

Abstract
The granulocyte-specific microRNA-223 (miR-223) has recently emerged as a negative regulator of NOD-like receptor 3 (NLRP3) expression, a central key player in chronic hepatic injuries such as fibrotic nonalcoholic steatohepatitis (NASH), as well as in other liver conditions including acute hepatitis. In this study, we evaluated the therapeutic effect of the synthetic miR-223 analog miR-223 3p in a murine model of lipopolysaccharide (LPS)/D-GalN-induced endotoxin acute hepatitis (EAH) or fibrotic NASH resultant of long-term feeding with a high-fat, fructose, and cholesterol (FFC) diet. miR-223 3p ameliorated the infiltration of monocytes, neutrophils, and early activated macrophages and downregulated the transcriptional expression of the pro-inflammatory cytokines Il6 and Il12 and the chemokines Ccl2, Ccl3, Cxcl1, and Cxcl2 in EAH. In fibrotic NASH, treatment with miR-223 3p led to a remarkable mitigation of fibrosis development and activation of hepatic stellate cells (HSCs). miR-223 3p disrupted the activation of the NLRP3 inflammasome by impairing the synthesis of cleaved interleukin-1β (IL-1β), mature IL-1β, and NLRP3, and the activation of caspase-1 p10 in both EAH and fibrotic NASH. Our data enlightens miR-223 3p as a post-transcriptional approach to treat acute and chronic hepatitis by silencing the activation of the NLRP3 inflammasome.
Keywords: inflammasome, microRNA, liver fibrosis, NLRP3, macrophage, monocyte, IL-1β, acute and chronic liver injury
Graphical Abstract
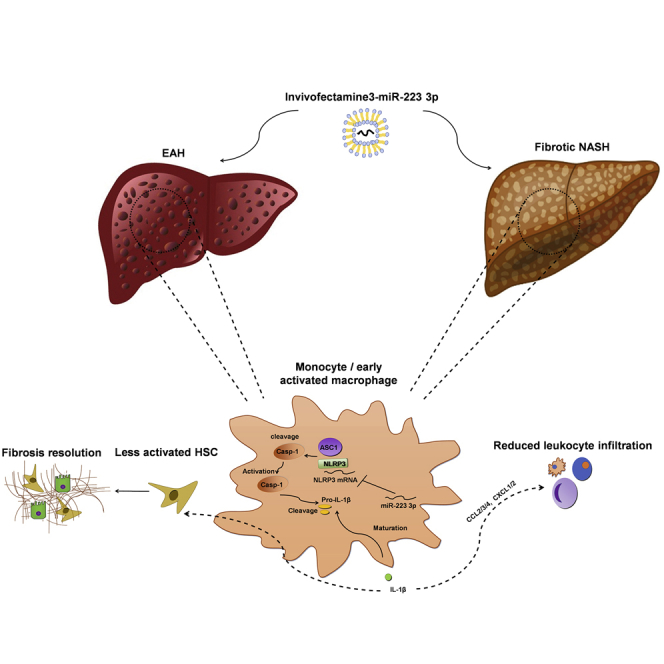
Calvente et al. present the synthetic miR-223 analog miR-223 3p as a highly translational approach that mitigates endotoxin hepatitis and fibrotic NASH—the two most prevalent instances of acute and chronic liver injuries, respectively—through post-transcriptional silencing of the NLRP3 inflammasome.
Introduction
Inflammation constitutes the central underlying pathology of most forms of acute and chronic liver injuries.1 Sustained, untreated liver inflammation often exacerbates tissue injury and results in an abnormal wound healing response that culminates in fibrosis and the latter cirrhosis.2 Ongoing evidence describes a pivotal role of the NOD-like intracellular receptor NLRP3 in the development of acute and chronic liver diseases. Upon activation, NLRP3 oligomerizes and recruits the adaptor protein apoptosis speck-like protein with a CARD domain (ASC) that initiates a prion-like oligomerization process with other ASC monomers, an essential step in the final assembling of the inflammasome. This NLRP3-ASC complex recruits and activates caspase-1 (p10), which in turn cleaves and matures the pro-inflammatory cytokines pro-interleukin-1β (IL-1β) and pro-IL-18 into their soluble forms,3 thus inducing the release of myeloid and leukocyte-specific chemoattractants that further promote inflammation.4, 5, 6, 7 Nevertheless, this chain of pro-inflammatory events can be interrupted by therapeutic interventions that silence upstream inflammasome pathways post-transcriptionally.
In the last decade, RNA therapy has emerged as a powerful tool to silence gene expression post-transcriptionally. Particularly, synthetic microRNA, a short non-coding ribonucleotide sequence that neutralizes the expression of target mRNAs,8 is gaining increasing attention as an inhibitory approach to mimic the mode of action of endogenous microRNAs (miRNAs) and therefore serve as a replacement, post-transcriptional therapy in a wide spectrum of genetic disorders.9 MicroRNA-223 (miR-223) is a granulocyte-enriched miRNA expressed in various organs, including the liver.10, 11 Its synthetic analog miR-223 3p has recently emerged as a potent anti-inflammatory ribonucleotide to treat various extra-hepatic inflammatory conditions through the silencing of Nlrp3 transcripts and blockade of the resultant inflammasome cascade and peripheral infiltration of pro-inflammatory cellular repertoires such as monocytes and granulocytes.4, 5, 6, 7, 12
Here, we present data supporting miR-223 3p as a mimic of the endogenous miR-223 and a potent, wide-spectrum approach to treat various forms of hepatic inflammation, including endotoxin acute hepatitis (EAH) and fibrotic nonalcoholic steatohepatitis (NASH), through inhibition of the NLRP3 inflammasome cascade.
Results
The potential anti-inflammatory effect of miR-223 3p, the strand 3p of the synthetic analog of the endogenous miR-223, was tested in a mouse model of EAH. To administer miR-223 3p prior to injury, we gave C57BL/6 mice a single intravenous (i.v.) injection of PBS (inflammation control mice), scrambled RNA (Scr-RNA controls), or miR-223 3p (miR-223 3p-treated group) at 0.6 mg/kg carried in Invivofectamine 3, a lipoid-based vehicle that enables safe delivery of ribonucleic acids to the liver and Kupffer cells.13, 14 Twenty-four hours later, EAH was induced with an intraperitoneal (i.p.) injection of lipopolysaccharide (LPS) in conjunction with D-galactosamine (LPS/D-GalN) at 40 μg/800 mg per kg body weight (B.W.) for 3 h (Figure 1A). Then, the mice were euthanized and the livers were harvested for biochemical and histological analysis.
Figure 1.

miR-223 3p Improves Acute Liver Inflammation
(A) Experimental design of miR-223 3p treatment in the acute liver inflammation (ALI) mouse model induced with LPS/D-GalN. (B) Representative microphotographs of liver slides stained with anti-F4/80, MPO or Ly6C antibodies, or TDT enzyme (TUNEL) specific for total macrophages, granulocytes, monocytes, or apoptotic cells, respectively. White or black arrows indicate MPO or Ly6C-positive cells, respectively. Scale bars, 100 μm. (C) Serum ALT levels measured by colorimetry. (D–F) Percentage of liver area stained positive for anti-F4/80 (D), MPO (E), or Ly6C (F) antibodies assessed in 10 randomly selected images by computerized image analyses. (G and H) Relative mRNA expression of Ccl2, Ccl3, and Cxcl1 (G) and Il6 and Il12 (H) measured by qRT-PCR. Blue-like bars refer to the scale of the right y axis. (I) Total hepatic apoptotic cells expressed as the percentage of area stained positive for TDT enzyme (TUNEL) in 10 aleatory selected images of liver slides. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA. Data are represented as means ± SD, n = 7−10/group.
The treatment with miR-223 3p significantly reverted the serum levels of alanine aminotransferase (ALT) to normal, compared to Scr-RNA controls (Figure 1C). Similarly, the population of total macrophages, as assessed by staining with anti-F4/80 antibody in paraffin-embedded sections, was notably less expanded in the livers of miR-223 3p-treated mice, relative to their respective Scr-RNA controls (Figures 1B–D). Similarly, immunohistochemical staining of acute inflammatory cells such as infiltrating hepatic macrophages (F4/80+) and neutrophils (myeloperoxidase [MPO]+) were significantly reduced in mice treated with miR-223 3p versus Scr-RNA controls (Figures 1B, 1E, and 1F). These results were confirmed when transcript levels of the monocyte and early activated macrophage chemokines Ccl2 and Ccl3, and the neutrophil chemoattractants Cxcl1 and Cxcl2 were all dramatically downregulated after miR-223 3p treatment versus Scr-RNA control (Figure 1G), suggesting an impaired de novo recruitment of acute pro-inflammatory cells to the liver. Furthermore, treatment with miR-223 3p led to a dramatic decrease of mRNA levels of the early pro-inflammatory cytokines Il6 and Il12 (Figure 1H) and a substantially lowered total dead cell population in liver sections (TUNEL staining positive), relative to Scr-RNA control (Figures 1B and 1I).
Jointly, these data indicate a protective, anti-inflammatory effect of miR-223 3p in EAH by mitigating the de novo infiltration of pro-inflammatory cells to the liver and mitigating hepatic cell death.
Three transcripts have been identified as targets of miR-223 3p:10, 12 Nlrp3 (NOD-like receptor 3), the initial signaling component of the inflammasome, the granulocyte maturation and myogenesis transcription factor Mef2c (Myocyte-Specific Enhancer Factor 2C), and the insulin growth and survival protein Igfr1 (insulin-like growth factor receptor 1); among these three target transcripts, Nlrp3 was the strongest downregulated after treatment with miR-223 3p, versus Scr-RNA (Figure S1A). Given the central role of NLRP3 as a fundamental triggering element in the liver innate immunity during acute injury,15 we focused on its expression and resultant activation cascade to elucidate the mechanism by which miR-223 3p exerts a protective, anti-inflammatory effect in EAH. We observed a notable decrease in the hepatic expression of the transcript and protein form of NLRP3 after miR-223 3p treatment, compared to the Scr-RNA control group (Figure 2A; Figures S2A and S2B).
Figure 2.

miR-223 3p Blocks the NLRP3 Inflammasome in EAH
(A and B) Relative mRNA expression of Nlrp3 (A) and Il1β (B) assessed by qRT-PCR. (C) Fold change of intensity of protein bands of cleaved IL-1β are shown in (D). Normalization by the housekeeping proteins GAPDH and measured by computerized ImageJ analysis. (D) Western blots of cleaved IL-1β and GAPDH proteins (original blots shown in Figure S2). **p < 0.01, ****p < 0.0001, one-way ANOVA. Data are represented as means ± SD, n = 6–10/group.
IL-1β plays a key role as a pro-inflammatory cytokine in acute inflammation by promoting the activation of infiltrating leukocytes such as monocytes and natural killer (NK) cells.16, 17 IL-1β constitutes the last element in the activation signaling cascade of the inflammasome after cleavage and maturation by active caspase-1.18 Strikingly, miR-223 3p treatment dramatically abolished the cleavage of IL-1β in the liver, compared to control Scr-RNA (Figures 2C and 2D; Figures S3C and S3D). This was confirmed by a very robust downregulation in the expression of Il1β mRNA (Figure 2B), indicating a remarkable hepatic anti-inflammatory effect.
To exclude off-target effects of miR-223 3p, we measured the expression of ASC, the adaptor protein of the inflammasome that complexes with NLRP3 and enables the activation of caspase-118 in the liver specimens of the mice described in Figure 1A. miR-223 3p-treated mice displayed negligible changes in expression levels of the ASC protein and transcript forms versus the control mice that received Scr-RNA (Figures S4A–S4C, S5A, and S5B). These results suggest the absence of NLRP3-inespecific effects of miR-223 3p in EAH.
To verify that miR-223 3p is responsible for the post-transcriptional silencing of NLRP3 and the resultant blockade of the inflammasome activation in EAH, we assessed the knockdown effectiveness of miR-223 3p toward Nlrp3 in immortalized macrophages primed with LPS and ATP. J744.2 macrophages were transfected with miR-223 3p or Scr-RNA in Lipofectamine 3000 at 90 nM, and 24 h later Nlrp3 expression was induced by priming the cells with 100 ng/mL of LPS overnight. Cells transfected with Scr-RNA served as transfection controls, whereas untreated cells represented the priming controls. The activation of the inflammasome was then induced by treating the cells with ATP for 1 h at 5 mM under serum-reduced conditions. Twenty-four hours of pre-priming transfection with miR-223 3p resulted in a very significant knockdown of Nlrp3 transcript and protein levels, compared to control cells treated with Scr-RNA (Figures S6A and S6B); these results correlated with a modestly reduced fluorescent area of the staining with anti-caspase-1 antibody (activation peptide subunit of caspase-1, p10) in the total miR-223 3p-transfected cell population versus Scr-RNA control cells (Figures S6D and S6E), indicating that miR-223 3p attenuates the activation of the inflammasome in immortalized macrophages.
Jointly, these results exhibit miR-223 3p as a mitigator of EAH by inhibiting the activation of the NLRP3 inflammasome without notable off-target effects.
To confirm that miR-223 3p mimics the anti-inflammatory effect and the Nlrp3-silencing activity of the endogenous miR-223 in EAH, we treated female wild-type (WT) or miR-223-deficient mice (miR-223−/−) with a single i.p. injection of PBS or LPS/D-GalN for 3 h, as detailed in Figure 1A (Figure 3A). WT mice given PBS (called untreated WT) served as non-inflammation controls for the group of miR-223−/− mice that were treated with PBS (termed untreated miR-223−/−). The group of WT mice injected with LPS/D-GalN (termed treated WT mice) served as inflammation controls for the miR-223−/− mice that received LPS/D-GalN (called treated miR-223−/−). Three hours after the LPS/D-GalN challenge, serum ALT levels were substantially elevated in miR-223−/− mice relative to their treated WT controls (Figure 3C), suggesting hepatic inflammation. This was consistent with a very significant increase of the total macrophage population in liver sections of untreated or treated miR-223−/− mice (versus their untreated or treated WT controls, correspondingly), as assessed by area stained positive for anti-F4/80 antibody (Figures 3B and 3D). Similarly, the staining of neutrophils (MPO+) and macrophages (F4/80+) was exacerbated in the treated or untreated miR-223−/− mice compared to their respective controls (Figures 3B, 3E, and 3F), confirming an aggravated acute inflammatory response in the absence of miR-223.
Figure 3.

miR-223 Deletion Worsens EAH
(A) Schematic representation of EAH induction in WT or miR-223-deficient (miR-223−/−) mice with LPS/D-GalN. (B) Staining of total macrophages (F4/80), monocytes (Ly6C), and neutrophils (MPO) with anti-F4/80, Ly6C, or MPO antibodies in liver slides. Scale bars, 100 μm. (C) Levels of ALT in serum. (D–F) Percentage of liver area stained positive for anti-F4/80 (D), MPO (E), or Ly6C (F) antibodies. ***p < 0.001, ****p < 0.0001, one-way ANOVA. Data show means ± SD, n = 5–7/group.
To confirm that the anti-inflammatory mechanism of miR-223 3p mirrors the mode of action of the endogenous miR-223, we assessed the NLRP3 inflammasome activation by staining of active caspase-1 with FAM-fluorescent labeled inhibitors of caspases (FAM-FLICA) in the livers of LPS/D-GalN-challenged miR-223−/− mice: the percentage of FAM-FLICA-positive area was significantly greater compared to their treated WT controls (Figures 4A and 4B), suggesting that endogenous miR-223 is capable of ameliorating the activation of the inflammasome under injurious conditions. This was in correlation with the mRNA expression of the intermediate and final inflammasome transcripts Nlrp3 and Il1β, respectively, that were both significantly upregulated in LPS/D-GalN-treated mice, relative to their treated WT controls (Figures 4C and 4D); given the enrichment of NLRP3 in macrophages3 and the reported delivery of various RNA analogs to Kupffer cells in Invivofectamine 3 nanoparticles,14 the in vivo transfection media of miR-223 3p, these results suggest a Nlrp3-targeted effect of miR-223 3p in liver macrophages.
Figure 4.

miR-223 Deficiency Exacerbates Activation of the NLRP3 Inflammasome in EAH
(A) Activation of caspase-1 measured by fluorescent in situ caspase activation (FLICA) assay in cryo-sectioned livers. Scale bars, 100 μm. (B) Percentage of fluorescence signal positive for FLICA in total liver area of 10 randomly selected images. (C and D) Relative mRNA expression of Nlrp3 (C) and Il1β (D) quantified by qRT-PCR. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant, one-way ANOVA. Results are shown as means ± SD, n = 5–7/group.
Although ultimate activation of the inflammasome was not observed in the untreated miR-223−/− mice, the transcription of both Nlrp3 and Il1β genes were notably augmented in these mice versus their untreated WT controls (Figures 4C and 4D), signifying that endogenous miR-223 can silence Nlrp3 transcripts and the resultant inflammasome cascade during acute hepatic stress. Conclusively, these results show that miR223 3p functionally and mechanistically mimics endogenous miR-223 in EAH.
To further test the potential of the anti-inflammatory and anti-fibrotic effect of miR-223 3p in other disease interplays beyond acute injury, we induced dietary chronic inflammation and fibrosis in a murine NASH model of high-fat/cholesterol diet combined with fructose/sucrose drinkable water (called fat, fructose, and cholesterol diet, FFC) for 26 weeks. During the last 2 weeks of feeding, two independent groups of WT mice were treated weekly with a single i.v. injection of Scr-RNA or miR-223 3p (called miR-223 3p-treated mice) at 1 mg/kg in Invivofectamine 3; Scr-RNA-treated mice served as inflammation controls, and a separate group fed with a normal chow diet (CD) constituted the non-inflamed controls (experimental design shown in Figure 5A).
Figure 5.

miR-R223 3p Mitigates Progression of Chronic Inflammation and Fibrosis in NASH
(A) Experimental design of miR-223 3p or Scr-RNA control treatment in a murine NASH model induced with a high-fat/cholesterol diet combined with sucrose/fructose in drinkable water (FFC diet). (B) Mouse body weight (B.W. expressed in g) during the course of the feeding with FFC diet and treatment with miR-223 3p or Scr-RNA. (C) Serum levels of ALT measured by colorimetry. (D) Staining of liver sections for examination of total macrophages (F4/80), neutrophils (MPO), activated stellate cells (α-SMA), and total collagen (Sirius Red). Scale bars, 100 μm. (E–H) Percentage of liver area positive for anti-F4/80 (E), MPO (F), or α-SMA (G) antibodies or Sirius Red chromogen (H) in 10 images selected aleatory. *p < 0.05, **p < 0.01, ****p < 0.0001; n.s., not significant, one-way ANOVA. Results represent means ± SD, n = 3–5/group.
After 26 weeks of feeding with FFC diet, the mice developed a notable increase in B.W., serum ALT levels, and total macrophage (F4/80+) and granulocyte (MPO+) hepatic infiltration, compared to feeding with normal CD diet (Figures 5B–5F). However, the treatment with miR-223 3p reduced the levels of serum ALT and total macrophage (F4/80+) and granulocyte (MPO+) hepatic populations compared to Scr-RNA, while maintaining steady B.W. (Figure 5B); these data point toward an improved inflammatory condition, likely without obvious side effects.
In the liver, sustained inflammation often leads to fibrosis, the deposition of interstitial collagen fibrils, and extracellular matrix (ECM) following activation of hepatic stellate cells (HSCs). Compared to feeding with normal CD, a 26-week diet course with FFC resulted in a significant hepatic collagen deposition and activation of HSCs (Figures 5D, 5G, and 5H), as measured by staining of total collagen with Sirius Red and activated HSCs with an anti-α-SMA antibody. By contrast, we observed an improved inflammatory response after treatment with miR-223 3p (Figures 5C–5F) that led to a lowered collagen production (Sirius Red+) and activation of HSCs (α-SMA+), versus treatment with control Scr-RNA (Figures 5G and 5H). These results exhibit an indirect anti-fibrotic effect of miR-223 3p during NASH progression.
The NLRP3 inflammasome is activated not only during early inflammation but also during chronicity. In NASH progression, activation of the inflammasome is essential for fibrosis development.18 In 26-week FFC-fed mice, treatment with miR-223 3p significantly shut down the expression of Nlrp3 mRNA, leading to an attenuation of the inflammasome activation versus Scr-RNA controls, as examined by FAM-FLICA staining for active caspase-1 (p10) and ELISA for mature IL-1β in serum (Figures 6C–6E). Strikingly, miR-223 3p did not affect the expression of the inflammasome adaptor protein ASC (compared to control Scr-RNA; Figures 6A and 6B), suggesting the absence of off-target effects associated with miR-223 3p treatment.
Figure 6.

miR-223 3p Ameliorates NLRP3 Inflammasome Activation in Fibrotic NASH
(A and B) Relative mRNA expression of Nlrp3 (A) and Asc (B) genes measured by qRT-PCR. (C) Active caspase-1 visualized by FLICA assay. Scale bars, 100 μm. (D) Percentage of liver area stained positive for FLICA in 10 randomly selected pictures. (E) Serum levels of mature IL-1β assessed by ELISA with monoclonal anti-IL-1 β antibody. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA. Results show means ± SD, n = 3–5/group.
Collectively, these results show evidence of a silenced expression and activation of the NLRP3 inflammasome and an improvement of inflammation and fibrosis after treatment with miR-223 3p in NASH progression.
Discussion
Inflammation is the most prevalent underlying pathology in both acute and chronic hepatic injury. Although it is self-limiting, inflammation is considered beneficial and contributes to tissue homeostasis. However, the inability to shut down the persistence of inflammation results in tissue damage and disease progression. Pro-inflammatory molecular events that are key in the onset of acute inflammation also play a central role in the development of chronic inflammatory diseases.19, 20, 21 Therefore, therapeutic targeting of early inflammatory events constitutes the approach of choice to prevent disease progression.
The NLRP3 inflammasome plays a pivotal role in both early and progressive inflammation.20, 22 Several compounds have emerged as blockers of the NLRP3 inflammasome cascade;19 nevertheless, only the synthetic miR-223 3p mimic has been shown to silence Nlrp3 post-transcriptionally and therefore mitigate de novo assembly of the inflammasome12, 23, 24 and subsequent tissue infiltration of pro-inflammatory cellular repertoires. Although miR-223 has recently emerged as a regulator of intestinal inflammation via the inhibition of the NLRP3 inflammasome,25 its anti-inflammatory effect during acute and chronic liver injury remains elusive. Here we employed miR-223 3p as an ample spectrum approach to treat inflammation in EAH and fibrotic NASH, the most prevalent instances of acute and chronic liver inflammatory diseases, respectively.26, 27
In this study, we showed that treatment with miR-223 3p prevented the development of LPS/D-GalN-induced endotoxin acute hepatitis in a remarkable manner: the Nlrp3-targeted silencing of miR-223 3p we observed resulted in a robust diminution of cleaved IL-1β, likely explaining the notable decrease in the transcription of the cytokines Il6, Il12, and Tnfα28 and their subsequent inducible chemokines Ccl2, Ccl3, Ccl4, Cxcl1, and Cxcl2. This promoted a reduced infiltration of monocytes, early activated macrophages, and neutrophils,29, 30 thus preventing the onset of inflammation. Strikingly, we found a substantial amelioration of cell death in these mice, suggesting a miR-223 3p-related protection from pyroptotic cell death as a result of caspase-1 inhibition.31, 32
Notably, miR-223 3p also resolved inflammation, activation of HSCs, and the latter fibrosis in a model of FFC-induced NASH, thus qualifying as an anti-inflammatory and anti-fibrotic therapy with potential translational implications.
A Nlrp3 mRNA-targeted specific outcome was modestly found after treatment with miR-223 3p. From the analyzed transcripts known as secondary targets of miR-223 3p (Igfr1 and Mef2c), Nlrp3 was the leading candidate. Taking the notion that the regulation of pro-inflammatory cytokines is not influenced by either Mf2c or Igfr1,33, 34 which are key regulators of granulopoiesis and insulin signaling in vascular smooth muscle cells (VSMCs) and granulocytes, respectively,10, 35 off-targeted anti-inflammatory effects and non-macrophage/monocyte-specific uptake of miR-223 3p are modestly excluded in our studies.
Invivofectamine 3 was employed in our studies to reach hepatic delivery of miR-223 3p as previously described.13 It has been shown to enable delivery of various RNA analogs to Kupffer cells;14 the increased Nlrp3 expression, caspase-1 activation, and cellular pro-inflammatory infiltration we found in miR-223−/− mice treated with LPS/D-GalN was not observed when challenged (LPS/D-GalN). WT mice were treated with miR-223 3p in an independent experiment, thus suggesting that Nlrp3-expresing liver macrophages are the main targets of miR-223 3p in this model of acute inflammation.
Significantly, we present miR-223 3p as a post-transcriptional neutralizer of Nlrp3 expression and a downstream blocker of the inflammasome complexation in both EAH and fibrotic NASH, enlightening miR-223 3p as a promising approach to treat an ample variety of liver diseases wherein the NLRP3 inflammasome plays a key role as a central injury sensor. Strikingly, unchallenged miR-223−/− mice did not display a blockade of the inflammasome formation, suggesting that miR-223 3p requires an exogenous stressor to indirectly block the assembly of the inflammasome.
Together, our data demonstrate that miR-223 3p is a highly translational and promising approach to treat acute and chronic liver diseases in which the NLRP3 inflammasome orchestrates the onset of inflammation.
Materials and Methods
Cells
J744.2 macrophages with low passages number (average of 12) were cultured at 37°C, 5% CO2, and 85%–98% humidity. Cells were grown until reaching 75% confluency in a T75 culture flask (Thermo Fisher Scientific, Carlsbad, CA) with 10% FBS-supplemented RPM1 media (GIBCO, Thermo Fisher Scientific).
Transfection of miR-223 3p to J744.2 Macrophages
About 8 × 105 J744.2 macrophages were allowed to grow in a 60 mm diameter Petri dish (Thermo Fisher Scientific) following the culture conditions described above. mirVana mmu-miR-223 3p mimic (mature sequence 5′-UGUCAGUUUGUCAAAUACCCCA-3′; Thermo Fisher Scientific) was transfected to J744.2 macrophages in Lipofectamine 3000. Twenty-four hours later, the cells were primed overnight with 100 ng/ml of LPS in the presence of 10% serum. Next, serum concentration was reduced in culture media to 2% and ATP was added at 5 mm for 1 h. The cells were then harvested for gene- and protein-expression analysis.
Mice
Mouse studies were performed under permission and guidelines of the Institutional Animal Care and Use Committee (IACUC) at UCSD. Eight-week-old female C57BL/6N and female B6 Cd45.1 mice at 7 to 8 weeks of age were purchased from Jackson Laboratories (Sacramento, CA). Eight-week-old female miR-223−/− mice purchased from Jackson Laboratories in a B6 Cd45.1 background were allowed to breed at 25°C housing with 12-h light/dark cycle.
Invivofectamine 3-miR-223 3p Packaging
0.6 mg/kg of mmu-miR-223 3p in vivo ready mirVana (mature sequence 5′-UGUCAGUUUGUCAAAUACCCCA-3′) was encapsulated in Invivofectamine 3 (Thermo Fisher Scientific, Carlsbad, CA) according to the manufacturer’s instructions to allow a safe systemic delivery.
miR-223 3p Treatment in LPS/D-GalN-Induced EAH
Eight-week-old female C57BL/6N mice were given a single i.v. injection of Scr-RNA or miR-223 3p loaded at 0.6 mg/kg B.W. in Invivofectamine 3 (Thermo Fisher Scientific). Eight hours after the injection, the mice were fasted overnight and EAH was induced with an i.p. injection of 40 μg/kg of LPS (from Escherichia coli strain 026:B6) in conjunction with 800 mg/kg D-GalN. Three hours later, the mice were euthanized, blood was collected, and the livers were harvested for histological, molecular, and biochemical analysis. Mice injected with PBS served as non-inflamed WT controls, mice treated only with LPS/D-GalN constituted the inflamed controls, and mice given Scr-RNA represented the inflamed miRNA delivery controls. Induction of EAH in miR-223−/− mice was performed as described above.
miR-223 3p Treatment in FFC Diet-Induced Fibrotic NASH
Eight-week-old male C57BL/6N mice were fed with CD and allowed to drink regular water for 1 week. Next, the diet was changed to a high-saturated fat/cholesterol diet (AIN-76 Western Diet, Test Diet, St. Louis, MO) and water was supplemented with sucrose/fructose at 42 g/L for 26 weeks. The combination of this diet with the sucrose/fructose water was termed FFC diet as previously described.36 During the last 2 weeks of the feeding, Scr-RNA or miR-223 3p loaded in Invivofectamine 3 at 1 mg/kg was injected once i.v. every 4 days. Forty-eight hours after the last injection, mice were fasted overnight and euthanized to collect blood and harvest the liver for analysis. A separate group of mice was kept on the CD diet to serve as non-injured WT controls. The mice given Scr-RNA constituted the inflammation controls.
Histology and Immunohistochemistry
5 μm thick formalin-fixed liver sections were preincubated with 3% hydrogen peroxide for 10 min and blocked with 5% BSA at room temperature (RT) for 2 h. Rat anti-Ly6C (1:100 dilution; Abcam, Cambridge, MA) or anti-F4/80 (1:50; Bio-Rad, Hercules, CA) and rabbit anti-MPO (1:100; Thermo Fisher Scientific) or anti-α-SMA (1:500, Abcam) antibodies were incubated on the blocked sections at 4°C in Dako Antibody Diluent (Odense, Denmark), except anti-Ly6C Ab that was diluted in PBS containing 1% BSA + 1% FBS. With the exception of anti-F4/80 Ab that required antigen retrieval in TBS-T with 2% BSA + 1% triton x-100 for 30 min at RT, the remaining Abs were antigen retrieved in near boiling citrate buffer (pH 6.0) for 20 min. Permeabilization with 0.2% tween-20 in PBS for 30 min was required before the blocking step in the sections to be incubated with anti-Ly6C Ab. After overnight incubation with primary Ab, the sections were washed in TBS-tween and incubated with ready-to-use HRP-linked anti-rat or rabbit secondary immunoglobulin G (IgG) antibody (Immpress HRP reagents; Vector Labs, Burlingame, CA) for 1 h at RT. Color was developed with DAB solution (Vector Labs) for up to 10 s. Nuclei were then counterstained with Mayer’s hematoxylin (Sigma-Aldrich, St. Louis, MO) for 2 min, followed by dehydration and embedding in glycerol. F4/80, Ly6C, or MPO-positive cells were quantified in 10 randomly selected fields (×200 magnification) imaged with a Nanozoomer 2.0HT Slide Scanner microscope (Hamamatsu Photonics K.K., Hamamatsu, Japan). The total stained area was analyzed by selecting brown areas using an unchanged threshold value in the macro function of ImageJ (NIH, Bethesda, MD). Results were represented as the percentage of total area occupied by positive cells per field in each specimen.
To identify collagen accumulation in liver tissue, we stained formalin-fixed hepatic sections of 5 μm thickness for 2 h at RT with an aqueous solution of saturated picric acid (Sigma-Aldrich) mixed with 0.1% Fast Green fetal calf serum (FCS) and 0.1% Direct Red Dye (Sigma-Aldrich). Ten randomly selected fields (×100 magnification) were imaged and photographed as above, and the percentage of Sirius Red-stained area was measured by ImageJ software with an adjusted unchanged threshold. Results are expressed as percentage of area occupied by Sirius Red-stained fibrils per field in each section.
Staining of overall apoptotic cells in 5 μm thick formalin-fixed liver sections was performed by the TUNEL assay according to the manufacturer’s instructions (ApopTag peroxidase in situ Apoptosis Detection kit, Millipore, Billerica, MA, USA). Images were also analyzed by ImageJ and results are represented as average percentage of TUNEL-positive area in each image.
Immunofluorescence
3 × 105 cells were plated overnight onto a polylysine-coated coverslip placed in a 6-well plate and were cultured, transfected, and primed as detailed above. After priming, the cells were washed and fixed with 4% paraformaldehyde for 10 min at RT. Next, the cells were washed, permeabilized (0.2% tween 20 in PBS 30 min), and incubated at 4°C with goat polyclonal anti-caspase-1 p10 (1:50; Santa Cruz Biotechnology, Santa Cruz, CA) in Dako Ab Diluent with Background Reducing Components. After overnight incubation, the cells were washed and treated with Alexa Fluor 598 anti-goat IgG Ab (1:1,000; Invitrogen, Carlsbad, CA) for 1 h at RT in the dark, followed by washing and 5 min nuclei staining with DAPI diluted at 1:200 in PBS. The coverslip was then carefully removed, and the cells were mounted onto a glass slide with Dako Fluorescent Mounting Media and analyzed with an Olympus FV1000 Confocal Microscope (Shinjuku, Tokyo, Japan) by repeat x-y scanning at 12.5 resolution pixels and unchanged lasers powers. Results were expressed as a percentage of anti-caspase-1 p10 normalized by total number of nuclear cells in each field (×450 magnification).
FAM-FLICA staining to determine the activation of caspase-1 in 5 μm cryo-sectioned liver biopsy was performed by diluting the FAM-FLICA reagent 1:200 and following the remaining steps as described in the manufacturer’s instructions.
Real-Time PCR
Total RNA was isolated with TRIzol reagent (Sigma-Aldrich). 1 μg of total RNA was reverse transcribed into cDNA using the iScript cDNA Synthesis Kit (Bio-Rad). The following mouse SYBR green primers were purchased from Integrated DNA Technologies (ITD, Skokie, IL): ASC forward 5′-CTTGTCAGGGGATGAACTCAAAA-3′; reverse 5′-GCCATACGACTCCAGATAGTAGC-3′; B2m forward 5′-CCCCACTGAGACTGATACATA CG-3′; reverse 5′-CGATCCCAGTAGACGGTCTTG-3′; Ccl2 forward 5′-TTAAAAACCTGGATCGGAACCAA-3′; reverse 5′-GCATTAGCTTCAGATTTACGGGT-3′; CCL3 forward 5′-TTCTCTGTACCATGACACTCTGC-3′; reverse 5′-CGTGGAATCTTCCGGCTGTAG-3′; Ccl4 forward 5′-TTCCTGCTGTTTCTCTTACACC-3′; reverse 5′-CTGTCTGCCTCTTTTGGTCAG-3′; CXCL1 forward 5′-CTGGGATTCACCTCAAGAACATC-3′; reverse 5`-CAGGGTCAAGGCAAGCCTC-3′; CXCL2 forward 5′-CCACCACCAGGCTACAGG-3′; Il1β forward 5`-GCAACTGTTCCTGAACTCAACT-3′; reverse 5′-ATCTTTTGGGGTCCGTCAACT-3′; Il6 forward 5`-TAGTCCTCCCTACCCCAATTTCC-3′; reverse 5′-TTGGTCCTTAGCCACTCCTTC-3′; Il12 p35 forward 5′-GAGGACTTGAAGATGTACCAG-3′; reverse 5′-TTCTATCTGTGTGAGGAGGGC-3′; Nlrp3 forward 5′-ATTACCCGCCCGAGAAAGG-3′; reverse 5′-TCGCAGCAAAGATCCACACAG-3′; Tnfα forward 5′-CCCTCACACTCAGATCATCTTCT-3′; and reverse 5′-GCTACGTGGGCTACAG-3′.
B2m or Hrtp1 was used to normalize data and to control for RNA integrity. cDNA amplification reactions were performed using a CFX96 real-time system (Bio-Rad). SYBR green reaction mixtures were purchased from Applied Biosystems. Gene-expression data were calculated by the ΔΔCT equation.
Immunoblot Analysis
Total protein fraction was extracted from approximately 100 mg of liver biopsy or 8 × 105 J744.2 macrophages, with RIPA lysis buffer for 30 min at 4°C. 20 or 40 μg of total tissue protein, or 10 μg of cellular protein (J744.2 macrophages), were loaded in 5% β-2-mercaptoethanol-contained SDS, boiled for 5 min, and electrophoresed via an Any kDa precast Tris-Glycine gel (Bio-Rad) at 60 v for 15 min, followed by 100 v until the end of the run. Tris-glycine-SDS at pH 8.3 was used as a running buffer. Twenty μg of protein was used to determine proteins with molecular weight (MW) higher than 90 kDa, whereas 40 μg was employed for proteins smaller than 60 kDa. After electrophoresis, the proteins were transferred from the gel to a nitrocellulose membrane in a Trans Blot Turbo Transfer system (Bio-Rad) for 7 min (for low MW proteins; <30 kDa) or 30 min (for mid and high MW proteins; >60 kDa) at 25 v. An ethanol-based solution was used as transfer buffer (Bio-Rad). Once proteins were transferred, blocking of the membrane was proceeded for 1 h in TBS-tween-diluted 5% dried milk. The membrane was then incubated with rabbit anti-cryopyrin (NLRP3, 1:1,000, Santa Cruz Biotechnology), anti-IL-1β (1:1,000, Abcam), anti-procaspase-1 (1:1,600, Abcam) or anti-ASC (1:1,000, Adipogen), and mouse anti-GAPDH (1:2,500; GeneTex, Irvine, CA, USA) or anti-α-tubulin (1:4,000, GeneTex) polyclonal antibodies at 4°C. After overnight incubation with primary antibody, the membrane was washed three times in TBS-tween and incubated with HRP-linked anti-mouse or anti-rabbit IgG antibodies for 1 h at RT. Finally, protein bands were visualized with an enhanced chemiluminescence reagent (Pico or Femto; Thermo Fisher Scientific) and digitized in a CCD camera (ChemiDoc, Bio-Rad). Intensity of bands was quantified with ImageJ. Results are expressed as arbitrary units of protein expression normalized against the housekeeping protein GAPDH or anti-α-tubulin.
Serum ALT
Mouse serum collected from 2 h-coagulated blood at RT was subjected to ALT analysis according to the manufacturer’s instruction (InfinityTM ALT; Thermo Fisher Scientific, Carlsbad, CA).
Mouse Serum IL-1β
Mouse IL-1β was quantified in 13 μl diluted serum (1:4) according to the manufacturer’s instructions (Mouse IL-10 ELISA Kit; Invitrogen), except 3,3',5,5'-tetramethylbenzidine (TMB) substrate addition that was replaced by chemiluminescent SuperSignal ELISA Femto (1:1 dilution; Thermo Fisher Scientific) incubated from 1 to 5 min in the dark. Results were calculated from luminescence units in linear regression standard curve.
Statistics
Significances of two group comparisons were determined with two-tailed Student’s t test. Significances of more than two groups were analyzed by one-way ANOVA. Significances of multiple comparisons between two or more groups were measured with two-way ANOVA. Tukey’s post hoc test was used to correct both one- and two-way ANOVA. p values of < 0.05 were considered significant. Error bars are represented as means ± SD. Experiments were repeated at least two or three times and assays were performed in duplicates or triplicates.
Author Contributions
C.J.C. performed and designed the experiments, analyzed the data, wrote the manuscript, and partially conceived the idea; H.D.P., M.T., and C.D.J. provided technical help; A.E.F. conceived the idea, helped on the design of the experiments, and critically revised the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
These studies were supported by the NIH (R01 DK113592 and R01 AA024206 to A.E.F.) and the microscopy core facility of the Department of Neuroscience at the UCSD (grant NS047101).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.09.013.
Supplemental Information
References
- 1.Del Campo J.A., Gallego P., Grande L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J. Hepatol. 2018;10:1–7. doi: 10.4254/wjh.v10.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman S.L. Liver fibrosis -- from bench to bedside. J. Hepatol. 2003;38(Suppl 1):S38–S53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 3.Jo E.K., Kim J.K., Shin D.M., Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016;13:148–159. doi: 10.1038/cmi.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim J.H., Um H.J., Park J.W., Lee I.K., Kwon T.K. Interleukin-1beta promotes the expression of monocyte chemoattractant protein-1 in human aorta smooth muscle cells via multiple signaling pathways. Exp. Mol. Med. 2009;41:757–764. doi: 10.3858/emm.2009.41.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parry G.C., Martin T., Felts K.A., Cobb R.R. IL-1beta-induced monocyte chemoattractant protein-1 gene expression in endothelial cells is blocked by proteasome inhibitors. Arterioscler. Thromb. Vasc. Biol. 1998;18:934–940. doi: 10.1161/01.atv.18.6.934. [DOI] [PubMed] [Google Scholar]
- 6.Amanzada A., Moriconi F., Mansuroglu T., Cameron S., Ramadori G., Malik I.A. Induction of chemokines and cytokines before neutrophils and macrophage recruitment in different regions of rat liver after TAA administration. Lab. Invest. 2014;94:235–247. doi: 10.1038/labinvest.2013.134. [DOI] [PubMed] [Google Scholar]
- 7.Nourshargh S., Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41:694–707. doi: 10.1016/j.immuni.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Dykxhoorn D.M., Novina C.D., Sharp P.A. Killing the messenger: short RNAs that silence gene expression. Nat. Rev. Mol. Cell Biol. 2003;4:457–467. doi: 10.1038/nrm1129. [DOI] [PubMed] [Google Scholar]
- 9.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 10.Johnnidis J.B., Harris M.H., Wheeler R.T., Stehling-Sun S., Lam M.H., Kirak O., Brummelkamp T.R., Fleming M.D., Camargo F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- 11.Cheung O., Puri P., Eicken C., Contos M.J., Mirshahi F., Maher J.W., Kellum J.M., Min H., Luketic V.A., Sanyal A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology. 2008;48:1810–1820. doi: 10.1002/hep.22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauernfeind F., Rieger A., Schildberg F.A., Knolle P.A., Schmid-Burgk J.L., Hornung V. NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 2012;189:4175–4181. doi: 10.4049/jimmunol.1201516. [DOI] [PubMed] [Google Scholar]
- 13.Eguchi A., De Mollerat Du Jeu X., Johnson C.D., Nektaria A., Feldstein A.E. Liver Bid suppression for treatment of fibrosis associated with non-alcoholic steatohepatitis. J. Hepatol. 2016;64:699–707. doi: 10.1016/j.jhep.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y., Rui M., Tang H., Xu Y. The distribution and cell uptake of ApoA1 modified lipid carriers of siRNA in mouse liver in vivo. A. J. P. S. 2013;8:228–233. [Google Scholar]
- 15.Guarda G., Zenger M., Yazdi A.S., Schroder K., Ferrero I., Menu P., Tardivel A., Mattmann C., Tschopp J. Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 2011;186:2529–2534. doi: 10.4049/jimmunol.1002720. [DOI] [PubMed] [Google Scholar]
- 16.Hadadi E., Zhang B., Baidžajevas K., Yusof N., Puan K.J., Ong S.M., Yeap W.H., Rotzschke O., Kiss-Toth E., Wilson H., Wong S.C. Differential IL-1β secretion by monocyte subsets is regulated by Hsp27 through modulating mRNA stability. Sci. Rep. 2016;6:39035. doi: 10.1038/srep39035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conti P., Dempsey R.A., Reale M., Barbacane R.C., Panara M.R., Bongrazio M., Mier J.W. Activation of human natural killer cells by lipopolysaccharide and generation of interleukin-1 alpha, beta, tumour necrosis factor and interleukin-6. Effect of IL-1 receptor antagonist. Immunology. 1991;73:450–456. [PMC free article] [PubMed] [Google Scholar]
- 18.Wree A., Eguchi A., McGeough M.D., Pena C.A., Johnson C.D., Canbay A., Hoffman H.M., Feldstein A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59:898–910. doi: 10.1002/hep.26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coll R.C., Robertson A.A.B., Chae J.J., Higgins S.C., Muñoz-Planillo R., Inserra M.C., Vetter I., Dungan L.S., Monks B.G., Stutz A. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015;21:248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim S.J., Lee S.M. NLRP3 inflammasome activation in D-galactosamine and lipopolysaccharide-induced acute liver failure: role of heme oxygenase-1. Free Radic. Biol. Med. 2013;65:997–1004. doi: 10.1016/j.freeradbiomed.2013.08.178. [DOI] [PubMed] [Google Scholar]
- 21.Szabo G., Petrasek J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015;12:387–400. doi: 10.1038/nrgastro.2015.94. [DOI] [PubMed] [Google Scholar]
- 22.Wu X., Dong L., Lin X., Li J. Relevance of the NLRP3 inflammasome in the pathogenesis of chronic liver disease. Front. Immunol. 2017;8:1728. doi: 10.3389/fimmu.2017.01728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Y., Hwang S., Cai Y., Kim S.J., Xu M., Yang D., Guillot A., Feng D., Seo W., Hou X., Gao B. 2019. MicroRNA-223 ameliorates nonalcoholic steatohepatitis and cancer by targeting multiple inflammatory and oncogenic genes in hepatocytes. Hepatol. Published online April 9, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye D., Zhang T., Lou G., Liu Y. Role of miR-223 in the pathophysiology of liver diseases. Exp. Mol. Med. 2018;50:128. doi: 10.1038/s12276-018-0153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neudecker V., Haneklaus M., Jensen O., Khailova L., Masterson J.C., Tye H., Biette K., Jedlicka P., Brodsky K.S., Gerich M.E. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017;214:1737–1752. doi: 10.1084/jem.20160462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan J., Li S., Li S. The role of the liver in sepsis. Int. Rev. Immunol. 2014;33:498–510. doi: 10.3109/08830185.2014.889129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ratziu V., Goodman Z., Sanyal A. Current efforts and trends in the treatment of NASH. J. Hepatol. 2015;62(1, Suppl):S65–S75. doi: 10.1016/j.jhep.2015.02.041. [DOI] [PubMed] [Google Scholar]
- 28.Arango Duque G., Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front. Immunol. 2014;5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J., Tian Y., Phillips K.L.E., Chiverton N., Haddock G., Bunning R.A., Cross A.K., Shapiro I.M., Le Maitre C.L., Risbud M.V. Tumor necrosis factor α- and interleukin-1β-dependent induction of CCL3 expression by nucleus pulposus cells promotes macrophage migration through CCR1. Arthritis Rheum. 2013;65:832–842. doi: 10.1002/art.37819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramos C.D.L., Canetti C., Souto J.T., Silva J.S., Hogaboam C.M., Ferreira S.H., Cunha F.Q. MIP-1alpha[CCL3] acting on the CCR1 receptor mediates neutrophil migration in immune inflammation via sequential release of TNF-alpha and LTB4. J. Leukoc. Biol. 2005;78:167–177. doi: 10.1189/jlb.0404237. [DOI] [PubMed] [Google Scholar]
- 31.Yu J., Nagasu H., Murakami T., Hoang H., Broderick L., Hoffman H.M., Horng T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA. 2014;111:15514–15519. doi: 10.1073/pnas.1414859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miao E.A., Rajan J.V., Aderem A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011;243:206–214. doi: 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Potthoff M.J., Olson E.N. MEF2: a central regulator of diverse developmental programs. Development. 2007;134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- 34.Holzenberger M., Dupont J., Ducos B., Leneuve P., Géloën A., Even P.C., Cervera P., Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 35.Delafontaine P., Song Y.H., Li Y. Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1 binding proteins in blood vessels. Arterioscler. Thromb. Vasc. Biol. 2004;24:435–444. doi: 10.1161/01.ATV.0000105902.89459.09. [DOI] [PubMed] [Google Scholar]
- 36.Charlton M., Krishnan A., Viker K., Sanderson S., Cazanave S., McConico A., Masuoko H., Gores G. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2011;301:G825–G834. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.