

Abstract
We demonstrate that the Knoevenagel condensation can be exploited in combinatorial synthesis on the solid phase. Condensation products from such reactions were structurally characterized, and their Michael reactivity with thiol and phosphine nucleophiles is described. Cyanoacrylamides were previously reported to react reversibly with thiols, and notably, we show that dilution into low pH buffer can trap covalent adducts, which are isolable via chromatography. Finally, we synthesized both traditional and DNA-encoded one-bead, one-compound libraries containing cyanoacrylamides as a source of cysteine-reactive reversibly covalent protein ligands.
Keywords: one bead one compound library, combinatorial chemistry, cyanoacrylamide, covalent, reversible, Knoevenagel condensation, DNA, DNA-encoded library
Graphical Abstract
INTRODUCTION
For some time there has been a strong bias in the pharmaceutical industry against the development of covalent protein ligands as drugs due to concerns about toxicity resulting from off-target reactions.1 This has lessened dramatically over the past few years.2 Many electrophiles, including most acrylamides, 2-chloropropionates, and several other groups, couple with cysteine thiols very slowly unless the reaction is templated by a small molecule–protein interaction.3–5 Effective drugs that contain these weakly reactive electrophiles are coming into clinical use, such as Ibrutinib, which alkylates a conserved cysteine proximal to the ATP-binding site in Bruton’s tyrosine kinase as well as certain mutants of the epidermal growth factor receptor.6,7 Such demonstrations of safety and efficacy have increased interest in the development of new covalent drugs.2 Compounds that bind to proteins covalently are also of great interest as probe molecules because their off-targets can be analyzed quantitatively on a proteome-wide basis using powerful, mass spectrometry-based techniques.8–10
Nonetheless, concerns remain that off-target alkylation reactions11 can cause toxicity and produce immunogenic products.12 For these reasons, there has been increasing interest in the development of molecules that bind to protein targets covalently but reversibly at physiological pH. Such molecules exhibit long-lived binding to a target protein so long as it is folded but dissociate rapidly if the protein is unfolded, thus eliminating the risk that a peptide–small molecule adduct will be presented to the immune system.
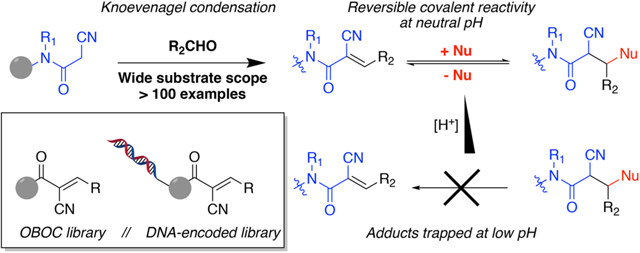
For this purpose, Taunton and co-workers have pioneered the application of 2-cyanoacrylamides that form reversible covalent adducts with cysteine thiols (Scheme 1).13–16 Incorporation of this functionality into a kinase-recognition scaffold resulted in compounds that dissociated from native RSK protein very slowly, but disruption of the protein’s three-dimensional folded structure resulted in rapid dissociation of the complex.13 Others have also demonstrated this strategy with inhibitors targeting mutant EGFR/cSrc and JAK3.17,18 Beyond kinases, cyanoacrylamides can also inhibit other thiol active-site targets such as cysteine proteases.19 5-Methylene-pyrroles were also recently demonstrated to react reversibly with thiols.20 Discovery of other reversible covalent inhibitors could be spurred by the development of software for the virtual screening of molecules containing these motifs.21–24
Scheme 1.

Knoevenagel Condensation and Reactivity of Cyanoacrylamides
We are interested in developing methods by which large numbers of compounds with these interesting electrophiles could be screened experimentally. Toward this end, we report here facile conditions to create one bead one compound (OBOC) libraries of oligomeric molecules capped by α-cyanoacrylamides with diverse functionality. In addition to contributing noncovalent interactions with the target, the cyanoacrylamide’s 3-substituent also modulates its Michael reactivity.25,26 These units are the product of a Knoevenagel condensation between cyanoacetamides and aldehydes (Scheme 1). As one of the oldest, mildest, and simplest carbon–carbon bond-forming reactions known to organic chemists,27 it was shown to be adaptable to the solid phase during the heyday of combinatorial chemistry in the 1990s.28,29 In 2003, Guo et al. reported the solid-phase split synthesis of a small library of Knoevenagel products.30 Their library was biased toward targeting tyrosine kinases, and as such, the substrate scope was limited to substituted benzaldehydes.
In this study, we show that a large variety of aliphatic and aromatic aldehydes react with resin-bound cyanoacetamide to form 3-substituted Knoevenagel products. We confirm that thiols (and phosphines) react reversibly with cyanoacrylamides under physiological conditions but demonstrate that these adducts are quite stable at lower pH and can be isolated if desired. This should be of utility in the characterization of ligand–protein adducts in the future. We also explore the reactivity of 2-carboxyacrylamide and α,β-unsaturated nitro compounds generated via similar solid-phase condensations. The former does not react with thiols or phosphines at neutral pH, whereas the latter reacts rapidly with even stoichiometric amounts of either nucleophile. Finally, we show that these synthetic conditions are mild enough to deploy in the creation of a DNA-encoded combinatorial library of cyanoacrylamides.
RESULTS
Optimized Solid-Phase Knoevenagel Conditions and Reaction Scope.
The previously reported DIC-activated coupling of cyanoacetic acid to a TentaGel resin-bound amine28,30 resulted in an exothermic reaction and a color change prior to addition to the resin. Under these conditions, the conversion to the desired product was poor and accompanied by side products that were not characterized. We reduced the concentration of reagents and added a slight excess of HOAt to the coupling solution (1.25 equiv relative to cyanoacetic acid and DIC). These conditions provided near quantitative conversion to the desired resin-bound cyanoacetamide and suppressed side product formation (Figure S1).
Next, we employed the previously described Knoevenagel condensation conditions (1 M aldehyde/piperidine in DMF overnight at room temperature) to examine the efficiency of coupling a variety of aldehydes with the immobilized cyanoacetamide. Under these general conditions, most aliphatic and aromatic aldehydes were suitable substrates, including those with branched alkyl groups (Figure 1), which have previously been described as underexploited diversity elements for cyanoacrylamides.25 However, these general conditions, when used with enolizable (i.e., R-CH2–CHO) aldehydes, produced multiple impurities in addition to the desired product. We suspected that this was the result of an off-pathway reaction of in situ-generated enamine. Replacing piperidine with the tertiary amine base triethylamine for these substrates gave the desired cyanoacrylamide as the major product. Ketones did not produce tetra-substituted alkenes under these conditions. In total, over 150 Knoevenagel reactions with different carbonyl substrates were tested (Table S1).
Figure 1.

Substrate scope of selected aldehydes suitable for Knoevenagel condensation with resin-bound cyanoacetamide. See methods for detailed experimental procedures. Purity determined by LCMS trace of crude cleaved material.
We also tested other immobilized substrates that could produce an “active methylene” under mild basic conditions. To that end, mono-tert-butyl malonic acid and 3-nitropropionic acid were coupled to TentaGel-displayed lysine (Boc-protected on the side chain). These lysine conjugates, as well as Lys-cyanoacrylamide, were reacted with 3-formylthiophene and base to provide products 1–3 (Figure 2) in excellent purity after acidic cleavage from the resin and good yield following HPLC purification. For example, 1 appeared to be produced in quantitative yield on resin, and an isolated yield of 69% was obtained upon isolation (the discrepancy presumably being to compound loss due to retention on the bead or upon HPLC purification). Although outside the scope of the present work, we anticipate that many other aldehydes would condense efficiently with these substrates.
Figure 2.

Michael acceptors synthesized via condensation of reactive methylene species with 3-thiophenecarbaldehyde.
Alkene Bond Geometry.
To determine the alkene bond geometries of compounds 1–3, we performed nuclear Overhauser effect spectroscopy (NOESY) and rotating frame NOESY (ROESY) (Figures S2–S6). For both 2 and 3, we observed positive through-space correlations that support the cis-bond geometry assignment with respect to the backbone amide. In the spectrum of 2, there is an NOE cross-peak between thiophene protons and the internal amide and lysine methine proton, which are only possible in a cis configuration (Figure S4). Compound 3’s extra backbone methylene protons were found to correlate with a thiophene proton, whereas no correlation was observed between the methylene and vinyl protons, which would be expected if the heterocycle was trans to the backbone amide (Figure S5). In the case of 1, the amide and vinyl signals nearly overlapped (8.23 and 8.22 ppm, respectively), complicating the assignment of multiple NOEs at those coordinates (Figure S2). After a D2O shake (Figure S3), only the NOE to the terminal amide was suppressed, indicating that the nonexchangeable signals are from the vinyl proton and consistent with trans alkene geometry. A second cyanoacrylamide, 4, bearing a benzyl-substituted alkene was synthesized and unequivocally characterized by ROESY as having trans geometry as well (Figure S6).
Characterization of Michael Reactivity.
We next determined the reactivity of these electrophiles. The reaction of cyanoacrylamides with thiols has been reported to be rapidly reversible,13,31 but the Michael reactivity of 2 and 3 remained to be determined. We were also interested in the reaction of the compounds with tris(carboxyethyl)phosphine (TCEP) because we employ this reagent in OBOC library synthesis. Because Michael addition ablates the UV-active chromophore, reactivity with nucleophiles can be monitored spectrophotometrically (Figure 3). We measured interactions among 1–3 with either β-mercaptoethanol (β-ME) or TCEP at physiological pH. Millimolar concentrations of βME reduced the UV activity of compound 1 (Figure 3A). From these data, we calculated a KD of 5.2 mM, which is in close agreement with the value reported previously for a structurally similar compound13 as well as for other cyanoacrylamides with glutathione.32 TCEP was found to be a more potent nucleophile, as half-maximal consumption of 1 was observed at 0.4 mM TCEP (Figure 3B). In stark contrast, addition of up to 100 mM β-ME or TCEP did not quench the chromophore of 2 (Figure 3C,D), indicating that this is a far less reactive Michael acceptor. The α,β-unsaturated nitro compound 3 was a highly reactive electrophile, forming adducts essentially stoichiometrically with both nucleophiles (Figure 3E,F). None of the compounds showed any reactivity toward ethanolamine at pH 7.4 (Figure S7).
Figure 3.

UV–vis spectrophotometry of compounds interacting with nucleophiles. (A–F) Interaction of 1–3 with βME or TCEP at pH 7.4. In A and C, the βME concentrations were 1.6, 3.13, 6.25, 12.5, 25, 50, and 100 mM. For B and D, the TCEP concentrations were 0.024, 0.096, 0.38, 1.5, 6.0, 24.0, and 98.0 mM. In E, the βME concentrations were 6.25, 12.5, 25, 50, 100, and 200 μM. In F, the TCEP concentrations were 6, 12, 24, 48, 96, and 191 μM.
For the reversibility of nucleophilic Michael addition to be probed with 1 and 3, enough βME or TCEP was added to convert most of the starting material to the covalent adduct. These solutions were then diluted 5-fold into neutral buffer. If the Michael addition is reversible, then the UV chromophore of 1 and 3 should be regenerated with kinetics corresponding roughly to the rate of βME or TCEP elimination, excluding any back reaction. Indeed, this is what was observed upon dilution of both the thiolated and phosphinylated adducts of cyanoacrylamide 1 (Figure 4A), meaning that the half-life of the complex is much shorter than the time required to dilute and perform the reading (~2 min). As a control, when βME or TCEP adducts of 1 were diluted into a solution that had additional nucleophile, their UV absorbance was not reconstituted (Figure 4A). This confirms the previously published conclusion that thiol addition to α-cyanoacrylamides is rapidly reversible under physiological conditions and extends this conclusion to phosphines as well. A different result was obtained when these experiments were repeated with the βME and TCEP adducts of α,β-unsaturated nitro compound 3. In this case, dilution of the compounds did not regenerate the chromophore, and we did not observe dissociation of nucleophilic adducts of 3 up to 3 h postdilution (Figure S8).
Figure 4.

Dilution experiments with compound 1. (A) Dilution of covalent complexes of 1 with either βME or TCEP causes a relative increase in absorbance. Note the shift of the red and green lines in the left graph to the teal and orange lines in the second graph, respectively. (B) Dilution experiments of thiolated (2–50 μM βME) 1 adducts into either neutral or low pH phosphate-citrate buffer.
The reversibility of thiol (and, as shown here, phosphine) addition to α-cyanoacrylamides is an attractive attribute for the development of these compounds as drugs but has a downside when viewed from the perspective of probe molecule development. Proteins interacting with irreversible covalent ligands can be characterized directly on a proteome-wide scale. This is extremely useful in evaluating the potential contributions of off-target interactions to whatever phenotype results from treatment of cells or animals with the probe. However, if a covalent adduct reverses rapidly upon dilution or unfolding of the protein, it cannot be characterized directly. This could perhaps be overcome by the application of the recently developed iso-TOP-ABPP technology,10 but it nonetheless would be of interest to be able to characterize adducts directly.
The reversibility of cyanoacrylamide-thiol Michael adducts is due to the acidity of the hydrogen alpha to the amide and nitrile residues, deprotonation of which leads to β-elimination of the thiolate.13 Therefore, we explored the possibility of stabilizing the Michael adduct by reducing the pH. When the βME adducts of 1 were diluted into a phosphate-citrate buffer at pH 2.6, the UV chromophore was not reconstituted (Figure 4B) in stark contrast to the result of diluting into a neutral pH solution. This observation was consistent over a range of βME concentrations with the relative change in absorbance being more extreme at βME concentrations above the KD (Table S2). Therefore, low pH stabilizes the thiolated conjugates.
This result encouraged us to attempt to observe the Michael products by liquid chromatography/mass spectrometry (LCMS). The low pH (2.5) of the 0.1% formic acid-supplemented running buffer should preserve intact thiol-cyanoacrylamide conjugates and allow their separation. Compound 1 was mixed with either βME or 3-mercaptopro-pionic acid (MPA) at concentrations above and below the KD followed by injection onto the LCMS. The relative abundance of each species was quantified via integration of their extracted ion chromatograms (Table 1). Notably, although the cyanoacrylamide eluted as a single peak, the thiolated conjugates eluted as multiple peaks characteristic of mixtures of diastereomers. We also performed competition experiments with both βME and MPA. MPA had a greater tendency to react with 1 than did βME, and the ratio of MPA to βME thiolated adducts was consistently 2:1 at concentrations both well above and well below the βME KD (Table 1). These results were confirmed with UV–vis as well (Figure S9). The order of thiol addition did not affect the distribution of products, providing further confirmation that thiol engagement is a rapid equilibrium process at neutral pH. Combined, these UV–vis and LCMS data demonstrate that cyanoacrylamide adducts can be trapped, isolated, and directly analyzed.
Table 1.
Quantification of Thiolated Conjugates from Mixed Thiol Incubation Experiments with Compound 1a
| sample | MA | [MA + BME] | [MA + MPA] |
|---|---|---|---|
| 200 μM 1 | 100% | n/a | n/a |
| 1 + 100 mM βME | 12.14% | 87.86% | n/a |
| 1 + 100 mM MPA | 6.49% | n/a | 93.51% |
| 1 + 50 mM βME, then 50 mM MPA | 8.23% | 30.61% | 61.16% |
| 1 + 50 mM MPA, then 50 mM βME | 7.46% | 29.84% | 62.70% |
| 1 + 100 μM βME | 98.30% | 1.70% | n/a |
| 1 + 100 μM MPA | 97.14% | n/a | 2.86% |
| 1 + 50 μM βME, then 50 μM MPA | 97.50% | 0.87% | 1.63% |
| 1 + 50 μM MPA, then 50 mM βME | 97.79% | 0.75% | 1.46% |
Table entries are relative values from integration of M + H extracted ion chromatograms.
MA = Michael acceptor. MA M + H = 307; [MA+βME] M + H = 385; [MA + MPA] M + H = 413.
Synthesis of OBOC Libraries Containing Cyanoacrylamides.
To demonstrate the utility of the Knoevenagel chemistry in combinatorial library synthesis, we designed the library shown in Figure 5. The library was created by split-and-pool synthesis on 90 μm TentaGel resin using standard Oxyma coupling conditions at the first two diversity positions,33 and condensation with 25 unique aldehydes to provide the cyanoacrylamide terminus. Mass orthogonal linkers (high-lighted in pink in Figure 5) encode the downstream stereochemistry of R2, permitting the use of both l- and d- amino acids at that position. This 6000-compound library is biased toward higher diversity at the terminal cyanoacrylamide. Postsynthesis, 24 beads were treated with cyanogen bromide to release compounds from the resin, and the resultant material was analyzed via MALDI MS and MS/MS. Twenty one of the samples displayed a clear molecular ion with the expected isotope splitting characteristic of the aryl bromide in the linker34 even though the N-terminal amide was found to be quite labile and fragmented under MS in-source ionization (Table S3, Figure S10). This phenomenon has been exploited in the past to identify structural features of peptides and proteins.35,36 Determination of the sequence from each of the 21 molecular ions by MS/MS was facile, and we conclude that the library is of high quality. Although the structure of each of the compounds in the library discussed above could be determined with MS, we hope to employ this chemistry in the construction of DNA-encoded OBOC libraries because they have many advantages.37 For the feasibility of this to be determined, a qPCR-based assay described previously by Malone and Paegel38 was employed to determine the amount of DNA that remained amplifiable by polymerase chain reaction (PCR) after being subjected to the Knoevenagel conditions we report here. When using either 3-thiophenecarbaldehyde or isobutyraldehyde, the amounts of PCR-amplifiable DNA remaining after the reactions were found to be 55 and 91%, respectively. For comparison, the conditions used for carboxylic acid coupling leave approximately half (51–57%) of the DNA in an amplifiable state.38 Thus, as hoped, the mild conditions employed for the Knoevenagel condensation are quite DNA compatible. This is in agreement with a previous report employing a variant of the Knoevenagel condensation in solution-phase DNA-encoded library synthesis.39
Figure 5.

Design of 6,000-member trimer peptoid-amino acid-cyanoacrylamide macrobead library.
This opened the door to incorporating cyanoacrylamides into a DNA-encoded OBOC library. The library was prepared on 10 μm TentaGel resin along with several thousand 160 μm beads that served as “sentinels” because they contain sufficient material for postsynthesis analysis of both the DNA and synthetic ligands. A variety of halo-acid and amine submonomer pairs account for a total of 70 different monomers at each of the first two positions. Some halo-acids would result in a sterically crowded N-terminus, creating a difficult substrate for subsequent acylation; thus, diamines were used exclusively to displace the halogen at these positions. Because DNA encoding removes the limitations of MS hit deconvolution, we broadened our monomer pool to increase diversity at all positions, especially at the cyanoacrylamide terminus (Figure 6A,C; Tables S4 and S5). Eighty-four unique aldehydes, many of which are isobaric, were condensed to form the cyanoacrylamide, resulting in a 411,600-member library.
Figure 6.

Design of a 411,600-member DNA-encoded cyanoacrylamide trimer library. (A) Partial representation of the diversity represented. Isobaric cyanoacrylamide substituents (dark red) are boxed together. Full set of diversity elements is available in Tables S4 and S5). (B) DNA quality control of library position R2 via NGS analysis of 4500 (1500 × 3 replicates) library beads post-synthesis. PICCO positions are shown in Figure S12. (C) Representative trimers assembled from the library elements.
The library’s sequenceability and synthetic quality were confirmed through Sanger sequencing and MALDI MS/MS of cleaved material from 23 individual 160 μm sentinel beads. Sequences expected from the encoding units were observed from 21 Sanger runs (Figure S11), and MS data consistent with the structures predicted by the encoding DNA were obtained for 15 of the 20 samples (Table S6; Figure S12). It should be noted that these library structures are not optimized for MALDI-MS detection, as was the previous MS-deconvolutable library. The DNA tags from 4500 10 μm library beads were amplified by PCR, and their amplicons were analyzed by next generation sequencing (NGS). All of the “codons” used were observed in the NGS data with approximately the expected frequency (Figure 6B, Figure S13, and Table S7). These data demonstrate that none of the encoding DNAs were rendered nonamplifiable as a result of the chemistry performed and, when taken together with the sentinel bead analysis, argue that the library is of high quality. Though other DNA-encoded covalent strategies have recently been reported,40–42 to the best of our knowledge this is the first report of a fully DNA-encoded library bearing reversibly covalent electrophilic groups.
DISCUSSION
We have demonstrated conditions that enable the use of the Knoevenagel condensation in OBOC library synthesis, including those encoded with DNA. The substrate scope is quite broad, allowing for the incorporation of significant diversity around the trans-substituted alkene. The mild general conditions for accessing cyanoacrylamides bearing aliphatic 3-substitutents are particularly noteworthy because these compounds are difficult to access in solution. Basu et al. noted that neat acetic acid at elevated temperatures (80 °C) was required to form aliphatic 3-substituents in 48–82% yields with only trace aldol products generated under milder conditions.17 Cyanoacrylamides 1 and 4 that we characterized by NMR were both exclusively trans isomers, but interestingly, others report that performing the Knoevenagel condensation at higher temperatures produces the cis isomer exclusively.43 Although our research was driven by combinatorial library applications, others may find these methods useful for medicinal chemistry when solution routes are inefficient.
These cyanoacrylamides react rapidly and reversibly with thiols as well as phosphines at neutral pH, as reported previously. Importantly, we found that these adducts are stable and observable by UV–vis and LCMS at low pH. These observations provide an explanation for previous reports of denatured protein-cyanoacrylamide adducts that surprised the authors based on the proposed rapid reversibility of the Michael reaction and led them to propose that this was due to nonspecific covalent interactions.17,43 Our findings provide a simple protocol that will allow investigators to characterize the site of binding of cyanoacrylamide-containing ligands to cysteine-containing proteins.
With respect to the other condensation products characterized in this work, α-carboxyacrylamide 2 showed no evidence of coupling with thiols or phosphines. This is likely due to the α-carboxylic acid being deprotonated at pH 7.4, thus reducing the electrophilic character of the acrylamide. This motif may not be useful as a weakly reactive electrophile in the development of reversible, covalent protein ligands but could be incorporated as a conformationally constrained structural feature in other OBOC libraries. Alternatively, we are currently investigating coupling of the free carboxylic acid with amines to provide α,α-bis-amides, which could confer tunable Michael reactivity based on the electronic nature of the amide substituent.5
On the other end of the spectrum, α,β-unsaturated nitro compound 3 reacted stoichiometrically with thiols and phosphines. Nitroalkenes have been characterized as potent Michael acceptors in synthetic applications in the past,44,45 but their characterization as biologically relevant electrophiles is more limited. One important recent example is nitrolinoleic acid, a product of nitric oxide signaling that reportedly binds reversibly with glutathione, cysteine, and human serum albumin.46,47 We did not observe retro-Michael regeneration of the chromophore of 3 following dilution of covalent complexes into neutral buffer, which would have demonstrated its reversibility. However, this experiment is not appropriate for very strong interactions, like that of 3 with nucleophiles, which may not be affected by concentration changes until the dilution factor is larger than the limit of detection. For our purposes, the hyper-reactivity of 3 precluded its deployment in OBOC libraries.
The Knoevenagel method described here for making electrophilic cyanoacrylamides now enters the repertoire of DNA-compatible chemistries. The amount of readable DNA tags postreaction is comparable with other reactions employed commonly to create DNA-encoded chemical libraries. This was important to us because the broad substrate scope of the reaction allows the use of building blocks that would be impossible to distinguish by mass spectrometry. Furthermore, DNA encoding enables miniaturization of the entire OBOC platform48 and facilitates screening of these libraries using a flow cytometer.49 Efforts to mine reversible covalent protein ligands from the libraries described here will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Hao Wu for his input throughout the project. We also thank Dr. Xiangming Kong for helpful advice in analyzing and interpreting NMR spectra. This research was supported by a grant from the National Institutes of Health (AG 054892). P.D. was supported by a fellowship kindly provided by the Klorfine Foundation.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscombsci.7b00169.
Supplementary data, tables, mass spectra, materials, methods, detailed procedures for the synthesis of the libraries and compounds, along with full NMR characterizations, and DNA-encoding protocols (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Potashman MH; Duggan ME Covalent Modifiers: an Orthogonal Approach to Drug Design. J. Med. Chem 2009, 52, 1231–1246. [DOI] [PubMed] [Google Scholar]
- (2).Singh J; Petter RC; Baillie TA; Whitty A The Resurgence of Covalent Drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- (3).Ward RA; Anderton MJ; Ashton S; Bethel PA; Box M; Butterworth S; Colclough N; Chorley CG; Chuaqui C; Cross DAE; Dakin LA; Debreczeni JÉ; Eberlein C; Finlay MRV; Hill GB; Grist M; Klinowska TCM; Lane C; Martin S; Orme JP; Smith P; Wang F; Waring MJ Structure- and Reactivity-Based Development of Covalent Inhibitors of the Activating and Gatekeeper Mutant Forms of the Epidermal Growth Factor Receptor (EGFR). J. Med. Chem 2013, 56, 7025–7048. [DOI] [PubMed] [Google Scholar]
- (4).Flanagan ME; Abramite JA; Anderson DP; Aulabaugh A; Dahal UP; Gilbert AM; Li C; Montgomery J; Oppenheimer SR; Ryder T; Schuff BP; Uccello DP; Walker GS; Wu Y; Brown MF; Chen JM; Hayward MM; Noe MC; Obach RS; Philippe L; Shanmugasundaram V; Shapiro MJ; Starr J; Stroh J; Che Y Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors. J. Med. Chem 2014, 57, 10072–10079. [DOI] [PubMed] [Google Scholar]
- (5).Cee VJ; Volak LP; Chen Y; Bartberger MD; Tegley C; Arvedson T; McCarter J; Tasker AS; Fotsch C Systematic Study of the Glutathione (GSH) Reactivity of N-Arylacrylamides: 1. Effects of Aryl Substitution. J. Med. Chem 2015, 58, 9171–9178. [DOI] [PubMed] [Google Scholar]
- (6).Ponader S; Chen S-S; Buggy JJ; Balakrishnan K; Gandhi V; Wierda WG; Keating MJ; O’Brien S; Chiorazzi N; Burger JA The Bruton Tyrosine Kinase Inhibitor PCI-32765 Thwarts Chronic Lymphocytic Leukemia Cell Survival and Tissue Homing in Vitro and in Vivo. Blood 2012, 119, 1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wang A; Yan X-E; Wu H; Wang W; Hu C; Chen C; Zhao Z; Zhao P; Li X; Wang L; Wang B; Ye Z; Wang J; Wang C; Zhang W; Gray NS; Weisberg EL; Chen L; Liu J; Yun C-H; Liu Q Ibrutinib Targets Mutant-EGFR Kinase with a Distinct Binding Conformation. Oncotarget 2016, 7, 69760–69769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cravatt BF; Wright AT; Kozarich JW Activity-Based Protein Profiling: From Enzyme Chemistry to Proteomic Chemistry. Annu. Rev. Biochem 2008, 77, 383–414. [DOI] [PubMed] [Google Scholar]
- (9).Lanning BR; Whitby LR; Dix MM; Douhan J; Gilbert AM; Hett EC; Johnson TO; Joslyn C; Kath JC; Niessen S; Roberts LR; Schnute ME; Wang C; Hulce JJ; Wei B; Whiteley LO; Hayward MM; Cravatt BF A Road Map to Evaluate the Proteome-Wide Selectivity of Covalent Kinase Inhibitors. Nat. Chem. Biol 2014, 10, 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Backus KM; Correia BE; Lum KM; Forli S; Horning BD; González-Páez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; Wolan DW; Cravatt BF Proteome-Wide Covalent Ligand Discovery in Native Biological Systems. Nature 2016, 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Engel J; Lategahn J; Rauh D Hope and Disappointment: Covalent Inhibitors to Overcome Drug Resistance in Non-Small Cell Lung Cancer. ACS Med. Chem. Lett 2016, 7, 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Uetrecht J; Naisbitt DJ Idiosyncratic Adverse Drug Reactions: Current Concepts. Pharmacol. Rev 2013, 65, 779–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Serafimova IM; Pufall MA; Krishnan S; Duda K; Cohen MS; Maglathlin RL; McFarland JM; Miller RM; Frödin M; Taunton J Reversible Targeting of Noncatalytic Cysteines with Chemically Tuned Electrophiles. Nat. Chem. Biol 2012, 8, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Krishnan S; Miller RM; Tian B; Mullins RD; Jacobson MP; Taunton J Design of Reversible, Cysteine-Targeted Michael Acceptors Guided by Kinetic and Computational Analysis. J. Am. Chem. Soc 2014, 136, 12624–12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).London N; Miller RM; Krishnan S; Uchida K; Irwin JJ; Eidam O; Gibold L; Cimermančič P; Bonnet R; Shoichet BK; Taunton J Covalent Docking of Large Libraries for the Discovery of Chemical Probes. Nat. Chem. Biol 2014, 10, 1066–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bradshaw JM; McFarland JM; Paavilainen VO; Bisconte A; Tam D; Phan VT; Romanov S; Finkle D; Shu J; Patel V; Ton T; Li X; Loughhead DG; Nunn PA; Karr DE; Gerritsen ME; Funk JO; Owens TD; Verner E; Brameld KA; Hill RJ; Goldstein DM; Taunton J Prolonged and Tunable Residence Time Using Reversible Covalent Kinase Inhibitors. Nat. Chem. Biol 2015, 11, 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Basu D; Richters A; Rauh D Structure-Based Design and Synthesis of Covalent-Reversible Inhibitors to Overcome Drug Resistance in EGFR. Bioorg. Med. Chem 2015, 23, 2767–2780. [DOI] [PubMed] [Google Scholar]
- (18).Forster M; Chaikuad A; Bauer SM; Holstein J; Robers MB; Corona CR; Gehringer M; Pfaffenrot E; Ghoreschi K; Knapp S; Laufer SA Selective JAK3 Inhibitors with a Covalent Reversible Binding Mode Targeting a New Induced Fit Binding Pocket. Cell Chem. Biol 2016, 23, 1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mohamed MF; Samir N; Ali A; Ahmed N; Ali Y; Aref S; Hossam O; Mohamed MS; Abdelmoniem AM; Abdelhamid IA Apoptotic Induction Mediated P53 Mechanism and Caspase-3 Activity by Novel Promising Cyanoacrylamide Derivatives in Breast Carcinoma. Bioorg. Chem 2017, 73, 43–52. [DOI] [PubMed] [Google Scholar]
- (20).Zhang Y; Zhou X; Xie Y; Greenberg MM; Xi Z; Zhou C Thiol Specific and Tracelessly Removable Bioconjugation via Michael Addition to 5-Methylene Pyrrolones. J. Am. Chem. Soc 2017, 139, 6146–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Krishnan S; Miller RM; Tian B; Mullins RD; Jacobson MP; Taunton J Design of Reversible, Cysteine-Targeted Michael Acceptors Guided by Kinetic and Computational Analysis. J. Am. Chem. Soc 2014, 136, 12624–12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).London N; Miller RM; Krishnan S; Uchida K; Irwin JJ; Eidam O; Gibold L; Cimermančič P; Bonnet R; Shoichet BK; Taunton J Covalent Docking of Large Libraries for the Discovery of Chemical Probes. Nat. Chem. Biol 2014, 10, 1066–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhang Y; Zhang D; Tian H; Jiao Y; Shi Z; Ran T; Liu H; Lu S; Xu A; Qiao X; Pan J; Yin L; Zhou W; Lu T; Chen Y Identification of Covalent Binding Sites Targeting Cysteines Based on Computational Approaches. Mol. Pharmaceutics 2016, 13, 3106–3118. [DOI] [PubMed] [Google Scholar]
- (24).Smith JM; Rowley CN Automated Computational Screening of the Thiol Reactivity of Substituted Alkenes. J. Comput.-Aided Mol. Des 2015, 29, 725–735. [DOI] [PubMed] [Google Scholar]
- (25).Bradshaw JM; McFarland JM; Paavilainen VO; Bisconte A; Tam D; Phan VT; Romanov S; Finkle D; Shu J; Patel V; Ton T; Li X; Loughhead DG; Nunn PA; Karr DE; Gerritsen ME; Funk JO; Owens TD; Verner E; Brameld KA; Hill RJ; Goldstein DM; Taunton J Prolonged and Tunable Residence Time Using Reversible Covalent Kinase Inhibitors. Nat. Chem. Biol 2015, 11, 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Krenske EH; Petter RC; Houk KN Kinetics and Thermodynamics of Reversible Thiol Additions to Mono- and Diactivated Michael Acceptors: Implications for the Design of Drugs That Bind Covalently to Cysteines. J. Org. Chem 2016, 81, 11726–11733. [DOI] [PubMed] [Google Scholar]
- (27).Knoevenagel E. Condensation Von Malonsäure Mit Aromati-schen Aldehyden Durch Ammoniak Und Amine. Ber. Dtsch. Chem. Ges 1898, 31, 2596–2619. [Google Scholar]
- (28).Zaragoza F. Carbon-Carbon Bond Formation on Solid Support: Synthesis of Monoacyl Piperazines by Knoevenagel-Type Condensation Reactions. Tetrahedron Lett. 1995, 36, 8677–8678. [Google Scholar]
- (29).Gordeev MF; Patel DV; Wu J; Gordon EGT Approaches to Combinatorial Synthesis of Heterocycles: Solid Phase Synthesis of Pyridines and Pyrido[2, 3-D]Pyrimidines. Tetrahedron Lett. 1996, 37, 4643–4646. [Google Scholar]
- (30).Guo G; Arvanitis EA; Pottorf RS; Player MR Solid-Phase Synthesis of a Tyrphostin Ether Library. J. Comb. Chem 2003, 5, 408–413. [DOI] [PubMed] [Google Scholar]
- (31).Pritchard RB; Lough CE; Currie DJ; Holmes HL Equilibrium Reactions of N-Butanethiol with Some Conjugated Heteroenoid Compounds. Can. J. Chem 1968, 46, 775–781. [Google Scholar]
- (32).Liu Z; Zhou X; Miao Y; Hu Y; Kwon N; Wu X; Yoon J A Reversible Fluorescent Probe for Real-Time Quantitative Monitoring of Cellular Glutathione. Angew. Chem., Int. Ed 2017, 56, 5812–5816. [DOI] [PubMed] [Google Scholar]
- (33).Subiros-Funósas R; Prohens R; Barbas R; El-Faham A; Albericio F Oxyma: an Efficient Additive for Peptide Synthesis to Replace the Benzotriazole-Based HOBt and HOAt with a Lower Risk of Explosion. Chem. - Eur. J 2009, 15, 9394–9403. [DOI] [PubMed] [Google Scholar]
- (34).Sarkar M; Pascal BD; Steckler C; Aquino C; Micalizio GC; Kodadek T; Chalmers MJ Decoding Split and Pool Combinatorial Libraries with Electron-Transfer Dissociation Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom 2013, 24, 1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Katta V; Chow DT; Rohde MF Applications of in-Source Fragmentation of Protein Ions for Direct Sequence Analysis by Delayed Extraction MALDI-TOF Mass Spectrometry. Anal. Chem 1998, 70, 4410–4416. [DOI] [PubMed] [Google Scholar]
- (36).Lennon JJ; Walsh KA Locating and Identifying Posttranslational Modifications by in-Source Decay During MALDI-TOF Mass Spectrometry. Protein Sci. 1999, 8, 2487–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).MacConnell AB; McEnaney PJ; Cavett VJ; Paegel BM DNA-Encoded Solid-Phase Synthesis: Encoding Language Design and Complex Oligomer Library Synthesis. ACS Comb. Sci 2015, 17, 518–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Malone ML; Paegel BM What Is a “DNA-Compatible” Reaction? ACS Comb. Sci 2016, 18, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tian X; Basarab GS; Selmi N; Kogej T; Zhang Y; Clark M; Goodnow RA Jr. Development and Design of the Tertiary Amino Effect Reaction for DNA-Encoded Library Synthesis. MedChemComm 2016, 7, 1316–1322. [Google Scholar]
- (40).Barluenga S; Zambaldo C; Ioannidou HA; Ciobanu M; Morieux P; Daguer J-P; Winssinger N Novel PTP1B Inhibitors Identified by DNA Display of Fragment Pairs. Bioorg. Med. Chem. Lett 2016, 26, 1080–1085. [DOI] [PubMed] [Google Scholar]
- (41).Chan AI; McGregor LM; Jain T; Liu DR Discovery of a Covalent Kinase Inhibitor From a DNA-Encoded Small-Molecule Library × Protein Library Selection. J. Am. Chem. Soc 2017, 139, 10192–10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zimmermann G; Rieder U; Bajic D; Vanetti S; Chaikuad A; Knapp S; Scheuermann J; Mattarella M; Neri D A Specific and Covalent JNK-1 Ligand Selected From an Encoded Self-Assembling Chemical Library. Chem. - Eur. J 2017, 23, 8152–8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Smith S; Keul M; Engel J; Basu D; Eppmann S; Rauh D Characterization of Covalent-Reversible EGFR Inhibitors. ACS Omega 2017, 2, 1563–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wade PA; Murray JK; Shah-Patel S; Palfey BA; Carroll PJ Tandem Nitroaldol–Dehydration Reactions Employing the Dianion of Phenylsulfonylnitromethane 1. J. Org. Chem 2000, 65, 7723–7730. [DOI] [PubMed] [Google Scholar]
- (45).Palomo C; Vera S; Mielgo A; Gómez Bengoa E Highly Efficient Asymmetric Michael Addition of Aldehydes to Nitroalkenes Catalyzed by a Simple Trans-4-Hydroxyprolylamide. Angew. Chem., Int. Ed 2006, 45, 5984–5987. [DOI] [PubMed] [Google Scholar]
- (46).Alvarez B; Turell L; Vitturi DA; Coitiño EL; Lebrato L; Moller MN; Sagasti C; Salvatore SR; Woodcock SR; Schopfer FJ Thiol Addition to Conjugated Nitrolinoleic Acid. FASEB J. 2017, 31, 605.1–605.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Turell L; Vitturi DA; Coitiño EL; Lebrato L; Moller MN; Sagasti C; Salvatore SR; Woodcock SR; Alvarez B; Schopfer FJ The Chemical Basis of Thiol Addition to Nitro-Conjugated Linoleic Acid, a Protective Cell-Signaling Lipid. J. Biol. Chem 2017, 292, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).MacConnell AB; Price AK; Paegel BM An Integrated Microfluidic Processor for DNA-Encoded Combinatorial Library Functional Screening. ACS Comb. Sci 2017, 19, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Mendes KR; Malone ML; Ndungu JM; Suponitsky-Kroyter I; Cavett VJ; McEnaney PJ; MacConnell AB; Doran TM; Ronacher K; Stanley K; Utset O; Walzl G; Paegel BM; Kodadek T High-Throughput Identification of DNA-Encoded IgG Ligands That Distinguish Active and Latent Mycobacterium Tuber-culosis Infections. ACS Chem. Biol 2017, 12, 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.