

Abstract
Exosomes are nanosized membranous vesicles secreted by a variety of cells. Due to their unique and pharmacologically important properties, cell-derived exosome nanoparticles have drawn significant interest for drug development. By genetically modifying exosomes with two distinct types of surface-displayed monoclonal antibodies, we have developed an exosome platform termed synthetic multivalent antibodies retargeted exosome (SMART-Exo) for controlling cellular immunity. Here, we apply this approach to human epidermal growth factor receptor 2 (HER2)-expressing breast cancer by engineering exosomes through genetic display of both anti-human CD3 and anti-human HER2 antibodies, resulting in SMART-Exos dually targeting T cell CD3 and breast cancer-associated HER2 receptors. By redirecting and activating cytotoxic T cells toward attacking HER2-expressing breast cancer cells, the designed SMART-Exos exhibited highly potent and specific anti-tumor activity both in vitro and in vivo. This work demonstrates preclinical feasibility of utilizing endogenous exosomes for targeted breast cancer immunotherapy and the SMART-Exos as a broadly applicable platform technology for the development of next-generation immuno-nanomedicines.
Keywords: antibodies, breast cancer, exosome, immunotherapy, protein engineering
Graphical Abstract
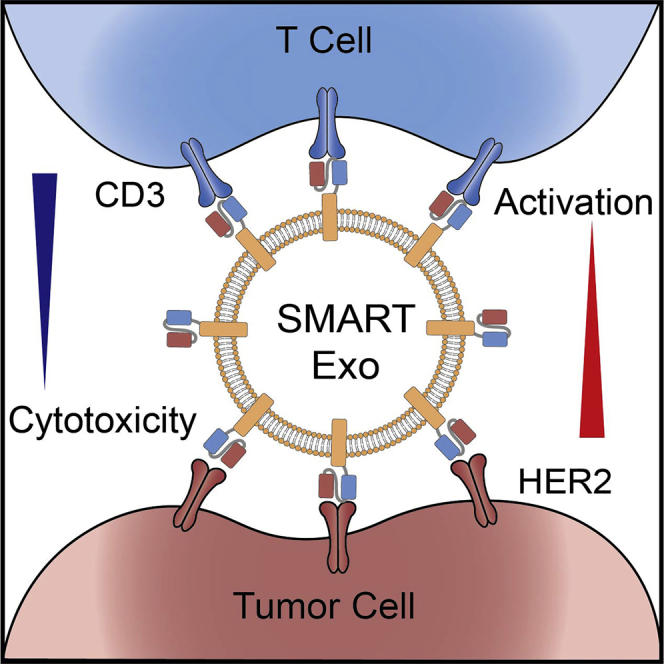
Zhang and colleagues report that reprogramming cell-derived exosomes with distinct types of monoclonal antibodies resulted in synthetic multivalent antibodies retargeted exosomes (SMART-Exos) displaying excellent potency and specificity in redirecting and activating T cells toward HER2-positive breast cancer cells for destruction, which may lead to an innovative class of exosome-based immunotherapeutics.
Introduction
Exosomes are phospholipid bilayer membranous vesicles secreted by various types of cells. By serving as nanocarriers of nucleic acids, proteins, and lipids, endogenous exosomes are known to play important roles in mediating intercellular communications through fusions with recipient cells, transfer of membrane receptors, and/or direct stimulations by surface receptor interactions.1, 2, 3, 4, 5, 6 In contrast with conventional nanoparticles, naturally occurring exosomes are characterized by abundant membrane proteins, high biocompatibility, and low immunogenicity, conferring unique and important properties for therapeutic development. For instance, tetraspanin CD9 abundantly expressed in exosomes can facilitate rapid membrane fusion with target cells, which enables direct cytosolic delivery of endogenous and exogenous therapeutic agents.7 Moreover, exosomal surface CD47 can protect exosomes from phagocytosis by the mononuclear phagocyte system.8 Clinically, human cell-derived exosomes are unlikely to have immunogenicity issues encountered by viral and synthetic nanocarriers.7
By leveraging these valuable characteristics, endogenously derived exosomes have been increasingly utilized for the delivery of various types of cargos.9, 10, 11, 12, 13, 14, 15, 16, 17 A number of preclinical and clinical studies demonstrate promising efficacy and safety for exosome-based therapeutics in the treatment of many human diseases, including cancer, immune disorders, ischemic diseases, liver diseases, and neurodegenerative diseases.9,13,15,18, 19, 20, 21, 22 Given their therapeutic potential, exosomes have drawn significantly growing interest for nanomedicine development. Recently, we reported the development of an exosome platform named synthetic multivalent antibodies retargeted exosome (SMART-Exo) that functions as an artificial modulator of cellular immunity to redirect immune effector cells and control their immunoreactivity.23 By genetically displaying two types of monoclonal antibodies on exosome surfaces, which are specific for human CD3 and epidermal growth factor receptor (EGFR), SMART-Exos simultaneously recognizing T cell surface CD3 and cancer cell-associated EGFR were generated and demonstrated to elicit highly potent in vitro and in vivo anti-cancer immunity in a controlled and directed fashion.
Herein, to explore the generality of this platform, we apply this approach to human epidermal growth factor receptor 2 (HER2)-expressing breast cancer, which accounts for 25%–30% of the most commonly diagnosed cancers among women worldwide.24 We rationally designed an innovative class of SMART-Exos (Figure 1), which feature genetically encoded anti-human CD3 and anti-human HER2 antibodies on the exosome surface. The resulting SMART-Exos dually targeting T cell CD3 and HER2 receptors were shown to not only recruit human T cells to HER2-positive breast cancer cells but also induce highly potent and specific killing of HER2-expressing breast cancer cells in the presence of non-activated human peripheral blood mononuclear cells (PBMCs). Importantly, in vivo studies using mouse xenograft models indicate excellent anti-tumor activities for the SMART-Exos. This study provides a SMART-Exos-based strategy for targeted immunotherapy of HER2-positive breast cancer and demonstrates SMART-Exos as a broadly applicable platform for the development of cell-free therapies.
Figure 1.

Schematic Representation of the Design and Application of αCD3-αHER2 SMART-Exos as a Targeted Breast Cancer Immunotherapy
Results
Design, Generation, and Characterization of SMART-Exos
We envisioned that by targeting T cell CD3 and HER2, which is frequently overexpressed in human breast cancers,25 the designed SMART-Exos may induce strong immune responses against HER2-positive breast cancer through redirecting and activating endogenous cytotoxic effector cells toward attacking tumor cells overexpressing HER2 receptor. To this end, we utilized the human platelet-derived growth factor receptor (PDGFR) transmembrane domain (TMD) as a fusion partner for genetic display of functional monoclonal antibodies on the exosomal surface. The TMD of PDGFR has been widely used to express functional proteins on mammalian cell surfaces and was also used to display functional proteins on exosome surfaces.13,26, 27, 28 To ensure co-expression of αCD3 and αHER2 antibodies on the same exosome nanoparticles and minimize decreased binding affinity resulting from potential steric hindrance between two antibody scaffolds, we fused single polypeptide encoding in-tandem single-chain variable fragments (scFvs) against human CD3 and HER2 receptors with the PDGFR TMD. A flexible (GGGGS)3 linker was inserted between two scFv antibodies. Because the orientation of individual scFvs may affect physicochemical and biological properties of the designed SMART-Exos, an anti-human CD3 UCHT1 scFv antibody was placed at the N or C terminus of the anti-human HER2 trastuzumab scFv, resulting in the αCD3-αHER2 and αHER2-αCD3 SMART-Exos (Figures 1 and S1).29 αCD3 and αHER2 SMART-Exos were also generated as controls by separately fusing the respective scFv antibodies with the PDGFR TMD (Figure S1). Each fusion construct included an N-terminal hemagglutinin (HA) epitope tag.
Following transfection of Expi293 cells with the generated expression constructs, secreted SMART-Exos in the chemically defined culture media without fetal bovine serum (FBS) were purified through differential centrifugation and ultracentrifugation.30,31 As a widely used method for isolation of extracellular vesicles, the differential ultracentrifugation usually results in intermediate recovery with intermediate purity.13,32,33 The overall yields for the expressed SMART-Exos were approximately 74 μg (5.4 × 109 particles) per 30 mL transfected cell culture. Immunoblot analysis showed expression of antibody-PDGFR TMD fusion proteins, as well as exosomal markers (CD9, CD81, and CD63) (Figure 2A). The binding of SMART-Exos to plate-coated human HER2-Fc was examined by enzyme-linked immunosorbent assay (ELISA) (Figures 2B and S2). No binding to HER2 was detected for αCD3 SMART-Exos. The αHER2 SMART-Exos showed the tightest binding to HER2, followed by αCD3-αHER2 SMART-Exos and αHER2-αCD3 SMART-Exos. This result was further confirmed by flow cytometric analysis with HER2-positive cell lines (Figure 2C). Furthermore, flow cytometric analysis revealed tight binding of the αCD3-αHER2 and αHER2-αCD3 SMART-Exos to both the CD3+ and HER2+ cell lines (Figures 2C and S3) and little binding to MDA-MB-468 cells (CD3− HER2−) (Figures S3 and S4), demonstrating functional display of dual scFv antibodies on exosome surface for targeting both CD3- and HER2-expressing cells. Consistent with ELISA results, the αCD3-αHER2 SMART-Exos displayed higher binding affinity to both the CD3+ and HER2+ cell lines in comparison with the αHER2-αCD3 SMART-Exos and were thus chosen for further in vitro and in vivo evaluation. Nanoparticle tracking analysis (NTA) of αCD3-αHER2 SMART-Exos showed a size distribution peaking at 109 nm in diameter, consistent with previous studies (Figure 2D).8,9,34,35 Transmission electron microscopy (TEM) analysis of αCD3-αHER2 SMART-Exos immunogold stained with anti-HA antibody confirmed the surface expression of antibody-PDGFR TMD fusion proteins (Figures 2E and S5). To examine whether αCD3-αHER2 SMART-Exos could recruit T cells toward HER2-expressing cancer cells, we analyzed SMART-Exos-mediated cell-cell interactions by confocal microscopy. The presence of αCD3-αHER2 SMART-Exos resulted in significant crosslinking (p < 0.0001; Figures 2F and 2G) of Jurkat and SK-BR-3 cells (HER2 3+) and Jurkat and HCC 1954 (HER2 2+) cells, but no crosslinking of Jurkat and MDA-MB-468 (HER2 0) cells.36 In contrast, few Jurkat cells were bound to these breast cancer cells in the presence of a mixture of αCD3 and αHER2 SMART-Exos or in the presence of native exosomes (Figures 2F, 2G, and S6). In addition to cell-cell crosslinking induced by αCD3-αHER2 SMART-Exos, confocal microscopic analysis revealed cellular uptake of the PKH67-labeled αCD3-αHER2 SMART-Exos by both Jurkat and HCC 1954 cells (Figure S7). These data demonstrate cell-cell interactions induced by αCD3-αHER2 SMART-Exos and suggest possible therapeutic cargo delivery by SMART-Exos to target cells.
Figure 2.

In Vitro Characterization of the SMART-Exos
(A) Immunoblot analysis of the SMART-Exos. (B) ELISA analysis of binding of the SMART-Exos to human HER2. Human HER2-Fc (0.5 μg/mL) was coated on plates overnight for ELISA analysis. Data are shown as mean ± SD of duplicates. (C) Flow cytometry analysis of the binding of the SMART-Exos to SK-BR-3 (HER2+ CD3−), HCC 1954 (HER2+ CD3−), and Jurkat (HER2− CD3+) cells. (D) Size distribution of the αCD3-αHER2 SMART-Exos. (E) TEM image of the αCD3-αHER2 SMART-Exos showing immunogold labeling of surface HA tags. Arrows indicate gold particles. Scale bars: 100 nm. (F) Confocal microscopy of the crosslinking of (left panel) SK-BR-3 (red) and Jurkat cells (green), (middle panel) HCC 1954 (red) and Jurkat cells (green), and (right panel) MDA-MB-468 (red) and Jurkat cells (green) mediated by the αCD3-αHER2 SMART-Exos. PBS and a mixture (1:1) of αCD3 and αHER2 SMART-Exos were included as controls. Scale bars: 50 μm. (G) Quantitative analysis of confocal microscopy of cell-cell crosslinking induced by SMART-Exos. Based on fluorescence color, cell numbers of each cell line were counted to calculate cell number ratios (Jurkat cell/target breast cancer cell) for each treatment group. Data are shown as mean ± SD (n = 5). ****p < 0.0001; nsp > 0.05. ns, not significant.
In Vitro Cytotoxicity of SMART-Exos
We next performed in vitro cytotoxicity assays using human PBMCs (effector cells) and HER2+ breast cancer cell lines (target cells). In the presence of non-activated human PBMCs, the αCD3-αHER2 SMART-Exos resulted in potent and specific killing of SK-BR-3 cells (HER2 3+) with an EC50 = 0.85 ± 0.23 ng/mL and exhibited no cytotoxicity for the MDA-MB-468 cells (HER2 0) (Figure 3A). Minimal killing effects were observed for the mixtures of αCD3 and αHER2 SMART-Exos on either cell type. In addition to SK-BR-3 (HER2 3+) cells, the αCD3-αHER2 SMART-Exos showed potent cytotoxicity for HCC 1954 (HER2 2+) with a half maximal effective concentration (EC50) of 50.20 ± 7.67 ng/mL (Figure 3B), indicating that the observed cytotoxicities of the αCD3-αHER2 SMART-Exos for different breast cancer cell lines correlate with their HER2 expression levels. In addition to the different levels of HER2 expression on breast cancer cell surface, the observed significant difference in cytotoxicity for SK-BR-3 and HCC 1954 cells could result from their differed sensitivity to immune attack. Using human PBMCs from five different healthy donors, αCD3-αHER2 SMART-Exos showed consistent efficacy in killing HCC 1954 cells (Figure S8). These results demonstrate excellent potency and specificity of the αCD3-αHER2 SMART-Exos for inducing HER2+ cancer cell-specific immune response. In addition, native exosomes displayed no cytotoxicity for HCC 1954 and MDA-MB-468 cells (Figure S9A).
Figure 3.

In Vitro Cytotoxicity of the αCD3-αHER2 SMART-Exos
(A) Cytotoxicity of the αCD3-αHER2 SMART-Exos for SK-BR-3 (HER2+) and MDA-MB-468 (HER2−) cells. (B) Cytotoxicity of the αCD3-αHER2 SMART-Exos for three breast cancer cell lines. Non-activated human PBMCs (effector cells) were incubated with breast cancer cells (target cells) at an effector:target (E:T) ratio of 10 for 48 h in the presence of αCD3-αHER2 SMART-Exos. A one-to-one mixture of αCD3 and αHER2 SMART-Exos was used as a control in (A). Following removal of human PBMCs suspensions, viabilities of target cells were determined with MTT assays. (C) In vitro cytotoxicity of αCD3-αHER2 SMART-Exos in the absence of human PBMCs. HCC 1954 cells (HER2 2+) and MDA-MB-468 cells (HER2 0) were incubated with various concentrations of αCD3-αHER2 SMART-Exos for 48 h, followed by measurements of cell viabilities by MTT assays. (D and E) In vitro cytotoxicity of αCD3-αHER2 SMART-Exos in the presence of human PBMCs or CD3-depleted PBMCs. (D) Flow cytometric analysis of CD3+ T cells in human PBMCs without and with CD3 depletion. (E) HCC 1954 cells (HER2 2+) were incubated with various concentrations of αCD3-αHER2 SMART-Exos in the presence of human PBMCs or CD3-depleted PBMCs for 48 h, followed by measurements of cell viability by MTT assays. The ratios for PBMCs/tumor cells were 10:1. EC50: 16.9 ± 3.3 ng/mL for PBMCs without depletion; not applicable (N.A.) for PBMCs with CD3 depletion. (F and G) In vitro cytotoxicity of αCD3-αHER2 SMART-Exos in the presence of human PBMCs or CD4- and/or CD8-depleted PBMCs. (F) Flow cytometric analysis of CD4+ and CD8+ T cells in human PBMCs without and with CD4 and/or CD8 depletion. (G) HCC 1954 cells (HER2 2+) were incubated with αCD3-αHER2 SMART-Exos (200 ng/mL) in the presence of human PBMCs or CD4- and/or CD8-depleted PBMCs for 48 h, followed by measurements of cell viability by MTT assays. The ratios for PBMCs/tumor cells were 10:1. Data are shown as mean ± SD of triplicates. **p < 0.01.
To confirm that the observed cytotoxicities induced by αCD3-αHER2 SMART-Exos are dependent on human CD3+ T cells, we examined cytotoxicities of αCD3-αHER2 SMART-Exos for breast cancer cells in the absence of human PBMCs or presence of CD3/CD4/CD8-depleted human PBMCs. It was shown that in the absence of human PBMCs, αCD3-αHER2 SMART-Exos have little effect on the viabilities of HCC 1954 and MDA-MB-468 cells at concentrations up to 1 μg/mL (Figure 3C). In contrast with their potent cytotoxicity against HCC 1954 cells in the presence of human PBMCs, αCD3-αHER2 SMART-Exos resulted in minimal cytotoxicity for those breast cancer cells at concentrations as high as 10 μg/mL in the presence CD3-depleted human PBMCs (Figures 3D and 3E). Furthermore, in vitro cytotoxicity assays for αCD3-αHER2 SMART-Exos with CD4- and/or CD8-depleted PBMCs as effector cells revealed significantly increased viabilities of target breast cancer cells relative to ones with non-depleted PBMCs (Figures 3F and 3G). These results indicated that αCD3-αHER2 SMART-Exos cause only minimal effects on the growth of breast cancer cells, and CD3+ T cells are required for the αCD3-αHER2 SMART-Exos-mediated cytotoxicity against HER2-expressing breast cancer cells.
SMART-Exos-Mediated T Cell Activation
In vitro T cell activation was then evaluated for αCD3-αHER2 SMART-Exos on the basis of T cell surface activation markers CD69 and CD25 and secreted interferon (IFN)-γ and granzyme B. Robust T cell activation was observed only in the presence of both HCC 1954 (HER2+) cells and αCD3-αHER2 SMART-Exos (Figures 4A–4D). Elevated granzyme B secretion indicated αCD3-αHER2 SMART-Exos-mediated engagement and activation of cytotoxic T cells toward HER2-positive target cells (Figure 4D). In the absence of HER2+ breast cancer cells or presence of MDA-MB-468 (HER2−) cells, the αCD3-αHER2 SMART-Exos resulted in no T cell activation. PBS, native exosomes, and mixtures of αCD3 and αHER2 SMART-Exos were unable to activate the T cells in the absence or presence of target breast cancer cells (HCC 1954 and MDA-MB-468) (Figures S9B–S9E). Moreover, the αCD3-αHER2 SMART-Exos exhibited dose-dependent activation of human T cells in the presence of HER2-expressing HCC 1954 cells (Figures 4E–4H). In addition, pre-treatments of human PBMCs with αCD3-αHER2 SMART-Exos revealed no effects on T cell activation induced by anti-CD3 and anti-CD28 antibodies (Figure S10). These results demonstrate robust T cell activation mediated by the αCD3-αHER2 SMART-Exos in a HER2+ cancer cell-dependent manner.
Figure 4.

T Cell Activation Induced by αCD3-αHER2 SMART-Exos
(A–D) HCC 1954 cell-dependent activation of T cells by αCD3-αHER2 SMART-Exos. (A) The percentages of CD69+ T cells. (B) The percentages of CD25+ T cells. (C) The levels of secreted IFN-γ. (D) The levels of secreted granzyme B. (E-H) Dose-dependent activation of T cells by αCD3-αHER2 SMART-Exos. (E) The percentages of CD69+ T cells. (F) The percentages of CD25+ T cells. (G) The levels of secreted IFN-γ. (H) The levels of secreted granzyme B. Non-activated human PBMCs were incubated with αCD3-αHER2 SMART-Exos or a one-to-one mixture of αCD3 and αHER2 SMART-Exos or PBS in the presence or absence of MDA-MB-468 (HER2−) or HCC 1954 (HER2+) cells at an E:T ratio of 10 for 24 h. The percentages of CD69+ and CD25+ T cells were analyzed by flow cytometry. The levels of secreted IFN-γ and granzyme B were measured by ELISA. Data are shown as mean ± SD of triplicates. ***p < 0.001; ****p < 0.0001.
In Vivo Stability, Efficacy, and Toxicity of SMART-Exos
The pharmacokinetics of αCD3-αHER2 SMART-Exos was then examined in mice. The intravenously administered αCD3-αHER2 SMART-Exos showed a characteristic two-phase clearance profile with an elimination half-life of 417.03 ± 126.63 min (Figure S11). Next, we evaluated in vivo efficacy of the αCD3-αHER2 SMART-Exos using human HER2+ breast cancer xenograft mouse models with tumors derived from HCC 1954 cells. According to our in vitro studies, HCC 1954 cells express high levels of HER2 on cell surface, and αCD3-αHER2 SMART-Exos induce potent cytotoxicity against HCC 1954 cells in the presence of human PBMCs. Subcutaneously implanted HCC 1954 cells can result in rapid growth of human tumors in mice. Thus, HCC 1954 cell-derived mouse xenograft models were used for in vivo efficacy studies. To evaluate anti-tumor immunity induced by αCD3-αHER2 SMART-Exos, immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice bearing tumors from HCC 1954 cells were engrafted with human PBMCs.
Mice treated with the αCD3-αHER2 SMART-Exos displayed significant inhibition of tumor growth (Figure 5A). In comparison, the PBS-treated mice showed rapid growth of tumors. These results demonstrate excellent efficacy of the αCD3-αHER2 SMART-Exos for HER2-positive tumors in mice. Moreover, no loss in body weight or the weights of major organs was observed for mice in the PBS- and SMART-Exos-treated groups (Figures 5B and 5C). In addition, the plasma levels of both alanine aminotransferase (ALT) (a liver damage marker) and creatinine (a kidney damage marker) at the end of the in vivo efficacy study were comparable between PBS- and SMART-Exos-treated groups, suggesting no liver or kidney damage (Figures 5D and 5E). Flow cytometric and immunohistofluorescence analysis indicated that in comparison with low levels of tumor-infiltrating T cells for tumors from PBS-treated mice, significant T cell infiltrations were observed in tumors of the SMART-Exos-treated mice (Figures 5F–5H), suggesting SMART-Exos-mediated recruitment of cytotoxic T cells to the tumor microenvironments. No significant differences in T cell abundance were detected in the other tissues examined, including the spleen, blood, and bone marrow (Figure 5F).
Figure 5.

In Vivo Evaluation of the αCD3-αHER2 SMART-Exos
(A) In vivo efficacy of the αCD3-αHER2 SMART-Exos. HCC 1954 cells were subcutaneously (s.c.) injected into the flank of female NSG mice (n = 5). In vitro activated and expanded human PBMCs from the same healthy donor were intraperitoneally (i.p.) injected into the mice on days 13 and 22 post tumor implantation. Two days post the first PBMCs injection, mice were intravenously (i.v.) injected with PBS or αCD3-αHER2 SMART-Exos (10 mg/kg, 1.5 × 1010 particles per mouse) every other day for a total of six times. Data are shown as mean ± SD. **p < 0.01. (B–E) Evaluation of systemic toxicity of SMART-Exo treatments. (B) Body weights of mice during in vivo efficacy study. Body weights of mice were measured every other day. Data are shown as mean ± SD. (C) Major organ weight indexes at the end of the in vivo efficacy study. The kidneys, livers, brains, lungs, and hearts from mice were collected and weighted, and organ weight indexes were calculated as organ weight (mg) per gram (g) of mouse body weight. (D) Plasma levels of kidney damage marker creatinine at the end of the in vivo efficacy study. (E) Plasma levels of liver damage marker alanine aminotransferase (ALT) at the end of the in vivo efficacy study. Data are shown as mean ± SD (n = 5 for body weights and organ weight indexes; n = 4 for plasma levels of ALT and creatinine). (F) In vivo T cell infiltration induced by the αCD3-αHER2 SMART-Exos. Blood, bone marrow, and single-cell preparations of residual tumors and spleens from the PBS- and SMART-Exos-treated groups were analyzed by flow cytometry. Data are shown as mean ± SD (n = 5; *p = 0.0195). (G) Representative immunohistofluorescence images of the margin and interior of frozen tumor sections from PBS- and SMART-Exos-treated mice. Blue: nuclei stained with DAPI. Green: CD3+ cells stained with the anti-CD3 antibody. Scale bars: 50 μm. (H) Quantitative representation of the number of CD3+ cells from each field of view along the margin and interior of each tumor from PBS- and SMART-Exos-treated groups (15 fields of view per region and two mice per group). Data are shown as mean ± SD. ****p ≤ 0.0001.
SMART-Exos-Displayed Antibody Molecules
SMART-Exos are featured with two distinct types of monoclonal antibodies displayed on the exosome surface. To determine the number of dual-scFvs on SMART-Exo nanoparticles and compare binding affinities of free bispecific scFv and SMART-Exos-displayed dual-scFvs, a bispecific αCD3-αHER2 scFv antibody with identical sequence and orientation was generated (Figure S12). Based on particle concentrations determined by NTA and the ELISA using purified bispecific αCD3-αHER2 scFv antibody as standards, it was estimated that on average each αCD3-αHER2 SMART-Exo particle carries approximately 1,180 ± 140 αCD3-αHER2 bispecific scFv molecules. Using plate-coated human HER2-Fc, ELISA-based binding assays indicated that in contrast with free bispecific αCD3-αHER2 scFv antibody with an EC50 of 241.5 ± 21.9 ng/mL, the accessible bispecific scFv molecules on αCD3-αHER2 SMART-Exos require concentrations of up to 8.3 ± 0.3 ng/mL for half-maximal binding (Figure S13), supporting significantly increased antibody avidity through multivalent expression on the exosome surface.
In addition, the in vitro cytotoxicity and T cell activation induced by αCD3-αHER2 SMART-Exo and bispecific αCD3-αHER2 scFv antibody were compared (Figures S14 and S15). Consistent with the increased antibody avidity, the αCD3-αHER2 SMART-Exos relative to free bispecific αCD3-αHER2 scFv antibody showed improved potency in inducing HER2-positive cell-specific cytotoxicity and T cell activation based on the concentrations of bispecific scFv and the molar concentrations of SMART-Exos. These results support enhanced efficacy for SMART-Exos with genetically displayed antibody molecules.
Discussion
Exosomes are cell-derived membranous vesicles known to mediate cell-cell communications. Extensive studies have been performed by utilizing exosomes as transport vehicles for drug delivery,9, 10, 11, 12 but fewer studies are done to develop exosome-based therapeutics through engineering their surface proteins, in particular for cancer immunotherapy. In this work, we genetically engineered exosomes to display dual scFv antibodies. As a result of multivalent expression of antibodies on exosome surface, the increased antibody avidity on spherical exosomes may enhance lateral stabilization of HER2 antigens and T cell receptor clustering and hence facilitate T cell activation. The designed αCD3-αHER2 SMART-Exos reveal excellent in vitro and in vivo efficacy in eliciting anti-tumor immunity against HER2-positive breast cancer cells in a controlled and directed manner.
Depending on the sources and states of parental cells, exosomes may possess immunomodulatory potential.37 To produce the αCD3-αHER2 SMART-Exos, we chose Expi293 cells that are derived from the HEK293 cell line. Extracellular vesicles from HEK293T cells were shown to have minimal toxicity and immunogenicity.38 No systemic cytotoxicity was observed for αCD3-αHER2 SMART-Exos in the animal studies. Further studies are needed to gain deeper understanding of the exosomal compositions and their effects on cellular functions and immune modulation for production of therapeutic exosomes with desired properties.
This work demonstrates preclinical feasibility of utilizing endogenous exosomes for targeted breast cancer immunotherapy. Future studies include immunogenicity of SMART-Exos, impacts of SMART-Exos administration on tumor growth and T cell functions, evolution of tumor growth following SMART-Exos treatment, and in vivo mechanism(s) of action. The therapeutic applications of the SMART-Exos could be further expanded by leveraging the broad scope of monoclonal antibodies, as well as by functionally displaying more than two types of monoclonal antibodies and/or effector proteins. Moreover, SMART-Exos could potentially carry various forms of endogenous and exogenous therapeutic cargos for enhanced efficacy through targeted delivery. Notably, the efficacy and safety for αCD3-αHER2 SMART-Exos need to be further evaluated in immunocompetent mice, which will involve the generation of surrogate mouse SMART-Exos and the use of murine syngeneic cancer models. Additionally, instead of using PBMCs from healthy donors, in vivo efficacy studies could be assessed for αCD3-αHER2 SMART-Exos by using PBMCs from donors with HER2-positive breast cancer, which could improve prediction of clinical outcomes.
In summary, genetically engineering exosomes with anti-CD3 and anti-HER2 antibodies resulted in SMART-Exos for potent and selective induction of HER2-expressing tumor-specific immunity, leading to the creation of an innovative class of immunotherapeutic candidates for HER2-positive breast cancer. Our results suggest that the SMART-Exos may provide a general and versatile approach for the development of next-generation immuno-nanomedicines.
Materials and Methods
Cell Culturing
SK-BR-3, HCC 1954, MDA-MB-468, and Jurkat cells were obtained from the American Type Culture Collection (ATCC) and cultured in RPMI 1640 medium (Corning, NY, USA) supplemented with 10% FBS (VWR International, PA, USA) at 37°C and 5% CO2. Expi293 cells (Thermo Fisher Scientific, MA, USA) were cultured in Expi293 expression medium with shaking at a speed of 125 rpm at 37°C and 8% CO2. Human PBMCs were obtained from HemaCare (Van Nuys, CA, USA).
Animal Study
All animal procedures were approved by the University of Southern California Institutional Animal Care and Use Committee (IACUC). Six- to eight-week-old female NSG mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and housed in sterilized cages for efficacy and T cell infiltration studies.
Molecular Cloning and Expression of SMART-Exos
Synthetic genes encoding the anti-HER2 and anti-CD3 scFvs, which were derived from anti-HER2 trastuzumab and anti-CD3 UCHT1 antibodies, respectively, were purchased from Integrated DNA Technologies (Skokie, IL, USA). The anti-HER2 scFv contained an N-terminal light chain variable region (VLαHER2) and C-terminal heavy chain variable region (VHαHER2) of the trastuzumab separated by a 218-peptide linker (GSTSGSGKPGSGEGS).39 The anti-CD3 scFv had an N-terminal light chain variable region (VLαCD3) and C-terminal heavy chain variable region (VHαCD3) of the anti-CD3 UCHT1 antibody connected by a flexible (GGGGS)3 linker. Through overlap extension PCR using primers listed in Table S1, gene fragments encoding anti-HER2-anti-CD3 dual-scFv and anti-CD3-anti-HER2 dual-scFv were generated. A flexible (GGGGS)3 linker was placed between the anti-HER2 and anti-CD3 scFvs. To generate expression constructs of SMART-Exos, single and dual-scFv gene fragments were cloned into pDisplay vector (Thermo Fisher Scientific, MA, USA) between BglII and SalI restriction enzyme sites, resulting in the fusion proteins of scFv antibody-TMD of human PDGFR. A HA-tag was located at the N terminus of the designed fusion proteins. The generated expression constructs were confirmed by DNA sequencing provided by GENEWIZ (South Plainfield, NJ, USA). The endotoxin-free plasmids were prepared using ZymoPURE Plasmid Maxiprep Kits (ZYMO Research, CA, USA), followed by transfection into Expi293 cells using ExpiFectamine 293 transfection kits (Thermo Fisher Scientific, MA, USA) by following the manufacturer’s instructions.
Molecular Cloning of Bispecific scFv
To generate the expression construct of anti-CD3-anti-HER2 bispecific scFv, the anti-CD3-anti-HER2 dual-scFv gene fragment was cloned into pFUSE vector (Invivogen, CA, USA) between EcoRI and NheI restriction enzyme sites. A HA-tag and a 6XHis-tag were placed at the N and the C terminus of the dual-scFv gene fragment, respectively. The resulting expression construct was confirmed by DNA sequencing provided by GENEWIZ (South Plainfield, NJ, USA). The endotoxin-free plasmids were prepared using ZymoPURE Plasmid Maxiprep Kits (ZYMO Research, CA, USA).
Purification of SMART-Exos
Culture media of Expi293 cells transfected with the expression constructs were collected at days 3 and 6 posttransfection. Expressed SMART-Exos were isolated by differential centrifugation and ultracentrifugation. In brief, cell cultures were centrifuged at 90 × g for 10 min at 4°C to pellet suspension cells. The collected supernatants were then centrifuged at 4,000 × g for 30 min and 24,000 × g for 40 min at 4°C, followed by ultracentrifugation at 265,000 × g for 120 min at 4°C in a Type 70 Ti rotor (Beckman Instruments, IN, USA). Exosome pellets were washed once with PBS, resuspended in PBS, and filtered with 0.2-μm filters. Protein concentrations of the exosomes were determined using Bradford assays.
Expression and Purification of Bispecific scFv
Anti-CD3-anti-HER2 bispecific scFv antibody was expressed through transient transfection into Expi293 cells using ExpiFectamine 293 transfection kits (Thermo Fisher Scientific, MA, USA) by following the manufacturer’s instructions. Culture media of Expi293 cells transfected with the expression construct were collected at days 3 and 6 posttransfection. Bispecific scFv was purified using HisPur Ni-NTA Resin (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions.
Immunoblot Analysis
Exosome samples (3 μg of protein) were boiled with 100 mM dithiothreitol (DTT) in NuPAGE LDS sample buffer (Thermo Fisher Scientific, MA, USA) at 98°C for 10 min, separated in 4%–20% ExpressPlus-PAGE gels (GeneScript, Piscataway, NJ, USA), and then transferred to Immun-Blot PVDF (polyvinylidene fluoride) membranes (Bio-Rad Laboratories, CA, USA) using a Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad Laboratories, CA, USA). The membranes were subsequently blocked with 5% BSA in PBST (PBS with 0.1% Tween 20) for 1 h at room temperature, followed by incubation with appropriate primary antibodies for 1 h at room temperature, which included anti-HA (2-2.2.14; Thermo Fisher Scientific, MA, USA), anti-CD9 (D8O1A; Cell Signaling Technology, MA, USA), anti-CD63 (H5C6; BioLegend, CA, USA), and anti-CD81 (1.3.3.22; Thermo Fisher Scientific, MA, USA). After 1-h incubation with appropriate secondary antibodies, including anti-mouse immunoglobulin G (IgG)-horseradish peroxidase (HRP) and anti-rabbit IgG-HRP (Thermo Fisher Scientific, MA, USA), the membranes were developed by additions of SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, MA, USA) and imaged using a ChemiDoc Touch Imaging System (Bio-Rad Laboratories, CA, USA).
TEM and NTA
For immunogold labeling of exosomes, 200-μm mesh grids were floated on 20 μL of exosome sample drop for 10 min and washed twice with water by floating on a drop of water for 30 s. The grids were then blocked with 1% goat serum for 30 min and were incubated with either the anti-HA antibody (2-2.2.14; Thermo Fisher Scientific, MA, USA) or the IgG1 control antibody (Thermo Fisher Scientific, MA, USA) diluted with 0.1% goat serum 1:10 for 2 h at room temperature, followed by incubation with a goat anti-mouse secondary antibody gold conjugate (1:10 dilution in 0.1% goat serum in PBS) for 2 h at room temperature. The grids were washed twice by floating on a drop of water for 30 s, and negatively stained with 2% uranyl acetate (UA) by floating on a drop of freshly filtered 2% UA for 30 s. The grids were blot dried with filter paper and imaged using JEOL1230 TEM (JEOL, Peabody, MA, USA).
The size distribution and particle concentration of the exosome samples were determined through NTA using a NanoSight LM10 (Malvern Instruments, UK) by following the manufacturer’s instructions. Ten replicates of analysis with 60 s for each were performed.
Bindings of SMART-Exos and Bispecific scFv to Human HER2 as Measured by ELISA
Ninety-six-well ELISA plates were coated with human HER2-Fc (0.5 and 2.5 μg/mL; R&D Systems, MN, USA) overnight at room temperature. Non-bound antigens were washed away with PBST (0.05% Tween 20) three times. The wells were blocked with PBS containing 1% BSA for 2 h, followed by washing with PBST. Various concentrations of SMART-Exos and bispecific scFv were added and incubated for 2 h, followed by washing. The anti-HA primary antibody (2-2.2.14; Thermo Fisher Scientific, MA, USA) was subsequently added for 2-h incubation, followed by washing and incubation with an anti-mouse IgG-HRP secondary antibody (Thermo Fisher Scientific, MA, USA) for 1 h. QuantaBlu Fluorogenic Peroxidase Substrate (Thermo Fisher Scientific, MA, USA) was then added after washing, and the fluorescence signals were measured using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader (BioTek, VT, USA).
Quantification of Surface scFv on αCD3-αHER2 SMART-Exos by ELISA
ELISA 96-well plates were coated with various concentrations of αCD3-αHER2 bispecific scFv and SMART-Exos overnight at room temperature. Non-bound antigens were washed away with PBST (0.05% Tween 20) three times. The wells were blocked with PBS containing 3% BSA for 2 h, followed by washing with PBST. The anti-HA primary antibody (2-2.2.14; Thermo Fisher Scientific, MA, USA) was subsequently added for 2-h incubation, followed by washing and incubation with an anti-mouse IgG-HRP secondary antibody (Thermo Fisher Scientific, MA, USA) for 1 h. QuantaBlu Fluorogenic Peroxidase Substrate (Thermo Fisher Scientific, MA, USA) was then added after washing, and the fluorescence signals were measured using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader (BioTek, VT, USA). Serial dilutions of αCD3-αHER2 bispecific scFv in PBS were used as standards to determine the protein concentrations of surface bispecific scFv on αCD3-αHER2 SMART-Exos. The numbers of surface αCD3-αHER2 bispecific scFv per SMART-Exos particle were calculated based on the measured protein concentrations of surface bispecific scFv and the particle concentrations of the same batch of αCD3-αHER2 SMART-Exos by NTA.
Flow Cytometry Analysis
The binding of SMART-Exos to human breast cancer cell lines (SK-BR-3, HCC 1954, and MDA-MB-468 cells) and Jurkat cells was analyzed by flow cytometry. Cells were incubated with exosomes (0.1 mg/mL) in PBS for 1 h at 4°C. Following three washes with PBS containing 2% FBS, cells were incubated with the anti-HA primary antibody (2-2.2.14; Thermo Fisher Scientific, MA, USA) for 30 min at 4°C. After washing three times with PBS containing 2% FBS, cells were incubated with the Alexa Fluor 488-conjugated anti-mouse IgG (H+L) secondary antibody (Thermo Fisher Scientific, MA, USA) for 30 min at 4°C. Following three washes, samples were analyzed using an LSR II flow cytometer (BD Biosciences, CA, USA). To evaluate cell surface expression levels of HER2 and CD3, we stained cultured SK-BR-3, HCC 1954, MDA-MB-468, and Jurkat cells with the trastuzumab followed by an anti-human IgG Fc-fluorescein isothiocyanate (FITC) antibody (Thermo Fisher Scientific, MA, USA) or an anti-CD3-FITC antibody (UCHT1; BioLegend, CA, USA). Samples were then analyzed using the LSR II flow cytometer. All flow cytometry data were processed with FlowJo_V10 software (Tree Star, OR, USA).
Confocal Microscopy of Cell Crosslinking
Breast cancer cells and Jurkat cells were stained with MitoSpy Red CMXRos (BioLegend, CA, USA) and carboxyfluorescein succinimidyl ester (CFSE; BioLegend, CA, USA), respectively, by following the manufacturer’s protocols. Jurkat cells (5 × 105) were incubated with SMART-Exos (100 μg/mL), native exosomes (100 μg/mL), or bispecific scFv (5.4 μg/mL) in 100 μL of PBS for 30 min at 4°C. In separate tubes, Jurkat cells were incubated with a mixture (1:1) of αCD3 and αHER2 SMART-Exos as controls. After washing with 1 mL of ice-cold PBS, the Jurkat cells were resuspended with 250 μL of RPMI 1640 media with 10% FBS, then mixed with SK-BR-3, HCC1954, or MDA-MB-468 cells (5 × 104) in the same media (250 μL). The cell mixtures were added onto glass coverslips in 24-well cell culture plates and incubated at 37°C with 5% CO2. After 4 h, wells were gently washed with PBS (500 μL) three times and imaged with a Leica SP8 confocal laser scanning microscope (Leica Microsystems, IL, USA) equipped with HC PL APO 40×/1.30 Oil CS2 and HC PL APO 63×/1.40 Oil CS2 oil immersion objective lenses using rhodamine (for MitroSpy Red) and FITC (for CFSE) filters. Images were processed using LAS X software (Leica Microsystems, IL, USA). Five images per group were randomly chosen, and cell numbers of Jurkat and breast cancer cells were counted to calculate ratios of Jurkat/breast cancer cells.
Confocal Microscopic Analysis of Cellular Internalization of αCD3-αHER2 SMART-Exos
The αCD3-αHER2 SMART-Exos were labeled with PKH67 dye by following the manufacturer’s protocols. HCC1954 cells (4 × 104) and Jurkat cells (2 × 105) were incubated with PKH67-labeled αCD3-αHER2 SMART-Exos (10 μg/mL) for 2 or 6 h at 37°C with 5% CO2. After incubation, cells were gently washed with PBS (500 μL) three times and imaged with a Leica SP8 confocal laser scanning microscope (Leica Microsystems, IL, USA) equipped with HC PL APO 40×/1.30 Oil CS2 and HC PL APO 63×/1.40 Oil CS2 oil immersion objective lenses using the FITC (for PKH67) filter. Images were processed using LAS X software (Leica Microsystems, IL, USA). Five images per group were randomly chosen, and fluorescence intensity of PKH67 was measured using ImageJ and normalized to cell number.
Human PBMCs Depletion and Isolation
CD3/CD4/CD8-depleted human PMBCs were isolated from blood of the healthy donors using RosetteSep Human CD3/CD4/CD8 Depletion Cocktail (STEMCELL Technologies, Canada) by following the manufacturer’s protocols. To isolate non-depleted PBMCs, blood was mixed with an equal volume of PBS and then added on top of 3/2 volume of Ficoll (GE Healthcare, IL, USA) and spun at 400 × g for 30 min without brake. The layer of PBMCs was transferred to a clean tube and washed with PBS three times before any applications. The isolated non-depleted or depleted PBMCs were stained with a FITC-labeled anti-human CD3 antibody (clone: UCHT1; BioLegend, CA, USA), allophycocyanin (APC)-labeled anti-human CD4 antibody (clone: OKT4; BioLegend, CA, USA), and phycoerythrin (PE)-labeled anti-human CD8 antibody (RPA-T8; BioLegend, CA, USA) and analyzed by the BD Fortessa X20 flow cytometer. Flow cytometry data were processed with FlowJo_V10 software (Tree Star, OR, USA).
In Vitro Cytotoxicity Assay
Cytotoxicity assays were performed using non-depleted or depleted non-activated PBMCs as effector cells and breast cancer cells as target cells. Target cells (1 × 104) were mixed with PBMCs (1 × 105) and incubated with various concentrations of SMART-Exos, native exosomes, or bispecific scFv for 48 h at 37°C with 5% CO2. Cells were washed with PBS to remove PBMC suspensions and then incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution for 2 h at 37°C. For cytotoxicity assays without effector cells, cancer cells (1 × 104) were incubated with various concentrations of SMART-Exos for 48 h at 37°C with 5% CO2. MTT solution (10 μL/well) was added and incubated for 2 h at 37°C. A total of 100 μL/well of lysis buffer (20% SDS in 50% dimethylformamide, 0.5% [v/v] 80% acetic acid, 0.4% [v/v] 1 N HCl, pH 4.7) was then added and incubated for 2 h at 37°C. The absorbance was measured at 570 nm using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader (BioTek, VT, USA). Percent cytotoxicity was calculated as follows:
T Cell Activation Analysis
Non-activated PBMCs (2 × 105) were incubated with target cells (2 × 104) in the presence of various concentrations of SMART-Exos, native exosomes, or bispecific scFv in 96-well cell culture plates. After 24 h, cells were stained with the anti-CD3-Alexa Fluor 488 (UCHT1; BioLegend, CA, USA), anti-CD25-PE (M-A251; BioLegend, CA, USA), anti-CD69-APC (FN50; BioLegend, CA, USA), and 7-aminoactinomycin D (7-AAD) (BioLegend, CA, USA), followed by flow cytometry analysis using a BD Fortessa X20 flow cytometer (BD Biosciences, CA, USA). The levels of secreted cytokines in the collected culture media were measured using appropriate ELISA kits (R&D System). Data are shown as mean ± SD of triplicates.
Effects of αCD3-αHER2 SMART-Exos Pre-treatment on T Cell Activation
Non-activated human PBMCs (effector cells) were incubated with 200 ng/mL of αCD3-αHER2 SMART-Exos or PBS (control) for 2 h at 37°C, followed by washing with PBS. PBMCs were then resuspended in RPMI 1640 medium with 10% FBS and stimulated with coated anti-human CD3 antibody (clone: OKT3; BioLegend, CA, USA) and soluble anti-human CD28 antibody (2 μg/mL) (clone: 28.2; BioLegend, CA, USA). After 24 h, cells were stained with the anti-CD3-Alexa Fluor 488 (clone: UCHT1; BioLegend, CA, USA), anti-CD25-PE (clone: M-A251, BioLegend, CA, USA), anti-CD69-APC (clone: FN50; BioLegend, CA, USA), and 7-AAD (BioLegend, CA, USA), followed by flow cytometry analysis using a BD Fortessa X20 flow cytometer (BD Biosciences, CA, USA). The levels of secreted cytokines in the collected culture media were measured using appropriate ELISA kits (R&D System). Data are shown as mean ± SD of duplicates.
Pharmacokinetics of αCD3-αHER2 SMART-Exos in Mice
The αCD3-αHER2 SMART-Exos (800 μg/mouse) were administrated by intravenous (i.v.) injection into BALB/c mice (n = 4). Blood samples were collected using Multivette 600 LH-Gel tubes (SARSTEDT, Germany) at 5, 30, 60, 120, 180, 240, and 360 min post i.v. injection, immediately followed by centrifugation at 10,000 × g for 5 min at room temperature. The plasma samples were diluted with PBS (1:50) and analyzed by sandwich ELISA with a mouse anti-HA monoclonal antibody (clone: 2-2.2.14; Thermo Fisher Scientific) as the capture antibody, a rabbit anti-HA polyclonal antibody (catalog number: 600-401-384; Rockland Immunochemicals, Limerick, PA, USA) as the detection antibody, and a goat anti-rabbit IgG (H+L) HRP (catalog number: 5220-0336; SeraCare, Milford, MA, USA). Serial dilutions of αCD3-αHER2 SMART-Exos in PBS with 2% mouse plasma were used as standards to quantify plasma concentrations of αCD3-αHER2 SMART-Exos. The plasma half-life of αCD3-αHER2 SMART-Exos in mice was determined by noncompartmental analysis using MATLAB.
In Vivo Efficacy and T Cell Infiltration Study
HER2-positive human breast cancer cell line HCC 1954 was used to evaluate the in vivo efficacy of the αCD3-αHER2 SMART-Exos. HCC 1954 cells (1.5 × 106 per mouse) in 50% Matrigel (BD Biosciences, CA, USA) were subcutaneously implanted into the right flank of NSG mice (6–8 weeks, female, n = 5). Freshly thawed human PBMCs were resuspended in RPMI 1640 medium with 10% FBS at 2 × 106 cells/mL and stimulated in flasks with coated anti-CD3 antibody (OKT3, BioLegend, CA, USA), soluble anti-CD28 antibody (2 μg/mL) (28.2; BioLegend, CA, USA), and recombinant human interleukin-2 (rhIL-2; 40 IU/mL; R&D Systems, MN, USA) for 3 days at 37°C with 5% CO2. Cells were then expanded in RPMI 1640 medium supplemented with 10% FBS and 40 IU/mL rhIL-2. When tumor sizes reached a volume of 80–100 mm3, mice received two intraperitoneal injections of activated human PBMCs (20 × 106 per mouse) with a 9-day interval. Two days following first human PBMCs injection, mice were administered intravenously with the αCD3-αHER2 SMART-Exos (10 mg/kg) or PBS every other day for a total of six times. Tumors were measured three times a week by a caliper. Tumor volumes were calculated as follows: mm3 = length × width2/2. At the end of the study, mice were euthanized. Tumors, major organs, and tissues were collected. The harvested blood and bone marrow were treated with the erythrocyte lysis solution (BioLegend, CA, USA) by following the manufacturer’s instructions. Tumors and spleens were cut into small pieces and subjected to mechanical disruption and separation, followed by passing through 70-μm strainers and treatment with the erythrocyte lysis solution. The resulting single-cell suspensions were stained with the FITC-labeled anti-human CD45 antibody and the Pacific Blue-labeled anti-human CD3 antibody, and analyzed by the BD Fortessa X20 flow cytometer. Flow cytometry data were processed with FlowJo_V10 software (Tree Star, OR, USA).
Immunohistofluorescence Analysis
Immunostaining of tumors was performed on 7-μm cryosections according to standard protocols. The tissues were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature and blocked with PBS containing 1% BSA and 5% goat serum for 1 h at room temperature. The tissue sections were then incubated with the anti-CD3 antibody (UCHT1; BioLegend, CA, USA) and the Alexa Fluor 488-conjugated anti-mouse IgG (H+L) secondary antibody (Thermo Fisher Scientific, MA, USA) sequentially for 1 h each at room temperature, followed by counterstaining for nuclei with DAPI. Images were acquired using a Leica SP8 confocal laser scanning microscope (Leica Microsystems, IL, USA) equipped with a HC PL APO 40×/1.30 Oil CS2 oil immersion objective lens using DAPI and FITC (for Alexa Fluor 488) filters. Images were processed using the LAS X software (Leica Microsystems, IL, USA). For quantification, 15 random, non-overlapping regions along the margin and interior of each tumor (n = 2 mice/group) were imaged.
ALT Activity Assay
At the end of the in vivo efficacy study, collected mouse plasma (10 μL) or standard solution (2 mM sodium pyruvate) was added to wells of clear 96-well plates, followed by additions of 50 μL of substrate solution (0.2 M alanine, 2 mM 2-oxoglutarate, pH 7.4) and incubation at 37°C for 30 min. Next, 50 μL of 2, 4-dinitrophenylhydrazine (2,4-DNPH) (1 mM) was added and incubated at 37°C for 10 min. Finally, 0.5 M sodium hydroxide was added, and the absorbance was measured at 510 nm using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader (BioTek, VT, USA). Amounts of pyruvate generated were calculated from the standard. The ALT activity was reported as nmol/min/mL = U/L, where 1 milliunit (mU) of ALT is defined as the amount of enzyme that generates 1.0 nmol of pyruvate per minute at 37°C.
Creatinine Colorimetric Assay
Working solutions were prepared by mixing picric acid (38 mM) with sodium hydroxide (1.2 M) at 1:1 ratio. Mouse plasma samples collected at the end of in vivo efficacy study were mixed with equal volume of trichloroacetic acid (TCA) (1.2 M) and centrifuged at 2,500 × g for 10 min. The collected supernatants and creatinine standards were added to 96-well plates, followed by additions of working solution. The absorbance was measured at 500 nm using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader (BioTek, VT, USA). Creatinine concentrations in mouse plasma were determined on the basis of standard curves.
Statistical Analysis
Statistical analyses were conducted with GraphPad Prism (GraphPad Software, CA, USA). Data are shown as mean ± SD. Statistical significance between two groups was determined using two-tailed Student’s t tests. One-way ANOVA analyses were carried out for comparisons of multiple groups. Significance of finding was defined as follows: not significant, nsp > 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Data Availability
The raw/processed data required to reproduce these findings are available from the authors upon request.
Author Contributions
Conceptualization, Y.Z.; Methodology, Y.Z., X.S., and Q.C.; Investigation, X.S., Q.C., T.H., and M.H.; Writing – Original Draft, Y.Z. and X.S.; Writing – Review & Editing, Y.Z. and X.S.; Funding Acquisition, Y.Z.; Resources, G.S., J.E.L., A.L.E., and H.-J.L.
Conflicts of Interest
The authors have filed a patent application on this work.
Acknowledgments
This work was supported in part by University of Southern California School of Pharmacy Start-Up Fund for New Faculty, University of Southern California Ming Hsieh Institute for Engineering Medicine for Cancer, the American Cancer Society (grant IRG-16-181-57), STOP CANCER Research Career Development Award (to Y.Z.), PhRMA Foundation Research Starter Grant in Translational Medicine and Therapeutics (to Y.Z.), grant P30CA014089 to the USC Norris Comprehensive Cancer Center, and grant P30DK048522 to the USC Research Center for Liver Diseases.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.11.020.
Supplemental Information
References
- 1.Colombo M., Raposo G., Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014;30:255–289. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- 2.Raposo G., Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camussi G., Deregibus M.-C., Bruno S., Grange C., Fonsato V., Tetta C. Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am. J. Cancer Res. 2011;1:98–110. [PMC free article] [PubMed] [Google Scholar]
- 4.Février B., Raposo G. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr. Opin. Cell Biol. 2004;16:415–421. doi: 10.1016/j.ceb.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Mathivanan S., Ji H., Simpson R.J. Exosomes: extracellular organelles important in intercellular communication. J. Proteomics. 2010;73:1907–1920. doi: 10.1016/j.jprot.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 6.Tian T., Zhu Y.L., Hu F.H., Wang Y.Y., Huang N.P., Xiao Z.D. Dynamics of exosome internalization and trafficking. J. Cell. Physiol. 2013;228:1487–1495. doi: 10.1002/jcp.24304. [DOI] [PubMed] [Google Scholar]
- 7.van den Boorn J.G., Schlee M., Coch C., Hartmann G. SiRNA delivery with exosome nanoparticles. Nat. Biotechnol. 2011;29:325–326. doi: 10.1038/nbt.1830. [DOI] [PubMed] [Google Scholar]
- 8.Kamerkar S., LeBleu V.S., Sugimoto H., Yang S., Ruivo C.F., Melo S.A., Lee J.J., Kalluri R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503. doi: 10.1038/nature22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 10.Bryniarski K., Ptak W., Jayakumar A., Püllmann K., Caplan M.J., Chairoungdua A., Lu J., Adams B.D., Sikora E., Nazimek K. Antigen-specific, antibody-coated, exosome-like nanovesicles deliver suppressor T-cell microRNA-150 to effector T cells to inhibit contact sensitivity. J. Allergy Clin. Immunol. 2013;132:170–181. doi: 10.1016/j.jaci.2013.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang S.C., Kim O.Y., Yoon C.M., Choi D.S., Roh T.Y., Park J., Nilsson J., Lötvall J., Kim Y.K., Gho Y.S. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano. 2013;7:7698–7710. doi: 10.1021/nn402232g. [DOI] [PubMed] [Google Scholar]
- 12.Munoz J.L., Bliss S.A., Greco S.J., Ramkissoon S.H., Ligon K.L., Rameshwar P. Delivery of Functional Anti-miR-9 by Mesenchymal Stem Cell-derived Exosomes to Glioblastoma Multiforme Cells Conferred Chemosensitivity. Mol. Ther. Nucleic Acids. 2013;2:e126. doi: 10.1038/mtna.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohno S., Takanashi M., Sudo K., Ueda S., Ishikawa A., Matsuyama N., Fujita K., Mizutani T., Ohgi T., Ochiya T. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013;21:185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun D., Zhuang X., Xiang X., Liu Y., Zhang S., Liu C., Barnes S., Grizzle W., Miller D., Zhang H.G. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010;18:1606–1614. doi: 10.1038/mt.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian Y., Li S., Song J., Ji T., Zhu M., Anderson G.J., Wei J., Nie G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383–2390. doi: 10.1016/j.biomaterials.2013.11.083. [DOI] [PubMed] [Google Scholar]
- 16.Wahlgren J., De L Karlson T., Brisslert M., Vaziri Sani F., Telemo E., Sunnerhagen P., Valadi H. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012;40:e130. doi: 10.1093/nar/gks463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhuang X., Xiang X., Grizzle W., Sun D., Zhang S., Axtell R.C., Ju S., Mu J., Zhang L., Steinman L. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011;19:1769–1779. doi: 10.1038/mt.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper J.M., Wiklander P.B., Nordin J.Z., Al-Shawi R., Wood M.J., Vithlani M., Schapira A.H., Simons J.P., El-Andaloussi S., Alvarez-Erviti L. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 2014;29:1476–1485. doi: 10.1002/mds.25978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fais S., O’Driscoll L., Borras F.E., Buzas E., Camussi G., Cappello F., Carvalho J., Cordeiro da Silva A., Del Portillo H., El Andaloussi S., Evidence-Based Clinical Use of Nanoscale Extracellular Vesicles in Nanomedicine Evidence-Based Clinical Use of Nanoscale Extracellular Vesicles in Nanomedicine. ACS Nano. 2016;10:3886–3899. doi: 10.1021/acsnano.5b08015. [DOI] [PubMed] [Google Scholar]
- 20.György B., Hung M.E., Breakefield X.O., Leonard J.N. Therapeutic applications of extracellular vesicles: clinical promise and open questions. Annu. Rev. Pharmacol. Toxicol. 2015;55:439–464. doi: 10.1146/annurev-pharmtox-010814-124630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oh K., Kim S.R., Kim D.K., Seo M.W., Lee C., Lee H.M., Oh J.E., Choi E.Y., Lee D.S., Gho Y.S., Park K.S. In Vivo Differentiation of Therapeutic Insulin-Producing Cells from Bone Marrow Cells via Extracellular Vesicle-Mimetic Nanovesicles. ACS Nano. 2015;9:11718–11727. doi: 10.1021/acsnano.5b02997. [DOI] [PubMed] [Google Scholar]
- 22.Wiklander O.P.B., Brennan M.A., Lötvall J., Breakefield X.O., El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019;11:eaav8521. doi: 10.1126/scitranslmed.aav8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng Q., Shi X., Han M., Smbatyan G., Lenz H.J., Zhang Y. Reprogramming Exosomes as Nanoscale Controllers of Cellular Immunity. J. Am. Chem. Soc. 2018;140:16413–16417. doi: 10.1021/jacs.8b10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross J.S., Slodkowska E.A., Symmans W.F., Pusztai L., Ravdin P.M., Hortobagyi G.N. The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist. 2009;14:320–368. doi: 10.1634/theoncologist.2008-0230. [DOI] [PubMed] [Google Scholar]
- 25.Shak S., Herceptin Multinational Investigator Study Group Overview of the trastuzumab (Herceptin) anti-HER2 monoclonal antibody clinical program in HER2-overexpressing metastatic breast cancer. Semin. Oncol. 1999;26(4 Suppl 12):71–77. [PubMed] [Google Scholar]
- 26.Beerli R.R., Bauer M., Buser R.B., Gwerder M., Muntwiler S., Maurer P., Saudan P., Bachmann M.F. Isolation of human monoclonal antibodies by mammalian cell display. Proc. Natl. Acad. Sci. USA. 2008;105:14336–14341. doi: 10.1073/pnas.0805942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho M., Nagata S., Pastan I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc. Natl. Acad. Sci. USA. 2006;103:9637–9642. doi: 10.1073/pnas.0603653103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koh E., Lee E.J., Nam G.H., Hong Y., Cho E., Yang Y., Kim I.S. Exosome-SIRPα, a CD47 blockade increases cancer cell phagocytosis. Biomaterials. 2017;121:121–129. doi: 10.1016/j.biomaterials.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 29.Zhu Z., Carter P. Identification of heavy chain residues in a humanized anti-CD3 antibody important for efficient antigen binding and T cell activation. J. Immunol. 1995;155:1903–1910. [PubMed] [Google Scholar]
- 30.El-Andaloussi S., Lee Y., Lakhal-Littleton S., Li J., Seow Y., Gardiner C., Alvarez-Erviti L., Sargent I.L., Wood M.J. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat. Protoc. 2012;7:2112–2126. doi: 10.1038/nprot.2012.131. [DOI] [PubMed] [Google Scholar]
- 31.Greening D.W., Xu R., Ji H., Tauro B.J., Simpson R.J. A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol. Biol. 2015;1295:179–209. doi: 10.1007/978-1-4939-2550-6_15. [DOI] [PubMed] [Google Scholar]
- 32.Hung M.E., Leonard J.N. Stabilization of exosome-targeting peptides via engineered glycosylation. J. Biol. Chem. 2015;290:8166–8172. doi: 10.1074/jbc.M114.621383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yim N., Ryu S.W., Choi K., Lee K.R., Lee S., Choi H., Kim J., Shaker M.R., Sun W., Park J.H. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat. Commun. 2016;7:12277. doi: 10.1038/ncomms12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gangalum R.K., Atanasov I.C., Zhou Z.H., Bhat S.P. AlphaB-crystallin is found in detergent-resistant membrane microdomains and is secreted via exosomes from human retinal pigment epithelial cells. J. Biol. Chem. 2011;286:3261–3269. doi: 10.1074/jbc.M110.160135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katsuda T., Tsuchiya R., Kosaka N., Yoshioka Y., Takagaki K., Oki K., Takeshita F., Sakai Y., Kuroda M., Ochiya T. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 2013;3:1197. doi: 10.1038/srep01197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao Y., Axup J.Y., Ma J.S., Wang R.E., Choi S., Tardif V., Lim R.K., Pugh H.M., Lawson B.R., Welzel G. Multiformat T-cell-engaging bispecific antibodies targeting human breast cancers. Angew. Chem. Int. Ed. Engl. 2015;54:7022–7027. doi: 10.1002/anie.201500799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang B., Yin Y., Lai R.C., Lim S.K. Immunotherapeutic potential of extracellular vesicles. Front. Immunol. 2014;5:518. doi: 10.3389/fimmu.2014.00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu X., Badawi M., Pomeroy S., Sutaria D.S., Xie Z., Baek A., Jiang J., Elgamal O.A., Mo X., Perle K. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles. 2017;6:1324730. doi: 10.1080/20013078.2017.1324730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whitlow M., Bell B.A., Feng S.L., Filpula D., Hardman K.D., Hubert S.L., Rollence M.L., Wood J.F., Schott M.E., Milenic D.E. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng. 1993;6:989–995. doi: 10.1093/protein/6.8.989. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw/processed data required to reproduce these findings are available from the authors upon request.