1091/mbc.E12-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z, Mir MA. 2015. Andes virus nucleocapsid protein interrupts protein kinase R dimerization to counteract host interference in viral protein synthesis. J Virol 89:1628–1639. doi: 10.1128/JVI.02347-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McInerney GM, Kedersha NL, Kaufman RJ, Anderson P, Liljestrom P. 2005. Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation. Mol Biol Cell 16:3753–3763. doi: 10.1091/mbc.e05-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White JP, Reineke LC, Lloyd RE. 2011. Poliovirus switches to an eIF2-independent mode of translation during infection. J Virol 85:8884–8893. doi: 10.1128/JVI.00792-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stockwell SR, Platt G, Barrie SE, Zoumpoulidou G, Te Poele RH, Aherne GW, Wilson SC, Sheldrake P, McDonald E, Venet M, Soudy C, Elustondo F, Rigoreau L, Blagg J, Workman P, Garrett MD, Mittnacht S. 2012. Mechanism-based screen for G1/S checkpoint activators identifies a selective activator of EIF2AK3/PERK signalling. PLoS One 7:e28568. doi: 10.1371/journal.pone.0028568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, Fulzele A, Wozniak JM, Gonzalez DJ, Kankel MW, Gao FB, Bennett EJ, Lecuyer E, Yeo GW. 2018. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172:590–604.e13. doi: 10.1016/j.cell.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. 2005. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parker R, Sheth U. 2007. P bodies and the control of mRNA translation and degradation. Mol Cell 25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Mir MA, Duran WA, Hjelle BL, Ye C, Panganiban AT. 2008. Storage of cellular 5’ mRNA caps in P bodies for viral cap-snatching. Proc Natl Acad Sci U S A 105:19294–19299. doi: 10.1073/pnas.0807211105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dauber B, Wolff T. 2009. Activation of the antiviral kinase PKR and viral countermeasures. Viruses 1:523–544. doi: 10.3390/v1030523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Vaheri A, Weber F, Plyusnin A. 2011. Old World hantaviruses do not produce detectable amounts of dsRNA in infected cells and the 5’ termini of their genomic RNAs are monophosphorylated. J Gen Virol 92:1199–1204. doi: 10.1099/vir.0.029405-0. [DOI] [PubMed] [Google Scholar]
- 23.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonin M, Oberstrass J, Lukacs N, Ewert K, Oesterschulze E, Kassing R, Nellen W. 2000. Determination of preferential binding sites for anti-dsRNA antibodies on double-stranded RNA by scanning force microscopy. RNA 6:563–570. doi: 10.1017/s1355838200992318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Son KN, Liang Z, Lipton HL. 2015. Double-stranded rna is detected by immunofluorescence analysis in RNA and DNA virus infections, including those by negative-stranded RNA viruses. J Virol 89:9383–9392. doi: 10.1128/JVI.01299-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee MH, Lalwani P, Raftery MJ, Matthaei M, Lutteke N, Kirsanovs S, Binder M, Ulrich RG, Giese T, Wolff T, Kruger DH, Schonrich G. 2011. RNA helicase retinoic acid-inducible gene I as a sensor of Hantaan virus replication. J Gen Virol 92:2191–2200. doi: 10.1099/vir.0.032367-0. [DOI] [PubMed] [Google Scholar]
- 27.Mayo CB, Cole JL. 2017. Interaction of PKR with single-stranded RNA. Sci Rep 7:3335. doi: 10.1038/s41598-017-03047-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel RC, Sen GC. 1998. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J 17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel CV, Handy I, Goldsmith T, Patel RC. 2000. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem 275:37993–37998. doi: 10.1074/jbc.M004762200. [DOI] [PubMed] [Google Scholar]
- 30.Singh M, Castillo D, Patel CV, Patel RC. 2011. Stress-induced phosphorylation of PACT reduces its interaction with TRBP and leads to PKR activation. Biochemistry 50:4550–4560. doi: 10.1021/bi200104h. [DOI] [PubMed] [Google Scholar]
- 31.Kok KH, Lui PY, Ng MH, Siu KL, Au SW, Jin DY. 2011. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 9:299–309. doi: 10.1016/j.chom.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Dabo S, Maillard P, Collados Rodriguez M, Hansen MD, Mazouz S, Bigot DJ, Tible M, Janvier G, Helynck O, Cassonnet P, Jacob Y, Bellalou J, Gatignol A, Patel RC, Hugon J, Munier-Lehmann H, Meurs EF. 2017. Inhibition of the inflammatory response to stress by targeting interaction between PKR and its cellular activator PACT. Sci Rep 7:16129. doi: 10.1038/s41598-017-16089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohshima D, Arimoto-Matsuzaki K, Tomida T, Takekawa M, Ichikawa K. 2015. Spatio-temporal dynamics and mechanisms of stress granule assembly. PLoS Comput Biol 11:e1004326. doi: 10.1371/journal.pcbi.1004326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruggieri A, Dazert E, Metz P, Hofmann S, Bergeest JP, Mazur J, Bankhead P, Hiet MS, Kallis S, Alvisi G, Samuel CE, Lohmann V, Kaderali L, Rohr K, Frese M, Stoecklin G, Bartenschlager R. 2012. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 12:71–85. doi: 10.1016/j.chom.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buchan JR. 2014. mRNP granules. Assembly, function, and connections with disease. RNA Biol 11:1019–1030. doi: 10.4161/15476286.2014.972208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchan JR, Kolaitis RM, Taylor JP, Parker R. 2013. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153:1461–1474. doi: 10.1016/j.cell.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu L, Ye L, Zhao R, Liu YF, Yang SJ. 2009. HSP70 induced by Hantavirus infection interacts with viral nucleocapsid protein and its overexpression suppresses virus infection in Vero E6 cells. Am J Transl Res 1:367–380. [PMC free article] [PubMed] [Google Scholar]
- 38.Mazroui R, Di Marco S, Kaufman RJ, Gallouzi IE. 2007. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol Biol Cell 18:2603–2618. doi: 10.1091/mbc.e06-12-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walters RW, Parker R. 2015. Coupling of ribostasis and proteostasis: Hsp70 proteins in mRNA metabolism. Trends Biochem Sci 40:552–559. doi: 10.1016/j.tibs.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hussein IT, Cheng E, Ganaie SS, Werle MJ, Sheema S, Haque A, Mir MA. 2012. Autophagic clearance of Sin Nombre hantavirus glycoprotein Gn promotes virus replication in cells. J Virol 86:7520–7529. doi: 10.1128/JVI.07204-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang K, Ma H, Liu H, Ye W, Li Z, Cheng L, Zhang L, Lei Y, Shen L, Zhang F. 2019. The glycoprotein and nucleocapsid protein of hantaviruses manipulate autophagy flux to restrain host innate immune responses. Cell Rep 27:2075–2091.e5. doi: 10.1016/j.celrep.2019.04.061. [DOI] [PubMed] [Google Scholar]
- 42.Plyusnin A. 2002. Genetics of hantaviruses: implications to taxonomy. Arch Virol 147:665–682. doi: 10.1007/s007050200017. [DOI] [PubMed] [Google Scholar]
- 43.Li XD, Lankinen H, Putkuri N, Vapalahti O, Vaheri A. 2005. Tula hantavirus triggers pro-apoptotic signals of ER stress in Vero E6 cells. Virology 333:180–189. doi: 10.1016/j.virol.2005.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McNulty S, Flint M, Nichol ST, Spiropoulou CF. 2013. Host mTORC1 signaling regulates Andes virus replication. J Virol 87:912–922. doi: 10.1128/JVI.02415-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, Parker R. 2017. The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol Cell 68:808–820.e5. doi: 10.1016/j.molcel.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mir MA, Panganiban AT. 2008. A protein that replaces the entire cellular eIF4F complex. EMBO J 27:3129–3139. doi: 10.1038/emboj.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stoltz M, Klingstrom J. 2010. Alpha/beta interferon (IFN-alpha/beta)-independent induction of IFN-lambda1 (interleukin-29) in response to Hantaan virus infection. J Virol 84:9140–9148. doi: 10.1128/JVI.00717-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lundkvist A, Fatouros A, Niklasson B. 1991. Antigenic variation of European haemorrhagic fever with renal syndrome virus strains characterized using bank vole monoclonal antibodies. J Gen Virol 72:2097–2103. doi: 10.1099/0022-1317-72-9-2097. [DOI] [PubMed] [Google Scholar]