

Abstract
The antiproliferative antimicrobial fungal metabolites known as the myrocins have been proposed to cross-link DNA by double nucleotide addition. However, the nature of the DNA-reactive species is ambiguous, as myrocins have been isolated as functionally distinct 5-hydroxy-γ-lactone and diosphenol isomers. Based on literature precedent, we hypothesized that the diosphenol 7 (assigned here the trivial name myrocin G) is the biologically active form of the representative isolate (+)-myrocin C (1). To probe this, we developed a short enantioselective route to 7. A powerful fragment-coupling reaction that forms the central ring of the target in 38% yield and in a single step was developed. In support of our hypothesis, 7 was efficiently transformed to the bis(sulfide) 6, a product previously isolated from reactions of 1 with excess benzenethiol. This work provides the first direct access to the diosphenol 7, sets the stage for elucidating the mode of interaction of the myrocins with DNA, and provides a foundation for the synthesis of other pimarane diterpenes.
Efforts to elucidate the mechanism of action of natural products are complicated when the metabolite can adopt two or more functionally distinct forms. This issue is exemplified by the antiproliferative antimicrobial metabolites myrocins C (1)1 and B (2),2,3 fungal isolates that contain a sensitive 5-hydroxy-γ-lactone residue (Scheme 1A, blue in 1 and 2). The literature indicates4 this substructure undergoes facile ring opening to the corresponding diosphenol under mildly acidic or basic conditions, raising uncertainty about its fidelity under biological conditions. Consistent with this, the diosphenol isomer of 2, (−)-myrocin A (3), has been identified in fungal cultures.5
Scheme 1.

(A) Structures of Myrocins A–C (1–3); (B) Structure of the Bis(sulfide) 6 and the Originally Proposed Mechanism for Its Formation; (C) Diosphenol 7, Which is Hypothesized to Be the Biologically-Active Form of 1; (D) Retrosynthetic Analysis of 7
Following their landmark total synthesis of (±)-myrocin C (1),6 Chu-Moyer and Danishefsky disclosed that treatment of synthetic (±)-1 with excess thiophenol and triethylamine generated the bis(sulfide) 6 (63%, Scheme 1B).7 The mechanism for formation of 6 was proposed to comprise SN2′ substitution of the tertiary hydroxyl group (1 → 4), isomerization to the diosphenol 5, and addition to the resulting activated cyclopropane. This reactivity led the authors to speculate that the myrocins cross-link DNA by sequential nucleotide addition reactions.8
The isolation of 3 suggests the existence of an analogous diosphenol isomer of 1, “myrocin G (7)” (Scheme 1C). Ring opening of a lactone structurally related to 1 has been reported to occur under acidic or basic conditions.4b The diosphenol isomer may be stabilized by a strong intramolecular hydrogen bond between the hydroxyl group and the adjacent carbonyl. Collectively, these data suggested to us an alternative order of events for 1 → 6 wherein ring opening of 1 to the diosphenol 7 precedes the initial alkylation.
Motivated by this analysis, we targeted 7 as the initial entry into this natural product family. In addition, we identified the diosphenol double bond as a strategic locus that could be converted retrosynthetically to the diketone 8 in a redox-neutral fashion (Scheme 1D). Further disconnection of the C9–C10 bond by a fragment-coupling reaction9 reveals the α,β-cyclopropylketone 9 and the unsaturated ketone 10 as two precursors of similar complexity. This synthetic strategy features the direct, stereocontrolled installation of the C9 alcohol, high modularity, and independent introduction of the peripheral C4 and C13 quaternary centers.
The coupling fragments 9 and 10 were prepared in three steps from known compounds (Scheme 2). Beginning with the Diels–Alder adduct 11,10 Wittig olefination [potassium bis(trimethylsilyl)amide, methyl triphenylphosphonium bromide], tandem enoxysilane hydrolysis and β-carbamate elimination (aqueous hydrochloric acid), and α-dehydroiodination (iodine, pyridine)11 provided the C-ring fragment 10 (22% over three steps, Scheme 2A). The A-ring fragment 9 was synthesized from the β-ketoester 12 (Scheme 2B).12 Stereoselective Robinson annulation13 between 12 and acrolein diethylacetal provided the enone 13 (32%, 92% ee). α-Dehydroiodination11 of 13 proceeded in 97% yield. Corey–Chaykovsky cyclopropanation14 (trimethylsulfoxonium iodide, sodium hydride) provided a 2.3:1 mixture of diastereomeric α,β-cyclopropylketones. The major (desired) diastereomer 9 was isolated in 64% yield after recrystallization.
Scheme 2. Synthesis of the Fragment Coupling Partners 9 and 10a.

aTMSE = 2-(trimethylsilyl)ethyl.
The fragment-coupling product 14 was obtained by activation of the iodocyclopropane 9 with iso-propylmagnesium chloride–lithium chloride complex,15 followed by addition of the α-iodoenone 10 (92%, 8.2:1 dr, Scheme 3). The stereoselectivity in the coupling was anticipated based on the known stereoelectronic preferences for nucleophilic addition to cycloalkanones16 and consideration of nonbonded interactions in the transition state. Notably, retro-aldol reaction of 14 is prevented by the geometric constraints imposed by the cyclopropane ring.
Scheme 3.

Synthesis of 19
The ring closure precursor 16 was prepared by a five-step sequence comprising Stille cross-coupling [tetrakis(triphenylphosphine)palladium(0), copper(I) iodide, cesium fluoride] with tributyl(1-ethoxyvinyl) tin, hydrolysis of the resulting vinyl ether (aqueous hydrochloric acid), tandem alcohol silylation–enoxysilane formation (trimethylsilyl trifluoromethanesulfonate, triethylamine), Rubottom oxidation (3-chloroperoxybenzoic acid), and conversion of the primary alcohol to an allyl carbonate (allyl chloroformate, pyridine, 37% overall). The intermediate Rubottom oxidation product was desilylated and the structure of the desilylated product was confirmed by X-ray analysis.17
After much experimentation, we found that treatment of the allyl carbonate 16 with sodium tert-butoxide in tetrahydrofuran at 0 °C generated the diosphenol 19 (64%). Mechanistic studies suggest that 19 is formed via aldol addition (16 → 17), carbonate migration (17 → 18), and β-elimination. The silyl migration product 20 was isolated separately in 15% yield.
This cyclization cascade provides expedient access to a protected form of myrocin G (7). After some consideration, we recognized that the fragment-coupling–ring-closure cascade could potentially be carried out in one flask by embedding a latent enolate nucleophile in the C-ring electrophile. Toward this end, we prepared the enoxysilane 24 by the sequence shown in Scheme 4. Beginning with the α-iodoenone 10, ketalization (ethylene glycol, triethylorthoformate, p-toluenesulfonic acid) followed by lithium–halogen exchange and addition of the Weinreb amide18 21 provided the α,β-unsaturated ketone 22 (70%, two steps). Removal of the silyl ether (tetra-n-butylammonium fluoride), installation of the allyl carbonate (allyl chloroformate, pyridine), and removal of the acetal (aqueous hydrochloric acid) generated the β-diketone 23 (46%, three steps). Site-selective deprotonation of 23 [lithium bis(trimethylsilyl)amide] and trapping of the resulting enolate with chlorotrimethylsilane provided the target enoxysilane 24 (59%).
Scheme 4.
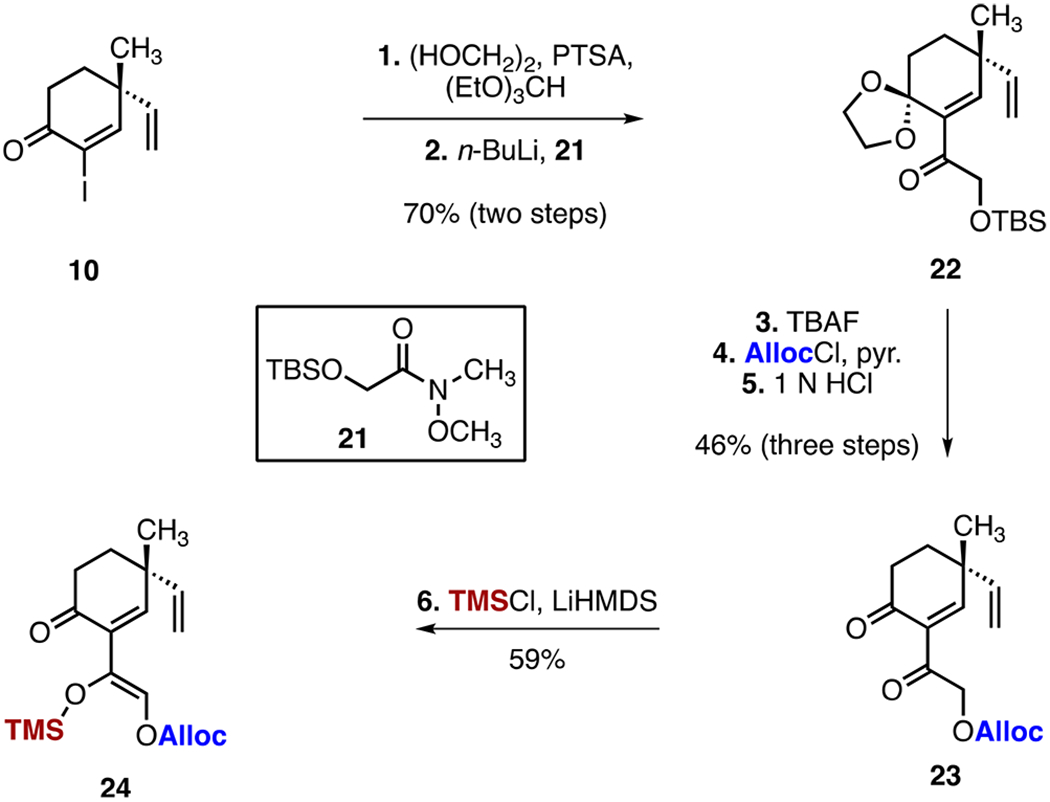
Synthesis of the Enoxysilane 24
Attempts to effect the fragment coupling of the enoxysilane 24 with the organomagnesium reagent derived from 9 were unsuccessful. We found, however, that lithium–halogen exchange (n-butyllithium, −78 °C), followed by immediate addition of the enoxysilane 24 and warming to 0 °C provided the fully annulated product 19 in 38% yield (Scheme 5). In this coupling reaction, the ring closure is triggered by migration of the trimethylsilyl substituent in the addition product 26. The modest yield of this transformation is offset to some extent by the rapid increase in molecular complexity achieved. Deprotection of 18 (tetra-n-butylammonium fluoride) then provided myrocin G (7, 64%). Subjecting synthetic 7 to the conditions disclosed by Chu-Moyer and Danishefsky7 generated the bis(sulfide) 6 (74%, Scheme 6). This result indicates that myrocin G (7) is a competent intermediate in the double nucleophilic addition of thiols and provides support for our hypothesis. We reasoned that (+)-myrocin C (1) itself might be accessible by temporarily disrupting the diosphenol hydrogen bond in 19 or 7, thereby making lactonization thermodynamically favorable. However, exploratory experiments toward this end have thus far been unsuccessful.
Scheme 5.

One-Step Synthesis of 19 from 9 and 24
Scheme 6.

Synthesis of the Bis(sulfide) 6 from the Diosphenol 7
In summary, we have developed a concise, enantioselective synthesis of myrocin G (7), the putative active form of the antiproliferative antimicrobial metabolite myrocin C (1). Key to the success of this approach was the development of a powerful annulation strategy that forges the central ring of the target in a single step from two synthetic precursors of similar complexity. With access to 7, we are now in position to probe its antiproliferative activity and determine its biological target. The fragment-coupling reaction we have developed should be amenable to the synthesis of other pimarane diterpenes.
Supplementary Material
ACKNOWLEDGMENTS
Financial support from the National Institutes of Health (Ruth L. Kirschstein National Research Service Award F31CA213964-03 to C.E.) and Yale University is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b10891.
X-ray structure determination for 007b-18079 (CIF) Detailed experimental procedures and characterization data for all new compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Hsu YH; Hirota A; Shima S; Nakagawa M; Nozaki H; Tada T; Nakayama M Structure of Myrocin C, A New Diterpene Antibiotic Produced by a Strain of Myrothecium sp. Agric. Biol. Chem 1987, 51, 3455. [Google Scholar]; (b) Nakagawa M; Hsu YH; Hirota A; Shima S; Nakayama M; Myrocin C, A New Diterpene Antitumor Antibiotic from Myrothecium Verrucaria. I. Taxonomy of the Producing Strain, Fermentation, Isolation and Biological Properties. J. Antibiot 1989, 42, 218. [DOI] [PubMed] [Google Scholar]; (c) Hsu YH; Hirota A; Shima S; Nakagawa M; Adachi T; Nozaki H; Nakayama M Myrocin C, A New Diterpene Antitumor Antibiotic from Myrothecium verrucaria. II. Physicochemical Properties and Structure Determination. J. Antibiot 1989, 42, 223. [DOI] [PubMed] [Google Scholar]
- (2).(a) Hsu YH; Nakagawa M; Hirota A; Shima S; Nakayama M Structure of Myrocin B, A New Diterpene Antibiotic Produced by Myrothecium verrucaria. Agric. Biol. Chem 1988, 52, 1305. [Google Scholar]; (b) Lehr N-A; Meffert A; Antelo L; Sterner O; Anke H; Weber RWS Antiamoebins, myrocin B and the basis of antifungal antibiosis in the coprophilous fungus Stilbella erythrocephala (syn. S. fimetaria). FEMS Microbiol. Ecol 2006, 55, 105. [DOI] [PubMed] [Google Scholar]
- (3).For a review of pimarane diterpenes, see:; Wang X; Yu H; Zhang Y; Lu X; Wang B; Liu X Bioactive Pimarane-type Diterpenes from Marine Organisms. Chem. Biodiversity 2018, 15, No. e1700276. [DOI] [PubMed] [Google Scholar]
- (4).(a) Langschwager W; Hoffmann HMR Ring-Chain Tautomerism Provides a Route to 7a-Hydroxy-3a-methyl-2,7-dioxoperhydrobenzofuran. Synthesis of the Hydroxy γ-Lactone Substructure of Myrocin and Other Bioactive Natural Products. Liebigs Ann. 1995, 797. [Google Scholar]; (b) Zander N; Langschwager W; Hoffmann HMR The Enol Lactone Approach to Protected Hydroxy γ-Lactones (5-Hydroxy-dihydro-furan-2-ones). Synth. Commun 1996, 26, 4577. [Google Scholar]
- (5).(a) Klemke C; Kehraus S; Wright AD; König GM New Secondary Metabolites from the Marine Endophytic Fungus Apiospora montagnei. J. Nat. Prod 2004, 67, 1058. [DOI] [PubMed] [Google Scholar]; (b) Tsukada M; Fukai M; Miki K; Shiraishi T; Suzuki T; Nishio K; Sugita T; Ishino M; Kinoshita K; Takahashi K; Shiro M; Koyama K Chemical Constituents of a Marine Fungus, Arthrinium sacchari. J. Nat. Prod 2011, 74, 1645. [DOI] [PubMed] [Google Scholar]
- (6).(a) Chu-Moyer MY; Danishefsky SJ A Remarkable Cyclopropanation: The Total Synthesis of Myrocin C. J. Am. Chem. Soc 1992, 114, 8333. [Google Scholar]; (b) Chu-Moyer MY; Danishefsky SJ; Schulte GK Total Synthesis of (±)-Myrocin C. J. Am. Chem. Soc 1994, 116, 11213. [Google Scholar]
- (7).Chu-Moyer MY; Danishefsky SJ The Mode of Action of Myrocin C: Evidence For a CC-1065 Connection. Tetrahedron Lett. 1993, 34, 3025. [Google Scholar]
- (8).For a review of natural products that damage DNA by nucleotide addition to an activated cyclopropane, see:; Wolkenberg SE; Boger DL Mechanisms of in Situ Activation for DNA-Targeting Antitumor Agents. Chem. Rev 2002, 102, 2477. [DOI] [PubMed] [Google Scholar]
- (9).This disconnection drew inspiration from a related elegant fragment-coupling employed by Baran and co-workers en route to (–)-maoecrystal V. See; Cernijenko A; Risgaard R; Baran PS 11-Step Total Synthesis of (–)-Maoecrystal V. J. Am. Chem. Soc 2016, 138, 9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Huang Y; Iwama T; Rawal VH Highly Enantioselective Diels-Alder Reactions of 1-Amino-3-siloxy-dienes Catalyzed by Cr(III)-Salen Complexes. J. Am. Chem. Soc 2000, 122, 7843. [Google Scholar]
- (11).Johnson CR; Adams JP; Braun MP; Senanayake CBW; Wovkulich PM; Uskokovic MR Direct α-Iodination of Cycloalkenones. Tetrahedron Lett. 1992, 33, 917. [Google Scholar]
- (12).(a) Ueda Y; Roberge G; Vinet V A simple method of preparing trimethylsilyl- and tert-butyldimethylsilyl-enol ethers of α-diazoacetoacetates and their use in the synthesis of a chiral precursor to thienamycin analogs. Can. J. Chem 1984, 62, 2936. [Google Scholar]; (b) Meng Z; Yu H; Li L; Tao W; Chen H; Wan M; Yang P; Edmonds DJ; Zhong J; Li A Total synthesis and antiviral activity of indolosesquiterpenoids from the xiamycin and oridamycin families. Nat. Commun 2015, 6, 6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Xu C; Zhang L; Luo S Asymmetric Enamine Catalysis with β-Ketoesters by Chiral Primary Amine: Divergent Stereocontrol Modes. J. Org. Chem 2014, 79, 11517. [DOI] [PubMed] [Google Scholar]
- (14).Corey EJ; Chaykovsky M Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J. Am. Chem. Soc 1965, 87, 1353. [Google Scholar]
- (15).(a) Ren H; Krasovskiy A; Knochel P Preparation of cyclic alkenylmagnesium reagents via an iodine/magnesium exchange. Chem. Commun 2005, 543. [DOI] [PubMed] [Google Scholar]; For a review, see:; (b) Klatt T; Markiewicz JT; Samann C; Knochel P Strategies to Prepare and Use Functionalized Organometallic Reagents. J. Org. Chem 2014, 79, 4253. [DOI] [PubMed] [Google Scholar]
- (16).Bürgi HB; Dunitz JD; Lehn JM; Wipff G Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 1974, 30, 1563. [Google Scholar]
- (17).See the Supporting Information.
- (18).Nahm S; Weinreb SM N-Methoxy-N-Mmethylamides as Effective Acylating Agents. Tetrahedron Lett. 1981, 22, 3815. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.