

Abstract
Adeno-associated virus (AAV) is a highly promising gene transfer vector, yet major cellular requirements for AAV entry are poorly understood. Using a genome-wide CRISPR screen for entry of evolutionarily divergent serotype AAVrh32.33, we identified GPR108, a member of the G protein-coupled receptor superfamily, as an AAV entry factor. Of greater than 20 divergent AAVs across all AAV clades tested in human cell lines, only AAV5 transduction was unaffected in the GPR108 knockout (KO). GPR108 dependency was further shown in murine and primary cells in vitro. These findings are further validated in vivo, as the Gpr108 KO mouse demonstrates 10- to 100-fold reduced expression for AAV8 and rh32.33 but not AAV5. Mechanistically, both GPR108 N- and C-terminal domains are required for transduction, and on the capsid, a VP1 unique domain that is not conserved on AAV5 can be transferred to confer GPR108 independence onto AAV2 chimeras. In vitro binding and fractionation studies indicate reduced nuclear import and cytosolic accumulation in the absence of GPR108. We thus have identified the second of two AAV entry factors that is conserved between mice and humans relevant both in vitro and in vivo, further providing a mechanistic understanding to the tropism of AAV gene therapy vectors.
Keywords: adeno-associated virus, AAV, receptor, CRISPR screen, GPR108, entry, endosomal escape, in vivo
Graphical Abstract
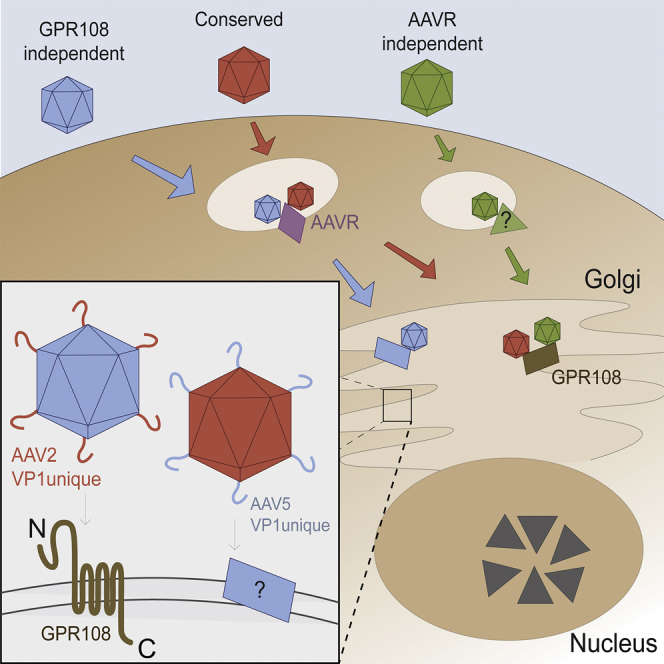
Adeno-associated virus (AAV) has been repurposed as a highly effective gene therapy vector, yet conserved cellular factors required for cellular entry are poorly understood. In this issue of Molecular Therapy, Vandenberghe and colleagues describe identification of a novel conserved AAV entry factor that is required in vivo for AAV transduction.
Introduction
Adeno-associated virus (AAV) is a non-pathogenic member of the dependovirus subfamily of parvovirus, which has been successfully engineered into an efficient gene therapy vector. AAV-based gene therapies have demonstrated great success in clinical trials1, 2, 3, 4 and more recently in the US Food and Drug Administration (FDA) approval of a gene therapy for an inherited type of blindness5 and a form of spinal muscular atrophy.3 Remarkably, how this family of viruses enters cells to efficiently deliver its cargo remains poorly understood. Cell and tissue tropism is determined by the capsid structure, which is composed of proteins VP1, VP2, and VP3. However, how different capsid variants differentially engage entry factors remains unclear as does how capsid engineering efforts can (re-)target vectors for improved performance.
For several AAV serotypes, attachment is mediated by capsid-specific sugar moieties on the cell surface, which have been studied extensively.6, 7, 8, 9, 10, 11, 12, 13 Post-attachment steps of AAV entry, and how these steps may differ between capsid variants, are less defined. Previous methodologies used to understand AAV entry have been technologically limited and prevented the characterization of major receptor and entry factor requirements shared among AAV serotypes. Indeed, experiments such as cDNA overexpression screens in poorly permissive cell lines led to the identification of some factors that increase transduction,14, 15, 16, 17, 18, 19 yet knockout (KO) of these proteins showed they are not an absolute requirement for AAV entry. Only recently, a haploid genetic screen identified a highly conserved AAV entry receptor, AAVR.20
In this study, in order to better understand the cellular entry determinants of a wide range of AAV variants, we undertook a genome-wide CRISPR screen using a divergent AAV serotype, rh32.33, which we previously reported to be AAVR-independent.21 We identified a second highly conserved entry factor, GPR108, which is required for entry of all AAV variants tested except for the highly divergent AAV5. Characterization of this G protein-coupled receptor-like protein reveals that it is localized to the trans-Golgi network (TGN) and its N- and C-terminal domains are essential for mediating AAV entry. Our studies indicate that GPR108 is not required for cellular attachment, yet it may play a role in endosomal escape of the virus, as GPR108 usage is dictated by a VP1 portion of capsid thought to be extruded from the particle only in the acidic conditions of the endosome.22 A decrease in the number of internalized genomes as well as a decrease in proportion of nuclear genomes in AAVR KO and GPR108 KO cells suggest that these proteins are required for a viral entry step upstream of nuclear import, and that AAV may be degraded in the absence of the required cellular entry factors. GPR108 was recently published to be an nuclear factor κB (NF-κB) activator that negatively regulates Toll-like receptor (TLR) signaling,23 and as such the molecular implications of GPR108 as an AAV entry factor in the context of cellular immune responses to AAV is of great interest.
We demonstrate that usage of this entry factor is conserved in mice, and that mouse GPR108 is highly functional in human cell lines. Importantly, in vivo transduction of the Gpr108 KO mouse model is decreased 10- to 100-fold. Based on cell fractionation data, and a required domain on the capsid for GPR108 dependency, we propose a model in which GPR108 is required for escape from an endosomal or TGN compartment. Following the identification of AAVR, we present GPR108 as a second highly conserved cellular entry factor required both in vitro and in vivo for the majority of primate AAVs. Indeed, most AAVs in current clinical use require both AAVR and GPR108 for entry, except for AAV5, which is AAVR-dependent yet GPR108-independent, and the converse is true for AAV4 and rh32.33.20,21,24
Results
Genome-wide CRISPR/Cas9 Screen Identifies Novel AAV Entry Factors
To better understand the major cell and tissue transduction determinants of AAV, we employed a highly stringent genome-wide CRISPR screen25, 26, 27, 28, 29 using the evolutionarily divergent and AAVR-independent rh32.33. We used a previously published genome-scale CRISPR KO (GeCKO) lentiviral guide RNA (gRNA) library,30 designed to target the majority of endogenous human protein-encoding genes and microRNAs (miRNAs), in order to identify entry requirements in Huh7 AAVR KO cells using multiple rounds of transduction and fluorescence-activated cell sorting (FACS) selection of GFP-negative cells after rh32.33 transduction with a GFP transgene (Figure 1A). Deep sequencing of single guide RNAs (sgRNAs) enriched in GFP-negative cells after two rounds of transduction highlighted several genes that were significantly enriched compared to unselected control cells, most notably GPR108 (Figure 1B). Gene ontology analysis demonstrated enrichment of several genes encoding transmembrane (TM) proteins (Figure 1B, group 6) as putative alternate pathway entry factors, as well as some involved in glycan biosynthesis such as NEU1, GCNT4, and CTSA.
Figure 1.

rh32.33 Entry Screen
(A) Huh7 AAVR KO cells undergo lentiCRISPR mutagenesis using a two-vector lentiviral GeCKO system. sgRNA library contains six sgRNAs per gene, targeting the entire human genome and 1,000 miRNAs to generate a cell line knockout library. Cells are subjected to multiple rounds of high MOI transduction and sgRNA deep sequencing to determine gene deletions enriched in GFP-negative cells to identify genes required for rh32.33 entry. (B) Entry screen hits from second round of GFP transduction. The x axis shows individual genes within the GeCKO library, grouped by gene ontology analysis. The y axis shows significance of hit based on RRA analysis. The bubble diameter indicates the number of individual sgRNAs per gene enriched in the selected population, relative to unselected control.
GPR108 Is Required for Entry of AAVR-Dependent and AAVR-Independent Serotypes in Multiple Cell Lines
We next interrogated AAV requirement of our most significantly enriched gene, GPR108 (Figure 1B). GPR108 is a protein of unknown function but has been described as structurally analogous to GPR107. Interestingly, GPR108 was also identified as a potential entry factor in the initial haploid screen that identified AAVR.20 This suggested to us that GPR108 may be important not only for rh32.33 entry, but also entry of other AAV serotypes. We therefore generated a GPR108 KO Huh7 cell line and tested a panel of natural and putative ancestral intermediate AAV capsids (Figure 2A) for GPR108 usage via a luciferase assay. Transduction of all tested serotypes except AAV5 was greater than 10- to 100-fold decreased in the GPR108 KO cells compared to wild-type (WT) cells (Figure 2B). AAV5 uniquely demonstrates similar transduction levels in WT and GPR108 KO cells, demonstrating that this loss of function is specific to certain AAV serotypes, and is not a general defect in endocytosis. Dependency on GPR108, or lack thereof, was unaffected by AAV co-infection with hAd5 in WT and GPR108 KO cells (Figure S2). Double KO of AAVR and GPR108 causes a complete loss of transduction of all known tested serotypes (Figure S2), highlighting the importance of both proteins for AAV transduction. Transient transfection of FLAG-tagged GPR108, but not its homolog GPR107, was able to rescue AAV transduction as shown by a roughly 100-fold increase in transduction by Anc80 relative to empty vector control (Figure 2C). KO of GPR108 in H1 HeLa cells causes highly reduced permissivity, an effect that can be rescued by stable re-introduction of GPR108 using a lentiviral vector (Figure 2D). These data indicate that GPR108 is both required and sufficient in HeLa and Huh7 cells for most of the primate AAVs, except for AAV5, which is unaffected in WT, KO, or rescue conditions (Figure 2).
Figure 2.

GPR108 Is a Highly Conserved AAV Entry Factor
(A) Phylogenetic tree of extant and putative evolutionary intermediate AAV serotypes, color-coded for AAVR and GPR108 dependence. Red, AAVR and GPR108; green, GPR108 only; blue, AAVR only. (B) Transduction of indicated AAV serotypes in WT or GPR108 KO Huh7 cells at 10,000 VG/cell with hAd5 helper virus. CMV.Luciferase.SVPA (AAVrh10, AAV8, AAVAnc82, AAV9, AAVAnc81, AAVAnc80, AAV3, AAV6.2, AAV1, AAVrh32.33, AAV4, AAV5) or CMV.eGFP.T2A.Luciferase.SVPA (AAVAnc83, AAVAnc110, AAV2) transgene. (C) Huh7 WT or GPR108 KO cells transfected with FLAG-tagged human GPR107 or GPR108 followed by transduction of the indicated serotype in the presence of hAd5 helper virus. 10,000 VG/cell CMV.Luciferase.SVPA transgene. (D) H1 HeLa cells deleted for GPR108, then stably rescued with GPR108 lentivirus, followed by transduction of the indicated serotypes at 10,000 VG/cell with hAd5 helper virus. CMV.Luciferase.SVPA transgene. All in vitro transductions are shown as SE measurement from minimum of three independent experiments, done in duplicate. Background is maximum observed value from untransduced control cells.
GPR108 Dependency of the Capsid Is Transferable and Requires a Motif on the VP1 Unique Domain
In order to further understand the function of GPR108 for entry and how it engages the capsid, we used chimeric capsids to identify the capsid domain that dictates GPR108 usage. Because AAV5 and AAV2 differ in their GPR108 usage but both require AAVR, we generated chimeras of these two serotypes (Figure 3A). Previous studies have shown that the structurally flexible VP1 and VP1/2 unique domains (VP1u and VP1/2u, respectively) can be swapped between serotypes,31 despite the structural constraints of the icosahedral capsid. We generated VP1u reciprocal chimeras between AAV2 and AAV5, and volume-matched crude viral preparations were used to transduce Huh7 WT, AAVR KO, GPR108 KO, or double-KO cells. As AAV2 and AAV5 both require AAVR, we observed the expected loss of transduction for all tested serotypes in the AAVR KO and double-KO cell lines (Figure 3B). Interestingly, the chimera containing the VP1 unique region of AAV5 (AAV5.2) was able to transduce GPR108 KO Huh7 cells to a similar level as WT cells, while the chimera containing AAV2 VP1 (AAV2.5) was unable to transduce GPR108 KO cells. GPR108 dependency on the AAV5 VP1u region was confirmed using equal titer transduction of iodixanol-purified AAV preparations in both primary GPR108 KO mouse embryonic fibroblast (MEF) cells (Figure 3C) as well as Huh7 KO cells lines in the presence and absence of hAd5 helper virus (Figure S2C), and functionality of the AAV chimeras was demonstrated by bioluminescence after 1011 genome copies (GC)/mouse was injected retro-orbitally into WT C57BL/6 mice (Figure 3D). These results demonstrate that the VP1u region of AAV dictates GPR108 usage, and that this cellular functionality is transferrable to other AAV serotypes.
Figure 3.

GPR108 Dependence Is Dictated by the VP1 Unique Region of Capsid and Is Transferrable to Other Serotypes
(A) Design of chimeric capsids used to determine GPR108 usage domain with amino acid number labeled above. vPLA, viral phospholipase domain. (B) Transduction of Huh7 WT, AAVR KO, GPR108 KO, or double-KO cells with the indicated WT or chimeric capsids (100 μL of crude vector preparation, plus hAd5 helper virus, CMV.eGFP.T2A.Luciferase transgene). (C) Transduction of MEF cells derived from WT or GPR108 KO C57BL/6 mice transduced with 10,000 VG/cell iodixanol-purified WT or chimeric capsids in the presence of hAd5 helper virus, CMV.Luciferase.SVPA transgene. (D) Bioluminescence quantification of iodixanol-purified CMV.Luciferase.SVPA packaged WT or chimeric capsids injected in WT C57BL/6 mice 7 days post-injection. n = 5 mice per group; ROI for quantification is the whole mouse.
Golgi-Localized Entry Factors AAVR and GPR108 Facilitate Entry at a Step Prior to Nuclear Import
As the VP1u region is shielded within the capsid prior to cellular internalization, we investigated whether GPR108 plays any role in attachment at the cell surface. We used a previously published cell-binding assay, which quantifies cell-bound viral genomes of different serotypes using qPCR,21 and quantified three different vectors with different entry factor requirements (Anc80, rh32.33, and AAV5) in WT, AAVR KO, GPR108 KO, or AAVR GPR108 double-KO cells. We observed no difference in the number of bound viral genomes per cell in any of the KO cell lines for any vector tested, yet we were able to detect differences in the number of bound viral genomes for different serotypes (Figure 4A). These data indicate that in the tested setting neither GPR108 nor AAVR is a major contributor in cellular attachment and consequently exert their role downstream of attachment as bona fide entry factors.
Figure 4.

Loss of AAV Entry Factors Causes an Entry Defect Upstream of Nuclear Import
(A) qPCR binding assay for cell-bound viral genomes in Huh7 WT, AAVR KO, GPR108 KO, or double-KO cells, assessed for the indicated capsid serotype after 1 h of binding on ice. Average of three independent experiments; error bars = SEM. (B) Western blot of cytoplasmic, membrane, or nuclear subcellular fractions from AAV entry/nuclear import assay (tubulin [α-tubulin], AIF [apoptosis-inducing factor 1], or H3 [histone H3]). (C) Percentage of total Anc80 (CMV.eGFP.WPRE) genomes quantified by ddPCR (CMV primer/probe) on subcellular fractions isolated from WT, AAVR KO, GPR108 KO, or AAVR KO GPR108 KO cells 48 h post-transduction at 10 VG/cell in 1 million cells. One representative experiment; error bars = SEM of three technical replicates. (D) Total number of recovered genomes from entry assay represented in (C). (E) Indirect immunofluorescence of FLAG-tagged GPR107 or GPR108 with anti-FLAG M2 primary antibody (green) or anti-TGN46 antibody as a trans-Golgi network marker (red) and DAPI staining of cell nuclei (blue).
We next aimed to determine where the entry defect exists in these cell lines by using Anc80, a vector dependent on both AAVR and GPR108 for transduction. Due to its high infectivity in Huh7, transduction with 10 GC/cell for 48 h allowed for a sensitive droplet digital PCR (ddPCR) readout of subcellular fractions in non-saturating conditions. Cytoplasmic, membrane, or nuclear fractions were cleanly separated, as determined by western blot for tubulin, apoptosis-inducing factor 1 (AIF), and histone H3, respectively (Figure 4B). In WT cells we were able to detect that between 60% and 70% of the total genome copies were present in the nucleus (Figure 4C), consistent with prior publications using a similar entry assay.32,33 A decrease in the percentage of nuclear genome copies is observed in AAVR KO (46.9%) and GPR108 KO (52.2%) cells, with a further decrease in the double AAVR-KO GPR108-KO (27%) cells. Interestingly, there is a roughly 10%–15% increase in membrane-associated genomes in all three of the KO cell lines, suggesting that the entry defect is not due to a loss of membrane attachment. Importantly, note that overall genome copies are decreased in all KO cell lines (Figure 4D), suggesting that in the absence of their specific entry pathway requirements the AAV particles or genomes may be degraded. While AAV genomes can still be detected in the nuclear fraction of KO cells, the overall decrease in intracellular genomes demonstrates that almost 20-fold fewer genomes make it into the nucleus of cells lacking AAVR and GPR108. These data suggest that both AAVR and GPR108 act either at the stage of, or upstream of, nuclear import. To determine the subcellular localization of GPR108 in relationship to the localization of AAVR, we designed FLAG-tagged GPR107 or GPR108 constructs containing a 3× glycine-alanine linker at the C terminus and subcloned these into pcDNA3.1(−). These constructs were transfected into Huh7 cells, and indirect immunofluorescence was used to determine its subcellular localization. As a control, we confirmed previous reports that GPR107 localizes to the TGN as shown by co-localization with TGN46 (Figure 4E).34 Co-localization patterns for GPR108 were shown to be comparable to GPR107 in the GPR108-FLAG-transfected cells, indicating that GPR108 is also primarily localized to the TGN (Figure 4E). The Golgi localization of GPR108 is consistent with a mode of action for entry that is at or upstream of the observed nuclear import defect.
GPR108 N- and C-Terminal Domains Are Required for Its Function as an AAV Entry Factor
Given the similar proposed domain structure and subcellular localization of GPR108 and GPR107, we made chimeras of these proteins to map which domains are functionally required for AAV entry. GPR107 and GPR108 are both relatively uncharacterized proteins predicted to have TM domains, with a large luminal N terminus and short cytoplasmic C terminus (Figure 5A). Chimeric constructs in which the N-terminal, TM, or C-terminal domains were swapped between GPR107 and GPR108, individually or in combination, were expressed in Huh7 cells and transduced with Anc80-expressing luciferase. Western blot detection in a cellular membrane protein fraction illustrated that all chimeric constructs were expressed, although expression levels and protein cleavage patterns appear to differ across constructs (Figure 5C). All constructs containing the GPR107 N-terminal domain demonstrate a lower molecular weight protein roughly 35 kDa in size corresponding to the TM and C-terminal domains (yellow arrows). Conversely, constructs containing the GPR108 N-terminal domain are observed at the molecular weight of the expected full-length GPR108 (no furin cleavage, black arrows), or in larger molecular weight isoforms, which are expected to be glycosylated or post-translationally modified isoforms (gray arrows) or protein aggregates (red arrows) due to the highly hydrophobic nature of the GPR108 N-terminal and TM domains. In this study, protein expression level does not correlate with the level of rescue, as GPR108 shows the highest level of rescue despite being expressed at lower levels than other constructs such as 108.107.107, and 108.108.107. This observation suggests that the observed rescue is not due to how much of the protein is present, but further understanding of the amount of GPR108 expression required for AAV entry warrants further characterization. The four constructs that partially rescued AAV transduction (demonstrating transduction levels between that of full-length GPR107 and GPR108) contained either the GPR108 N- or C-terminal domains (Figure 5B). Only the chimera comprised of the GPR107 N- and C-terminal domains with the GPR108 TM portion did not rescue AAV entry. Conversely, the construct containing N- and C-terminal domains of GPR108 and the GPR107 TM portion rescued transduction to similar levels as full-length GPR108. This suggests that the GPR107 TM domain is similarly functional to the GPR108 TM domain in an AAV entry factor context. Notably, all chimeras co-localized with TGN46, similarly to the full-length GPR107 and GPR108 controls, suggesting that the observed entry deficits are not due to protein mislocalization (Figure 5C). Overexpression of either GPR107 or GPR108 shows disruption of specific membrane staining of TGN46 (Figure S3), and as such we are unable to accurately quantify co-localization in this system. These data show that both the N- and C-terminal domains of GPR108 are important for optimal AAV entry, and defects of a single domain give a partial functional loss.
Figure 5.

GPR108 N- and C-Terminal Domains Are Required for AAV Entry
(A) Predicted membrane topology of nonfunctional GPR107, with indicated disulfide bond location, furin cleavage site, alanine-glycine linker, and functional GPR108. (B) Rescue of Anc80 transduction by transient transfection with FLAG-tagged GPR107/GPR108 chimeric proteins or transfected empty vector (EV). Cells transduced with 10,000 VG/cell, CMV.Luciferase.SVPA transgene after hAd5 helper virus co-infection. (C) Corresponding anti-FLAG western blot from cellular membrane fractions demonstrating protein expression in cells transduced in (B). Black arrows point to expected full-length protein; gray arrows point to expected glycosylation states; red arrows point to protein aggregates; and yellow arrows point to expected N-terminal domain cleavage product. AIF (apoptosis-inducing factor 1) was used as a membrane protein loading control. (D) Indirect immunofluorescence of chimeric proteins using anti-FLAG antibody as described in Figure 4E.
GPR108 AAV Usage Is Conserved in Mice and Humans
As multiple animal models are used for AAV-based gene therapies, it is important to understand how species-specific variation of AAV entry factors may influence AAV entry of different capsid variants. We re-introduced expression constructs with FLAG-tagged mouse cDNAs of GPR108 or GPR10735 into Huh7 GPR108 KO cells, and, as with their human homologs, we observed rescue with mGPR108 but not mGPR107 (Figure 6A). This indicates that both human and murine GPR108 are capable of mediating AAV entry. Anti-FLAG western blot demonstrates expression of both human and mouse GPR107 and GPR108 in Huh7 cells (Figure 6B). The human GPR107 is known to contain a furin cleavage site in its N-terminal domain,34 resulting in detection of a 34-kDa C-terminally-tagged fragment (Figure 3B), a fragment that is also observed for mouse GPR107. GPR108 migrates at the expected molecular weight of the full-length protein, suggesting that GPR108 does not contain an analogous cleavage site in its N-terminal domain. Additionally, distinct bands at higher molecular weights are observed for both human and mouse GPR108, suggesting that both human and mouse GPR108 may undergo post-translational modification (Figure 6B).
Figure 6.

GPR108 Usage Is Conserved in Mice
(A) Transduction of Anc80, AAV9, AAV9.PHP-B, rh32.33, AAV4, and AAV5 in Huh7 WT or GPR108 KO cells transfected with FLAG-tagged mouse GPR107 or GPR108 (10,000 VG/cell CMV.Luciferase.SVPA transgene plus hAd5 helper virus). (B) Anti-FLAG western blot of cell membrane fraction of human or mouse GPR107- or GPR108-transfected or empty vector (EV)-transfected Huh7 cells. (C) WT and GPR108 KO Hepa cells transduced with AAV9, AAV9.PHP-B, rh32.33, AAV4, and AAV5 (10,000 VG/cell CMV.Luciferase.SVPA transgene).
Because murine GPR108 is functional in human cells, we interrogated whether it was necessary for AAV entry in the murine hepatoma-derived Hepa cell line. AAV5 transduces WT Hepa cells well, while some other serotypes such as rh32.33 and AAV4 are less efficient (Figure 6C). Similar to the human cell lines tested, we observed that all AAVs other than AAV5 demonstrated a loss in transduction of approximately 100-fold in the absence of mGPR108. Interestingly, we also tested AAV9 and its peptide insertion variant, AAV9.PHP-B, which is reported to have the unique ability to cross the blood-brain barrier into the brain of C57BL/6 mice upon systemic injection.36,37 While AAV9.PHP-B was able to transduce WT Hepa cells (derived from C57BL/6 mice) to a high level, GPR108 deletion caused a greater than 100-fold decrease in transduction (Figure 6C). This clearly demonstrates that the unique targeting property of AAV9.PHP.B is not due to altered GPR108 usage. The functionality of mGPR108 in a human context and AAV’s requirement for GPR108 in mouse cells indicate that GPR108 usage is conserved between species that are permissive to AAV transduction, an important implication for the development of AAV-based gene therapy models.
Most Engineered Capsid Variants Require GPR108
After determining that the peptide insertion variant AAV9-PHP.B required GPR108 in mouse cells, we wanted to assess the GPR108 requirement in other peptide insertion and other engineered capsid variants. We tested three peptide insertion variants (Figure S4A) whose parental capsid serotype we previously demonstrated to require GPR108 (Figure 2B) in human (Huh7) GPR108 KO cells and all showed a loss of transduction in GPR108 KO cells (Figure S4B), demonstrating that the unique cellular tropism observed by these variants is not due to altered GPR108 requirement. Additionally we examined a rationally designed AAV2 variant, AAV2 HSPG,38 which is unable to bind its attachment glycan heparan sulfate proteoglycan (HSPG). The HSPG variant had overall decreased transduction in all cell lines, yet transduction of the GPR108 KO cells were 10- to 100-fold decreased compared to their WT counterpart (Figure S4C), suggesting that GPR108 usage is independent of glycan attachment.
GPR108 Is Required in Primary Cells and In Vivo
Although several cellular proteins have been described to influence AAV transduction in immortalized cell lines,14, 15, 16, 17, 18, 19,39,40 thus far only the AAV receptor, AAVR, has been shown to dictate AAV entry in primary cells and in a KO mouse model.20 Additionally, these cellular factors have been determined to be specific for only one or a small subset of AAVs. We therefore tested a large capsid panel in MEF primary cells derived from either WT or Gpr108 KO mice.23 All tested serotypes other than AAV5 required GPR108, as shown by a 10- to 100-fold decrease in transduction in Gpr108 KO cells (Figure 7A). We next assessed the requirement for GPR108 in vivo using bioluminescence after systemic injection of CMV.luciferase.SVPA packaged AAV8, rh32.33, or AAV5 control in a Gpr108 KO mouse (Figure 7B). Quantification of bioluminescence demonstrates an average of 10-fold (AAV8) or 100-fold (rh32.33) decrease in transduction at all measured time points in the Gpr108 KO mouse compared to the WT mouse (Figure 7C). Although there is not a complete loss of transduction by AAV8, a 10-fold decrease demonstrates that this is still a major factor influencing AAV8 transduction in vivo. This decrease in transduction is specific to GPR108-independent serotypes, as AAV5 shows similar or slightly higher transduction in the Gpr108 KO mouse compared to the WT mouse. These data demonstrate the highly conserved nature of GPR108 usage both in vitro and in vivo.
Figure 7.

GPR108 Is Required for Transduction in Primary Cells and In Vivo
(A) Mouse embryonic fibroblast (MEF) cells derived from WT or GPR108 KO mice transduced with the indicated serotype. The same transduction protocol was used as described in Figure 2B. (B) In vivo bioluminescence of WT or GPR108 KO mice injected retro-orbitally with 1011 VG/mouse of CMV.Luciferase.SVPA packaged AAV8, rh32.33, or AAV5 at 14 days post-injection. (C) Quantification of bioluminescence at 7, 14, 28, or 56 days post-injection from mice in (B). n = 3 mice per group; uninjected is maximum observed luminescence from uninjected mice across all time points. Error bars represent SEM.
Discussion
While AAV continues to gain traction as a viral vector platform for gene therapy, its mechanism of action remains poorly understood. This poor mechanistic basis limits the control and understanding of safety, targeting, and other properties of this novel class of drugs. The glycan specificity of various AAVs, the recent identification of AAVR, and the variable AAVR dependencies of AAV variants20,21 provide a first set of data toward what makes up the complex tropism of AAV, particularly in vivo. Identification of GPR108 as a second highly conserved AAV entry factor further enhances our understanding of AAV biology.
Since most primate AAVs require both AAVR and GPR108 for transduction, and the fact that the sum of the quantitative effect of knockdown in vitro and in vivo on infectivity is greater than 100%, both proteins appear part of a common AAV transduction pathway. The natural diversity within AAV provides us with AAV5 (GPR108-independent, yet AAVR-dependent) and AAV4 or rh32.33 (GPR108-dependent, yet AAVR-independent), demonstrating that GPR108 acts independently in this pathway, without any evidence of cooperativity. These data support a model in which AAVR acts upstream or downstream of GPR108, possibly through a direct hand-off mechanism or through as yet undefined co-factors. Figure 8 illustrates our proposed model for primate AAV entry of different AAV variants. While subject to validation in further mechanistic studies, this model is supported by AAV entry factor transduction, subcellular localization, and other molecular datasets reported herein, and in previous reports. Briefly, the primary pathway used by most AAVs (red) requires AAVR and GPR108, with GPR108 here being proposed as a downstream factor to AAVR given (1) the data supporting a (modest) localization of AAVR on the cell surface,20 (2) the late endosome and TGN localization of GPR108 (Figure 4E), and (3) the location of a required domain for GPR108 activity on the VP1u region (Figure 3), which is only extruded in the acidic endosome. A small subset of AAVs (green) use an alternate receptor in place of AAVR, which has yet to be identified (Figure 8B). A single known AAV serotype, AAV5 (blue), uses an alternate entry factor in place of the highly conserved GPR108 (Figure 8C). More in-depth studies are required to confirm the temporal relationship of GPR108 and AAVR and which other co-factors are required for entry and post-entry.
Figure 8.

Summary of Capsid Variant Entry Factor Requirements and Model
(A) Three different entry pathway classes exist in which capsids require both AAVR and GPR108 (red), AAVR only (blue), or GPR108 only (green). (B) Green capsids use an as-of-yet unidentified receptor or receptor complex in place of AAVR, dictated by the VP3 outer surface of intact capsids, while blue and red capsids both require AAVR (purple). (C) Blue capsids use an as-of-yet unidentified entry factor in place of GPR108, dictated by the internalized VP1-unique N terminus of capsid, while red and green capsids require GPR108 (brown).
The intracellular localization of both AAVR and GPR108 at the Golgi at steady-state levels highlights the importance of this subcellular compartment for AAV entry and suggests that AAV may be shielded in an endosome/lysosome until deep within the endomembrane system, and not released into the cytoplasm after attachment. It has been shown that functional transduction of multiple AAV serotypes requires retrograde transport to the TGN,41 and AAVR chimeras have demonstrated that AAVR trafficking to the TGN is necessary for AAV entry.20 The identification and characterization of GPR108 as a TGN protein (Figure 4E) highlight the pivotal importance of the Golgi for AAV entry, as all AAVs tested to date require either AAVR, GPR108, or both AAVR and GPR108 for transduction.
Multiple studies have demonstrated direct AAVR binding to intact capsid,20,24 and recent publications indicate that AAV2-AAVR co-crystal structures present a binding domain on the outer surface,42,43 within the VP3 portion. This suggests that AAVR binds the intact capsid and traffics to the TGN, at which point endosomal acidification triggers a conformational change in capsid and extrusion of the VP1/2 unique region of capsid,44 then allowing engagement of GPR108 via the VP1u region. It has been demonstrated that VP1 is internalized prior to entry both in cell lines, using a VP1-specific antibody for immunofluorescence, and in vitro, using dot-blot assays after heat treatment of the capsid,22 and this conformational change is required prior to nuclear entry, as microinjection of AAV to the nucleus does not allow productive infection.44 A major known functional activity of the VP1 unique domain is its phospholipase activity,45 suggesting that GPR108 may facilitate endosomal escape in some way, potentially by stabilizing the extruded VP1u domain or by generating a pore in the endosomal membrane through which the genome can be extruded. Lipid products of phosphatidylcholine, which is cleaved by the AAV2 phospholipase domain,45 have been shown to play a role in post-Golgi transport. Importantly, genome-containing capsids can assemble from VP3 only and can traffic to the TGN, yet they are completely defective for entry in the absence of VP1/2.45,46 There have been reports of basic repeat regions within the VP1/2 region of capsid that are required for transduction and nuclear import,47,48 and it is of interest to determine the effect that mutagenesis of these regions may have on VP1 engagement of GPR108. It is additionally of interest to identify the cellular factor that AAV5 uses in place of GPR108 for this entry step. Because the VP1 requirement for entry and nuclear import functions is conserved within other parvoviruses,49,50 the highly divergent AAV5 may use similar cellular factors to other parvoviruses for these entry functions.
Our understanding of GPR108 in the cell is limited, yet our studies shed some light on its mechanistic role in AAV entry. The lack of major differences in cellular attachment of AAV to GPR108 KO cells (Figure 4A) suggests that the role of GPR108 is internal, and as such is a bona fide entry factor. Indeed, a decrease in intracellular AAV genomes (Figure 4D) and a decrease in the proportion of nuclear genomes (Figure 4C) in AAVR KO or GPR108 KO cells demonstrate that AAVR and GPR108 act upstream of nuclear import. We therefore propose a novel multifactor entry mechanism in which most AAVs bind AAVR and require it for proper trafficking, followed by a requirement of GPR108 for endosomal escape at the TGN (Figure 8).
It is curious that both the N and C termini of GPR108 are required for its function but that the TM domain can be exchanged with GPR107 and remain fully functional (Figure 5B). While the sequence is more highly conserved within this domain than the N- or C-terminal domains, only 72% of sequence identity is conserved between the TM domain of GPR107 and GPR108 (compared to 50% and 40% sequence identity for N- and C-terminal domains, respectively). This suggests that although the sequences are fairly unique, the TM domains may have a similar function in a cellular context that can be facilitated through either the GPR107 or GPR108 TM domain. Recent reports demonstrate that a single cytoplasmic loop within the GPR108 TM domain determines its NF-κB activation activity,23 so it is curious that the entire TM domain is exchangeable between GPR107 and GPR108 (Figure 5B). This suggests that the TM domains of these two proteins have redundant cellular functions or that GPR108 functions as an AAV entry factor that is completely independent of its function as an NF-κB-related protein, and further investigation of GPR108 functional domains will be informative in teasing apart these possibilities. It is unclear whether this poorly characterized class of G protein-coupled receptor-like proteins possesses any signaling functions, yet this and other studies on the role of GPR107 in endocytosis,51 in particular of Pseudomonas aeruginosa exotoxin A34 and ricin,52 suggest that these proteins may play a role in retrograde transport of specific cargo and that the N- and C-terminal domains appear to dictate this specificity. The presence of N-terminal domain furin cleavage sites and potential post-translational modifications (Figures 5C and 6B) may influence these cellular functionalities.
Historically, animal models have been used to predict utility of AAV in humans. While mice and other large animal models are AAV permissive, questions remain whether the cellular co-factors are the same, and/or whether their engagement between species is equally efficient. Here, a conserved functionality of mouse GPR108 in human cells (Figure 6A) and, reciprocally, the GPR108 requirement in mouse cells was demonstrated (Figure 6C). While GPR108 KO causes a 10- to 100-fold decrease in transduction of all tested serotypes (except AAV5), some highly transducing serotypes are still able to achieve appreciable levels of luciferase expression in a GPR108 KO context. This can be seen for highly transducing serotypes AAV2 and AAV3 in both human-derived Huh7 cells (Figure 2B) as well as mouse-derived MEF cells (Figure 7A). However, some serotypes, such as AAV6.2, can specifically transduce human GPR108 KO cells (although still 100-fold less than WT levels) (Figure 2B), and AAV4 can specifically transduce GPR108 KO MEF cells (Figure 7A). This suggests that some serotypes may be able to use alternate entry factors at a high MOI in certain cell types, as has been described previously in AAVR KO cells,21 yet it is unclear whether this alternate entry is facilitated by AAV serotype-specific proteins. Further investigation into cellular determinants of AAV entry and efficiency in different species may help explain cellular tropism differences and may be relevant to how doses translate across species to inform the eventual human application.
GPR107 KO is embryonic lethal,51 yet a viable GPR108 KO mouse was recently generated.23 Both tested GPR108-dependent serotypes demonstrate a significant decrease in overall transduction, as shown by bioluminescent imaging after systemic injection (Figures 7B and 7C). In vivo rh32.33 appears to be more highly dependent on GPR108 than AAV8, as rh32.33 transduction is decreased to background levels and AAV8 transduction is only decreased 10-fold. These data are in line with this and previous reports on AAVR,21 that when forced, some AAVs may be able to enter cells, although inefficiently, through alternative pathways and/or co-factors that in vitro studies to date have been unable to define.
Our studies suggest a revised understanding of AAV entry: previous models proposed that AAV is released into the cytosol early in the endolysosomal system after cellular attachment, followed by karyopherin-specific import of the capsid through a nuclear pore complex via basic repeat regions in VP1/2.48,53 Recent studies suggest instead that the AAV particle is shielded until deep within the endocytic pathway, maintaining its status within a membrane-bound cellular compartment until reaching the TGN, and potentially gaining access to the nucleus through this subcellular compartment. This hypothesis is consistent with previous reports of perinuclear accumulation of AAV virions during the AAV entry process.41,45,54 If this suggested entry mechanism is correct, it may help explain the unique lack of innate immune activation from AAVs that would be expected of other viruses that undergo endosomal escape earlier in the endosomal system and cause activation of cytoplasmic DNA sensors.
The in vivo tropism of AAV serotypes is highly divergent and provides an opportunity for gene therapy applications that aim to match the appropriate targeting properties with the requirements for the indication and therapeutic approach. These studies provide the opportunity to characterize and consider novel AAVs with improved targeting. Remarkably, little is known what molecular host determinants and which structures on AAV drive tropism, limiting our ability to control or engineer its properties or to determine the conservation (i.e., translatability) across species of the novel properties introduced on an AAV. Here, we augment the body of work that aims to address this complex tropism question, by adding a second proteinaceous entry factor, GPR108, to our understanding of AAV transduction. Through exploration and characterization of the primate AAV diversity, we identified varying dependencies on AAVR and/or GPR108, which help shape an updated model of AAV entry and transduction biology.
Materials and Methods
Cell Culture and Transfection
All cell lines were maintained in DMEM (Corning) supplemented with 10% fetal bovine serum (FBS; GE Healthcare) and 100 IU/mL penicillin/streptomycin (Corning) in a humidified incubator with 5% CO2 at 37°C. Huh7 WT, Huh7 AAVR KO, and H1 HeLa cell lines were previously published.20 Hepa cells were acquired from ATCC. WT and Gpr108 KO MEF cells were generated and cultured as previously described.23. Cells were transfected using PolyJet In Vitro DNA Transfection Reagent (SignaGen, catalog no. SL100688) using the standard protocol. Expression of FLAG-tagged constructs was determined by western blotting of cellular membrane fractions.
Cloning and Plasmid Constructs
FLAG-tagged GPR107 and GPR108 constructs containing flanking NotI and BamHI restriction sites were synthesized by Genewiz, followed by restriction enzyme subcloning into pcDNA3.1(−) plasmid using NotI and BamHI (NEB) restriction sites. GenBank sequences used for synthesis were as follows: mouse GPR107, BAC26961; mouse GPR108, NP_084360; human GPR107, AAK57695; human GPR108, XP_290854. Capsid chimeras were generated from AAV2 and AAV5 nucleotide sequences at the VP1 junction as demonstrated in Excoffon et al.55 Capsid chimeras were synthesized by Genewiz and subcloned into pAAVector2 using HindIII and SpeI restriction sites. Lentivial plasmids were purchased from Addgene or Sigma. LentiCas9-blast (52962), psPAX2 (12260), pCMV-VSV-G (8454), and GeCKO V2A and GeCKO V2B (1000000048 and 1000000049) were purchased from Addgene. Individual sgRNA lentivirus constructs targeting an individual gene used for screen validation and KO experiments were purchased from Sigma as QuickPick glycerol stock clones in the Sigma LV04 vector backbone.
Protein Extraction and Western Blot
For stability of GPR proteins in solution, cellular membrane fractions were isolated using a Cell Signaling Technology cell fractionation kit (catalog no. 9038). Membrane fractions corresponding to 100,000 total cells per sample per well were boiled for 2 min at 90°C in Laemmli buffer with β-mercaptoethanol, run on 4%–12% NuPage Bis-Tris gels, and transferred to a polyvinylidene fluoride (PVDF) membrane. FLAG-tagged proteins were detected using mouse anti-FLAG M2 primary antibody (Sigma, catalog no. F3165), and membrane fraction loading control AIF was detected using rabbit anti-AIF (Cell Signaling Technology, catalog no. 5318). Western blot subcellular fraction purity was analyzed using mouse anti-tubulin (Abcam ab7291), rabbit anti-AIF (Cell Signaling Technology, catalog no. 5318S), or rabbit anti-histone H3 (Cell Signaling Technology, catalog no. 4499S).
Lentivirus Production
Lentivirus was produced from HEK293T cells (ATCC, Manassas, VA, USA) by transient transfection using PolyJet In Vitro DNA Transfection Reagent (SignaGen, catalog no. SL100688) using the manufacturer’s protocol for lentiviral production. LentiCas9-blast and individual sgRNA-containing lentiviruses were produced in HEK293T cells seeded overnight at 4 × 106 cells per 10-cm dish. 1 h prior to transfection, medium was changed to fresh pre-warmed D10, followed by transfection of psPAX2, pLentiCas9-Blast or LV04, and pCMV-VSV-G at a 10:10:1 ratio. Medium was changed to fresh D10 6 h after transfection, and supernatant virus was harvested 48 h later, clarified by centrifugation at 2,000 rpm for 5 min in a Sorvall tabletop centrifuge, and filtered through a 0.45-μm filter. Large-scale GeCKO lentivirus was produced as previously described.56 Briefly, V2A and V2B were produced as individual lentiviral library preparations using a large-scale transfection of the protocol described above, in Corning HYPERflask culture vessels. Supernatant virus was collected at day 2 and day 3 post-transfection, filtered through a 0.45-μm filter, and concentrated by ultracentrifugation at 24,000 rpm for 2 h at 4°C in a SW-28 rotor. Concentrated lentiCRISPR library was titered on Huh7 AAVR KO Cas9 cells by determining % transduced cell survival after 2 days of puromycin selection, relative to untransduced control cells in the absence of puromycin.
Generation of Stable Cell Lines
Cell lines were seeded at 1 × 106 cells per well of a six-well plate the night prior to transduction. Cells were transduced by spinfection for 30 min at 25°C and 2,500 rpm in a tabletop centrifuge using 1 mL per well of supernatant lentivirus in the presence of 8 μg/μL Polybrene (Thermo Fisher Scientific, catalog no. TR1003G). Medium was changed to fresh D10 following spinfection, and 1 day later stably transduced cells were selected using 5 μg/μL puromycin (Sigma-Aldrich, catalog no. P9620) for 2 days.
AAV Production and Purification
High-titer vectors were produced, purified, and titrated by the MEEI/SERI Gene Transfer Vector Core (https://www.vdb-lab.org/vector-core/). Large-scale vector preparations were generated by polyethylenimine (Polysciences, catalog no. 24765-2) triple transfection of pAdDeltaF6, a gift from James M. Wilson (Addgene plasmid no. 112867), pAAVector2[Cap], and pCMV.Luciferase.SVPA, pCMV.eGFP.T2A.Luciferase, or pCMV.eGFP.WPRE.bGH transgenes in a 2:1:1 ratio. 520 μg of total DNA was transfected in 10-layer HYPERFlasks using a PEI Max/DNA ratio of 1.375:1 (w/w). 3 days after transfection, vectors were concentrated by tangential flow filtration and purified by iodixanol gradient ultracentrifugation as previously described.57 Chimeric and point mutant viral vectors were produced on a smaller scale as crude viral preparations by the same transfection method in 10-cm cell culture plates. Three days after transfection, cells and supernatant were collected, subjected to three freeze-thaw cycles, and then crude virus preparation was clarified by centrifugation for 10 min at 10,000 rpm in a Thermo Scientific FIBERLite F15-8 × 50cy rotor at 4°C.
AAV Vector Titration
DNase1-resistant viral genomes of iodixanol-purified vector preparations were quantified by TaqMan qPCR (Thermo Fisher, catalog no. 4304449) using primer/probe set detecting CMV promoter. Vector purity was assessed by SDS-PAGE electrophoresis.
AAV Transduction
All luciferase transduction assays were done by seeding 10,000 cells per well in black-bottom 96-well plates overnight. When indicated, cells were pre-incubated with 200 plaque-forming units (PFU)/cell of WT hAd5 (University of Pennsylvania Vector Core) in D10 for 2 h, and then hAd5-containing medium was removed prior to transduction. Cells were transduced with the indicated AAV variant either at 1 × 104 VG/cell in 50 μL of serum-free DMEM (iodixanol-purified vectors) for 1 h at 37°C, after which D10 was added to a total volume of 200 μL, or 100 μL per well of crude virus preparation (Figure 3B only) that was added for 1 h at 37°C, removed, and then D10 was added. Transduction levels were analyzed by luciferase assay 48 h post-transduction.
Luciferase Assays
2 days post-transduction, cell culture medium was removed and cells were lysed in 20 μL per well of 1× Reporter Lysis Buffer (Promega), then frozen at −80°C. After thawing, firefly luciferase (ffLuc) expression was measured in relative light units/s on a Synergy H1 hybrid multi-mode microplate reader using 100 μL of luciferin buffer (200 mM Tris [pH 8], 10 mM MgCl2, 300 μM ATP, 1× firefly luciferase signal enhancer [Thermo Fisher, catalog no. 16180], and 150 μg/mL d-luciferin).
Entry Screen
Huh7 AAVR KO Cas9 cells were transduced with concentrated V2A or V2B lentivirus at an MOI of 0.3 in six-well plates by spinfection as described above for 30 min at 25°C with 8 μg/μL Polybrene, followed by incubation at 37°C and 5% CO2 for 1.5 h, after which fresh D10 media were added. Puromycin was added at a concentration of 5 μg/μL 24 h post-transduction to select sgRNA-expressing cells. Cells were cultured with puromycin for 1 week to carry out selection and allow editing to occur before selection with AAV. 30 million cells from each half of the mutagenized library (V2A and V2B cells) were transduced with 100,000 VG/cell rh32.33CMV.eGFP.WPRE. Cells were transduced in a total volume of 10 mL serum-free DMEM in each of two 15-cm plates for 1 h followed by addition of 10 mL of DMEM/20% FBS, and cells were split the following day. Three days post-transduction cells were collected for FACS sorting by trypsinization, spun in a table-top centrifuge at 2,000 rpm for 5 min, and then resuspended in PBS (without calcium and magnesium) with 5 mM EDTA. FACS sorting was done at the Massachusetts General Hospital Flow Cytometry Core (Simches Research Building) on a BD FACSAria Fusion cell sorter instrument. Cells were collected into DMEM supplemented with 20% FBS and penicillin/streptomycin. Selected cells were expanded and genomic DNA was extracted from a total of 107 cells per sample. GFP-negative cells from each half of the library were split in half and either sequenced or subjected to a second transduction and FACS sort using the same transduction protocol.
Genome Extraction, Illumina Barcode Addition, and Deep Sequencing
Genomic DNA from control (unselected) or selected cells was extracted using a QIAGEN blood and cell culture DNA midi kit (catalog no. 13343). Barcode addition and Illumina adaptor addition were carried out as previously described.56 Briefly, a two-step PCR was carried out using sample-specific primers to specifically amplify sgRNA sequence and distinguish samples during multiplexed sequencing on an Illumina MiSeq machine as described.56
Next-Generation Sequencing Data Analysis
Sequencing data were first processed with Cutadapt to remove adaptor sequences58 and subsequently analyzed through the MAGeCK software package, version 0.5.7.59 First, trimmed fastq files from both halves of all experimental and control libraries were converted to tables of sgRNA counts through the MAGeCK count command, using the non-targeting sgRNA controls to normalize the counts. Next, the normalized data from the two halves of the library were combined into a single summary file for further analysis. We then tested for enrichment of sgRNAs in the experimentally selected samples relative to unselected controls through the robust ranking algorithm (RRA) as implemented in MAGeCK. sgRNAs with counts of zero were removed prior to statistical analysis.
Entry Screen Analysis
After sequencing, raw reads were mapped to known sgRNA sequences using the MAGeCK analysis pipeline. Significance values were determined for the entire library after normalization to control population within each half of the library (V2A and V2B), and data is reported as raw p value without multiple test correction.
Visualization
Analyzed data were visualized through an in-house script written in Python through use of the Matplotlib visualization library.60 For the purposes of visualization only, genes were sorted by gene ontology terms.61, 62, 63 The hits (by p value) were then plotted according to their uncorrected p value along the y axis and were arbitrarily scattered within their categories along the x axis. The size of the dot was determined according to the formula 5 × N3, where N is the number of sgRNAs determined to be enriched by upstream analysis.
Isolation of Single-Cell KO Clones
Cas9 cells were transduced with lentivirus expressing individual targeting sgRNA (LV04 constructs) as described in “Generation of Stable Cell Lines” above. After at least 1 week of puromycin selection, individual cell clones were plated by limiting dilution in 96-well plates in DMEM supplemented with 20% FBS plus non-essential amino acids and penicillin/streptomycin to increase cell survival. 2–3 weeks after plating, single-cell clones were expanded and screened for KO.
Cell-Binding Assay
The indicated cell lines were plated on 24-well plates at 5 × 104 cells per well overnight. Cells were placed on ice for 10 min, and then 109 VG per well prechilled vector was added in a total volume of 200 μL per well. Vectors were allowed to bind cells on ice on an orbital shaker platform for 1 h. Following binding, cells were washed three times with ice-cold PBS with Mg2+ and Ca2+ and then 100 μL of PBS per well was added and plates were frozen at −80°C. Binding assay plates underwent three freeze-thaw cycles, prior to resuspension and viral genome quantification by qPCR as described above using CMV primer/probe.
Cell Entry/Fractionation Assay
106 Huh7 single or double-KO cells were transduced with 10 VG/cell Anc80 (CMV.eGFP.WPRE) in six-well plates for 48 h and then harvested with PBS/EDTA (5 μM). Subcellular fractionation was done using Cell Signaling Technology cell fractionation kit (catalog no. 9038) using the recommended protocol with slight modification as follows: nuclear fractions were processed by addition of 1 vol of QuickExtract buffer (Lucigen, QE09050) and passaged through a 27-gauge needle. AAV genome concentration in each fraction was quantified by ddPCR using CMV primer/probe.
Indirect Immunofluorescence
Cells were imaged 24 h post-transfection on glass coverslips cultured in 48-well culture dishes. Cells were fixed in 4% PFA for 20 min at room temperature, washed twice with PBS, and then permeabilized using 20% ice-cold methanol. Slides were blocked using 10% BSA for 30 min at 4°C. Blocked coverslips were then incubated for 1 h at room temperature on a rotary shaker with mouse anti-FLAG M2 primary antibody (Sigma, catalog no. F1804) at 1:1,000 dilution and rabbit anti-TGN46 antibody (Novus Biologicals, catalog no. NBP1-49643) at 1:20 dilution in 3% BSA in PBS. Cells were washed three times for 10 min at room temperature on a rotary shaker prior to incubation with anti-rabbit Alexa Fluor 594 and anti-mouse Alexa Fluor 488 at 1:1,000 with DAPI for 1 h at room temperature on a rotary shaker. Cells were washed three times for 10 min prior to mounting with Dako fluorescence mounting medium (Dako, catalog no. S3023).
Imaging
For indirect immunofluorescence and co-localization studies, cells were imaged at ×63 with oil immersion on a Zeiss Axio Imager M2. Merged images were generated using ImageJ. Direct GFP fluorescence in Figure S4 was imaged 2 days post-transduction with a ×10 objective on an Evos FL cell imaging system (Thermo Fisher).
Animal Studies
In vivo analysis of chimeric AAV capsids were done by injection of 1011 VG/mouse in a total of 100 μL of PBS retro-orbitally into C57BL/6 mice under Schepens Eye Research Institute Institutional Animal Care and Use Committee approved animal protocol no. S-519-1021. Bioluminescent images were quantified using IVIS analysis software with the region of interest (ROI) designated as the whole mouse. Gpr108 KO mice were generated as described previously23 and were injected with 1011 VG/mouse in a total of 100 μL of PBS via tail vein alongside littermate-matched WT C57BL/6 control mice.
Author Contributions
A.M.D., E.Z., S.P., J.Z., J.E.C., and L.H.V. were responsible for the experimental design. A.M.D. performed the entry screen with intellectual input from S.P. and J.E.C. E.Z. performed entry screen analysis. A.M.D. performed the generation of Huh7 and Hepa GPR108 KO cell lines, designed and produced chimeric capsids, and conducted cellular binding and entry assays, cell fractionation, western blots, and transduction assays in cell lines. S.P. generated H1 HeLa GPR108 KO and rescue cell lines, and J.Z. characterized the H1 HeLa GPR108 KO cell line. N.Z., J.S.F., R.E., C.P., and G.L.Z. performed and analyzed the in vivo studies. N.Z. performed transduction in MEF cells. A.M.D. wrote the manuscript with input from all authors.
Conflicts of Interest
J.E.C. and S.P. are co-inventors on a Stanford University-owned patent about the use of AAVR for modulating AAV infection. J.E.C. is the recipient of the Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. L.H.V. is an inventor on several patents related to AAV gene therapy, including AncAAV variants, AAV9, and method patents, which are licensed to several biopharma companies. L.H.V. further receives funding from Lonza Houston, Selecta Biosciences, and Solid Biosciences, licensors to AncAAV technology. L.H.V. is a consultant to Nightstar, Selecta, Akousos, and Exonics and a Founder of Akouos. L.H.V. has a financial interest in TDTx, a company developing AAV gene therapies. He is an inventor of technology related to AAV gene therapy, a founder of the company, and also serves on its Board of Directors. L.H.V.’s interests were reviewed and are managed by MEE and Partners HealthCare in accordance with their conflicts of interest policies.
Acknowledgments
This work was supported by NIH grant R01 AI130123 (to J.E.C.) and by funding from Lonza Houston and Giving/Grousbeck (to L.H.V.). The authors would like to thank Brian Seed, PhD, and current and former members of the Vandenberghe and Carette laboratory, including Eva Andreas-Mateos and Andreas Puschnick for discussions and support. We thank the MEEI/SERI Gene Transfer Vector Core members Ru Xiao, Trisha Barungi, Allison Cucalon, Haiyan Qiu, and Julio Sanmiguel for viral vector production and titration. We thank David Dombkowski (MGH) for FACS sorting and analysis.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.11.005.
Supplemental Information
References
- 1.Samulski R.J., Chang L.S., Shenk T. Helper-free stocks of recombinant adeno-associated viruses: normal integration does not require viral gene expression. J. Virol. 1989;63:3822–3828. doi: 10.1128/jvi.63.9.3822-3828.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 4.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., Pasi K.J. AAV5-factor VIII gene transfer in severe hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 5.Voretigene neparvovec-rzyl (Luxturna) for inherited retinal dystrophy. Med. Lett. Drugs Ther. 2018;60:53–55. [PubMed] [Google Scholar]
- 6.Wu Z., Miller E., Agbandje-McKenna M., Samulski R.J. α2,3 and α2,6 N-linked sialic acids facilitate efficient binding and transduction by adeno-associated virus types 1 and 6. J. Virol. 2006;80:9093–9103. doi: 10.1128/JVI.00895-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaludov N., Brown K.E., Walters R.W., Zabner J., Chiorini J.A. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. J. Virol. 2001;75:6884–6893. doi: 10.1128/JVI.75.15.6884-6893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng R., Govindasamy L., Gurda B.L., McKenna R., Kozyreva O.G., Samulski R.J., Parent K.N., Baker T.S., Agbandje-McKenna M. Structural characterization of the dual glycan binding adeno-associated virus serotype 6. J. Virol. 2010;84:12945–12957. doi: 10.1128/JVI.01235-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Summerford C., Samulski R.J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 1998;72:1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kern A., Schmidt K., Leder C., Müller O.J., Wobus C.E., Bettinger K., Von der Lieth C.W., King J.A., Kleinschmidt J.A. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J. Virol. 2003;77:11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Opie S.R., Warrington K.H., Jr., Agbandje-McKenna M., Zolotukhin S., Muzyczka N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 2003;77:6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen S., Bryant K.D., Brown S.M., Randell S.H., Asokan A. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J. Biol. Chem. 2011;286:13532–13540. doi: 10.1074/jbc.M110.210922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bell C.L., Vandenberghe L.H., Bell P., Limberis M.P., Gao G.P., Van Vliet K., Agbandje-McKenna M., Wilson J.M. The AAV9 receptor and its modification to improve in vivo lung gene transfer in mice. J. Clin. Invest. 2011;121:2427–2435. doi: 10.1172/JCI57367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qing K., Mah C., Hansen J., Zhou S., Dwarki V., Srivastava A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 1999;5:71–77. doi: 10.1038/4758. [DOI] [PubMed] [Google Scholar]
- 15.Kashiwakura Y., Tamayose K., Iwabuchi K., Hirai Y., Shimada T., Matsumoto K., Nakamura T., Watanabe M., Oshimi K., Daida H. Hepatocyte growth factor receptor is a coreceptor for adeno-associated virus type 2 infection. J. Virol. 2005;79:609–614. doi: 10.1128/JVI.79.1.609-614.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurzeder C., Koppold B., Sauer G., Pabst S., Kreienberg R., Deissler H. CD9 promotes adeno-associated virus type 2 infection of mammary carcinoma cells with low cell surface expression of heparan sulphate proteoglycans. Int. J. Mol. Med. 2007;19:325–333. [PubMed] [Google Scholar]
- 17.Akache B., Grimm D., Pandey K., Yant S.R., Xu H., Kay M.A. The 37/67-kilodalton laminin receptor is a receptor for adeno-associated virus serotypes 8, 2, 3, and 9. J. Virol. 2006;80:9831–9836. doi: 10.1128/JVI.00878-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Pasquale G., Davidson B.L., Stein C.S., Martins I., Scudiero D., Monks A., Chiorini J.A. Identification of PDGFR as a receptor for AAV-5 transduction. Nat. Med. 2003;9:1306–1312. doi: 10.1038/nm929. [DOI] [PubMed] [Google Scholar]
- 19.Weller M.L., Amornphimoltham P., Schmidt M., Wilson P.A., Gutkind J.S., Chiorini J.A. Epidermal growth factor receptor is a co-receptor for adeno-associated virus serotype 6. Nat. Med. 2010;16:662–664. doi: 10.1038/nm.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pillay S., Meyer N.L., Puschnik A.S., Davulcu O., Diep J., Ishikawa Y., Jae L.T., Wosen J.E., Nagamine C.M., Chapman M.S., Carette J.E. An essential receptor for adeno-associated virus infection. Nature. 2016;530:108–112. doi: 10.1038/nature16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dudek A.M., Pillay S., Puschnik A.S., Nagamine C.M., Cheng F., Qiu J., Carette J.E., Vandenberghe L.H. An alternate route for adeno-associated virus (AAV) entry independent of AAV receptor. J. Virol. 2018;92 doi: 10.1128/JVI.02213-17. e02213-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kronenberg S., Böttcher B., von der Lieth C.W., Bleker S., Kleinschmidt J.A. A conformational change in the adeno-associated virus type 2 capsid leads to the exposure of hidden VP1 N termini. J. Virol. 2005;79:5296–5303. doi: 10.1128/JVI.79.9.5296-5303.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong D., Zhou H., Na S.Y., Niedra R., Peng Y., Wang H., Seed B., Zhou G.L. GPR108, an NF-κB activator suppressed by TIRAP, negatively regulates TLR-triggered immune responses. PLoS ONE. 2018;13:e0205303. doi: 10.1371/journal.pone.0205303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pillay S., Zou W., Cheng F., Puschnik A.S., Meyer N.L., Ganaie S.S., Deng X., Wosen J.E., Davulcu O., Yan Z. AAV serotypes have distinctive interactions with domains of the cellular receptor AAVR. J. Virol. 2017;91:e00391-17. doi: 10.1128/JVI.00391-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng J., Zhou Y., Zhu S., Wei W. High-throughput screens in mammalian cells using the CRISPR-Cas9 system. FEBS J. 2015;282:2089–2096. doi: 10.1111/febs.13251. [DOI] [PubMed] [Google Scholar]
- 27.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koike-Yusa H., Li Y., Tan E.P., Velasco-Herrera Mdel.C., Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014;32:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Y., Zhu S., Cai C., Yuan P., Li C., Huang Y., Wei W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509:487–491. doi: 10.1038/nature13166. [DOI] [PubMed] [Google Scholar]
- 30.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mays L.E., Wang L., Tenney R., Bell P., Nam H.J., Lin J., Gurda B., Van Vliet K., Mikals K., Agbandje-McKenna M., Wilson J.M. Mapping the structural determinants responsible for enhanced T cell activation to the immunogenic adeno-associated virus capsid from isolate rhesus 32.33. J. Virol. 2013;87:9473–9485. doi: 10.1128/JVI.00596-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salganik M., Aydemir F., Nam H.J., McKenna R., Agbandje-McKenna M., Muzyczka N. Adeno-associated virus capsid proteins may play a role in transcription and second-strand synthesis of recombinant genomes. J. Virol. 2014;88:1071–1079. doi: 10.1128/JVI.02093-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aydemir F., Salganik M., Resztak J., Singh J., Bennett A., Agbandje-McKenna M., Muzyczka N. Mutants at the 2-fold interface of adeno-associated virus type 2 (AAV2) structural proteins suggest a role in viral transcription for AAV capsids. J. Virol. 2016;90:7196–7204. doi: 10.1128/JVI.00493-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tafesse F.G., Guimaraes C.P., Maruyama T., Carette J.E., Lory S., Brummelkamp T.R., Ploegh H.L. GPR107, a G-protein-coupled receptor essential for intoxication by Pseudomonas aeruginosa exotoxin A, localizes to the Golgi and is cleaved by furin. J. Biol. Chem. 2014;289:24005–24018. doi: 10.1074/jbc.M114.589275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar A.J. Human GPR107 and murine Gpr108 are members of the LUSTR family of proteins found in both plants and animals, having similar topology to G-protein coupled receptors. DNA Seq. 2007;18:235–241. doi: 10.1080/10425170701207182. [DOI] [PubMed] [Google Scholar]
- 36.Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.L., Yang B., Huber N., Pasca S.P., Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hordeaux J., Wang Q., Katz N., Buza E.L., Bell P., Wilson J.M. The neurotropic properties of AAV-PHP.B are limited to C57BL/6J mice. Mol. Ther. 2018;26:664–668. doi: 10.1016/j.ymthe.2018.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vandenberghe L.H., Wang L., Somanathan S., Zhi Y., Figueredo J., Calcedo R., Sanmiguel J., Desai R.A., Chen C.S., Johnston J. Heparin binding directs activation of T cells against adeno-associated virus serotype 2 capsid. Nat. Med. 2006;12:967–971. doi: 10.1038/nm1445. [DOI] [PubMed] [Google Scholar]
- 39.Blackburn S.D., Steadman R.A., Johnson F.B. Attachment of adeno-associated virus type 3H to fibroblast growth factor receptor 1. Arch. Virol. 2006;151:617–623. doi: 10.1007/s00705-005-0650-6. [DOI] [PubMed] [Google Scholar]
- 40.Ling C., Lu Y., Kalsi J.K., Jayandharan G.R., Li B., Ma W., Cheng B., Gee S.W., McGoogan K.E., Govindasamy L. Human hepatocyte growth factor receptor is a cellular coreceptor for adeno-associated virus serotype 3. Hum. Gene Ther. 2010;21:1741–1747. doi: 10.1089/hum.2010.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nonnenmacher M.E., Cintrat J.C., Gillet D., Weber T. Syntaxin 5-dependent retrograde transport to the trans-Golgi network is required for adeno-associated virus transduction. J. Virol. 2015;89:1673–1687. doi: 10.1128/JVI.02520-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang R., Cao L., Cui M., Sun Z., Hu M., Zhang R., Stuart W., Zhao X., Yang Z., Li X. Adeno-associated virus 2 bound to its cellular receptor AAVR. Nat. Microbiol. 2019;4:675–682. doi: 10.1038/s41564-018-0356-7. [DOI] [PubMed] [Google Scholar]
- 43.Meyer N.L., Hu G., Davulcu O., Xie Q., Noble A.J., Yoshioka C., Gingerich D.S., Trzynka A., David L., Stagg S.M., Chapman M.S. Structure of the gene therapy vector, adeno-associated virus with its cell receptor, AAVR. eLife. 2019;8:e44707. doi: 10.7554/eLife.44707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sonntag F., Bleker S., Leuchs B., Fischer R., Kleinschmidt J.A. Adeno-associated virus type 2 capsids with externalized VP1/VP2 trafficking domains are generated prior to passage through the cytoplasm and are maintained until uncoating occurs in the nucleus. J. Virol. 2006;80:11040–11054. doi: 10.1128/JVI.01056-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Girod A., Wobus C.E., Zádori Z., Ried M., Leike K., Tijssen P., Kleinschmidt J.A., Hallek M. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J. Gen. Virol. 2002;83:973–978. doi: 10.1099/0022-1317-83-5-973. [DOI] [PubMed] [Google Scholar]
- 46.Venkatakrishnan B., Yarbrough J., Domsic J., Bennett A., Bothner B., Kozyreva O.G., Samulski R.J., Muzyczka N., McKenna R., Agbandje-McKenna M. Structure and dynamics of adeno-associated virus serotype 1 VP1-unique N-terminal domain and its role in capsid trafficking. J. Virol. 2013;87:4974–4984. doi: 10.1128/JVI.02524-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grieger J.C., Snowdy S., Samulski R.J. Separate basic region motifs within the adeno-associated virus capsid proteins are essential for infectivity and assembly. J. Virol. 2006;80:5199–5210. doi: 10.1128/JVI.02723-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson J.S., Li C., DiPrimio N., Weinberg M.S., McCown T.J., Samulski R.J. Mutagenesis of adeno-associated virus type 2 capsid protein VP1 uncovers new roles for basic amino acids in trafficking and cell-specific transduction. J. Virol. 2010;84:8888–8902. doi: 10.1128/JVI.00687-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vihinen-Ranta M., Wang D., Weichert W.S., Parrish C.R. The VP1 N-terminal sequence of canine parvovirus affects nuclear transport of capsids and efficient cell infection. J. Virol. 2002;76:1884–1891. doi: 10.1128/JVI.76.4.1884-1891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tullis G.E., Burger L.R., Pintel D.J. The minor capsid protein VP1 of the autonomous parvovirus minute virus of mice is dispensable for encapsidation of progeny single-stranded DNA but is required for infectivity. J. Virol. 1993;67:131–141. doi: 10.1128/jvi.67.1.131-141.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou G.L., Na S.Y., Niedra R., Seed B. Deficits in receptor-mediated endocytosis and recycling in cells from mice with Gpr107 locus disruption. J. Cell Sci. 2014;127:3916–3927. doi: 10.1242/jcs.135269. [DOI] [PubMed] [Google Scholar]
- 52.Elling U., Taubenschmid J., Wirnsberger G., O’Malley R., Demers S.P., Vanhaelen Q., Shukalyuk A.I., Schmauss G., Schramek D., Schnuetgen F. Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell Stem Cell. 2011;9:563–574. doi: 10.1016/j.stem.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nicolson S.C., Samulski R.J. Recombinant adeno-associated virus utilizes host cell nuclear import machinery to enter the nucleus. J. Virol. 2014;88:4132–4144. doi: 10.1128/JVI.02660-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nonnenmacher M., Weber T. Adeno-associated virus 2 infection requires endocytosis through the CLIC/GEEC pathway. Cell Host Microbe. 2011;10:563–576. doi: 10.1016/j.chom.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Excoffon K.J., Koerber J.T., Dickey D.D., Murtha M., Keshavjee S., Kaspar B.K., Zabner J., Schaffer D.V. Directed evolution of adeno-associated virus to an infectious respiratory virus. Proc. Natl. Acad. Sci. USA. 2009;106:3865–3870. doi: 10.1073/pnas.0813365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joung J., Konermann S., Gootenberg J.S., Abudayyeh O.O., Platt R.J., Brigham M.D., Sanjana N.E., Zhang F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017;12:828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lock M., Alvira M., Vandenberghe L.H., Samanta A., Toelen J., Debyser Z., Wilson J.M. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum. Gene Ther. 2010;21:1259–1271. doi: 10.1089/hum.2010.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10. [Google Scholar]
- 59.Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hunter J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- 61.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45(D1):D331–D338. doi: 10.1093/nar/gkw1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carbon S., Ireland A., Mungall C.J., Shu S., Marshall B., Lewis S., AmiGO Hub. Web Presence Working Group AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25:288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.