

Abstract
The amyloid cascade model of Alzheimer’s disease posits the primacy of amyloid beta deposition preceding tau-mediated neurofibrillary tangle formation. The amyloid-tau-neurodegeneration biomarker-only diagnostic framework similarly requires the presence of amyloid beta for a diagnosis on the Alzheimer’s continuum. However, medial temporal lobe tau pathology in the absence of amyloid beta is frequently observed at autopsy in cognitively normal individuals, a phenomenon that may reflect a consequence of aging and has been labelled ‘primary age-related tauopathy’. Alternatively, others argue that this tauopathy reflects an early stage of the developmental continuum leading to Alzheimer’s disease. We used positron emission tomography imaging to investigate amyloid beta and tau positivity and associations with cognition to better inform the conceptualization of biomarker changes in Alzheimer’s pathogenesis. Five hundred twenty-three individuals from the Alzheimer’s Disease Neuroimaging Initiative who had undergone flortaucipir positron emission tomography imaging were selected to derive positron emission tomography positivity thresholds using conditional inference decision tree regression. A subsample of 301 individuals without dementia (i.e. those with normal cognition or mild cognitive impairment) had also undergone florbetapir positron emission tomography imaging within 12 months and were categorized into one of the four groups based on cortical amyloid and Braak stage I/II tau positivity: A−/T−, A+/T−, A−/T+, or A+/T+. Tau positivity in the absence of amyloid beta positivity (i.e. A−/T+) comprised the largest group, representing 45% of the sample. In contrast, only 6% of the sample was identified as A+/T−, and the remainder of the sample fell into A−/T− (22%) or A+/T+ (27%) categories. A−/T− and A+/T− groups had the best cognitive performances across memory, language and executive function; the A−/T+ group showed small-to-moderate relative decreases in cognition; and the A+/T+ group had the worst cognitive performances. Furthermore, there were negative associations between Braak stage I/II tau values and all cognitive domains only in the A−/T+ and A+/T+ groups, with strongest associations for the A+/T+ group. Among our sample of older adults across the Alzheimer’s pathological spectrum, 7-fold fewer individuals have positron emission tomography evidence of amyloid beta pathology in the absence of tau pathology than the converse, challenging prevailing models of amyloid beta’s primacy in Alzheimer’s pathogenesis. Given that cognitive performance in the A−/T+ group was poorer than in individuals without either pathology, our results suggest that medial temporal lobe tau without cortical amyloid beta may reflect an early stage on the Alzheimer’s pathological continuum.
Keywords: tau imaging, amyloid imaging, Alzheimer’s disease, mild cognitive impairment, biomarkers
Despite the primacy of amyloid beta within many theoretical frameworks of pathological progression leading to Alzheimer’s dementia, we found a high prevalence of tau positron emission tomography in the absence of amyloid that conferred subtle cognitive deficits consistent with prodromal Alzheimer’s disease. Results indicate that tau pathology may appear earlier than previously thought and have a significant role in the Alzheimer’s prodrome independently of amyloid beta.
Graphical Abstract
Graphical Abstract.
Introduction
Amyloid beta (Aβ) plaques and tau neurofibrillary tangles represent two of the original defining neuropathological features of Alzheimer’s disease (AD; Alzheimer, 1907). Although initially no distinction was made in the relative causal contributions of these two pathologies, the prevailing view of Alzheimer’s disease pathogenesis later shifted to an Aβ-centric perspective that considered Aβ to be the primary harbinger of the disease state, referred to as the amyloid cascade model (Glenner and Wong, 1984; Hardy, 2017). The deterministic gene mutations for AD result in increased accumulation of the Aβ protein (Blacker and Tanzi, 1998) and are generally regarded as confirmatory of the primacy of beta-amyloidosis in AD pathogenesis, although a study by Oxtoby et al. (2018) in early-onset familial AD gene mutation carriers demonstrated identical cortical PiB Aβ and CSF p-tau abnormality rates. Furthermore, the recent failures of large-scale clinical trials targeting Aβ have called into question the verity of the amyloid cascade model (Egan et al., 2018; Honig et al., 2018; Selkoe, 2019). Consequently, the role of Aβ as the primary driving force in the pathogenesis of AD has recently come under scrutiny (Ricciarelli and Fedele, 2017; Morris et al., 2018).
A recent re-conceptualization of the AD diagnostic framework has built upon the amyloid cascade hypothesis by proposing a biomarker characterization of the disease in terms of Aβ pathology (A), tau pathology (T), and neurodegeneration (N), thus comprising the amyloid-tau-neurodegeneration or ‘AT(N)’ framework (Jack et al., 2016, 2018a,b). Although this framework purports agnosticism with regard to the temporal emergence of abnormal biomarkers, it retains an Aβ-centric perspective by necessitating that abnormal Aβ must be present to constitute ‘Alzheimer’s pathologic change’ (A+/T−), followed by a subsequent pathological change in tau that transitions the diagnosis to ‘Alzheimer’s disease’ (A+/T+). In contrast, tau in the absence of Aβ (A−/T+), either with or without neurodegeneration, is labelled as ‘non-Alzheimer’s pathologic change’ within this framework. Such nomenclature suggests that individuals in this category are not on the Alzheimer’s continuum but rather on an alternative pathological trajectory (Burnham et al., 2016; Gordon et al., 2016; Mormino et al., 2016). Controversy remains over the designation of tau in the absence of Aβ as a non-Alzheimer’s disease process, however, as some studies indicate that this group (A−/T+) is indistinguishable from Aβ-positive groups on a host of purportedly ‘non-Alzheimer’s disease’ clinical and biological factors (Knopman et al., 2013).
Analogous to the AT(N) framework’s ‘non-Alzheimer’s disease pathologic change’ is the concept of primary age-related tauopathy (PART), which postulates that medial temporal lobe (MTL) tau in the absence of Aβ reflects a feature of aging separate from the Alzheimer’s continuum that is ‘observed in cognitively normal individuals’, with ‘severe PART’ associated with amnestic changes only (Crary et al., 2014). Similar controversy exists as to whether PART should be considered a distinct pathological entity (Crary, 2016; Bell et al., 2019) or whether it is better represented as an early Alzheimer’s process given its phenomenological similarity to AD (Braak and Del Tredici, 2014; Duyckaerts et al., 2015) and evidence of cognitive decline despite the absence of Aβ-positivity (Jefferson-George et al., 2017; Josephs et al., 2017). Although PART has typically been studied using post-mortem histopathology, recent advances in positron emission tomography (PET) imaging for tau (Jagust, 2018) now allow for in vivo investigation of MTL tau in the absence of Aβ that can clarify the characteristics of PART and its separability from—or inclusion on—the Alzheimer’s continuum.
Accordingly, we examined the proportion of older adults without dementia [i.e. cognitively normal (CN) and mild cognitive impairment (MCI)] who had MTL tau in the absence of Aβ (A−/T+) relative to individuals with Aβ in the absence of tau (A+/T−), with the expectation that there will be a higher prevalence of A−/T+ individuals based on the neuropathological staging studies of Braak and Del Tredici (2014, 2015). Furthermore, given prior suggestions by Crary et al. (2014) that A−/T+ represents a common feature of aging in primarily CN individuals (i.e. PART), we explored this notion by comparing the biomarker groupings on neuropsychological function. In concert with findings from Braak and colleagues, our results may have implications for the amyloid cascade hypothesis and AT(N) framework by indicating the need for the recognition of MTL tauopathy as a primary substrate of the Alzheimer’s continuum or, at the very least, agnosticism regarding the temporal sequence of pathological events in AD.
Materials and methods
Participants
Initially, data were assessed from 523 participants of the Alzheimer’s Disease Neuroimaging Initiative (ADNI) who had flortaucipir data available to create tau standardized uptake variable ratio (SUVR) positivity thresholds. Of these 523 participants with available flortaucipir data, 350 participants also had available florbetapir data acquired within 12 months of the flortaucipir data to create Aβ SUVR positivity thresholds. Subsequently, a subset of 301 individuals without dementia (i.e. CN and MCI) who had data from both PET measures acquired within 12 months of one another were included in analyses investigating the concurrence of Aβ and tau positivity (i.e. A/T grouping) and subsequent group comparisons in clinical and cognitive characteristics.
Amyloid positron emission tomography processing
Processing methods for ADNI florbetapir (18F-AV-45) data have been previously described (Landau et al., 2013; Landau and Jagust, 2015). Briefly, all PET images were smoothed to the resolution of the lowest resolution scanners (8-mm full-width half-maximum; see http://adni.loni.usc.edu/methods/pet-analysis-method/pet-analysis/ for a detailed description of PET preprocessing methods). Preprocessed PET images were co-registered with the individual’s native-space structural magnetic resonance imaging that was collected within the closest proximity to the PET scan (typically 3 months or less) to obtain Freesurfer-defined regional standardized uptake variable values. A cortical summary measure was derived by calculating a volume-weighted average across frontal, cingulate, lateral parietal and lateral temporal regions. All SUVs were intensity normalized by dividing each value by the whole cerebellum to derive SUVRs, as recommended in ADNI’s florbetapir processing methods (Landau and Jagust, 2015). Although a positivity threshold of 1.11 is recommended for the intensity normalized cortical summary measure (Joshi et al., 2012; Landau and Jagust, 2015), we derived a new threshold using the procedures described below to maintain methodological consistency with the derivation of a tau positivity threshold (see also below). Notably, florbetapir data were selected based on the acquisition date closest in time to the baseline flortaucipir scan acquisition date within a 12-month period. On average, acquisition of the florbetapir scan followed the flortaucipir scan by 0.54 months with an SD of 1.69 months in either direction.
Tau positron emission tomography processing
Processing methods for ADNI flortaucipir (18F-AV-1451) data have been previously described (Landau and Jagust, 2016; Maass et al., 2017). As above, preprocessed PET images were smoothed to 8-mm full-width half-maximum and co-registered with the structural magnetic resonance imaging to obtain Freesurfer-defined regional standardized uptake variable values. Flortaucipir data were partial volume-corrected using the Geometric Transfer Matrix approach (Baker et al., 2017). Regional partial volume-corrected SUVR values were combined to approximate Braak stages I/II, III/IV and V/VI; specific Freesurfer-defined regions included within each Braak stage can be found in Maass et al. (2017). Lastly, partial volume-corrected Braak stage composite ROIs were intensity normalized using the inferior cerebellar grey matter as a reference region to create SUVRs, as recommended in ADNI’s flortaucipir processing methods (Landau and Jagust, 2016).
Amyloid and tau positivity thresholding
Thresholds for the determination of Aβ and tau positivity were derived with conditional inference classification trees using the ctree() function from the party package in R version 3.5.0 (https://cran.r-project.org/), similar to methods used in previous ADNI studies of tau PET (Schöll et al., 2016; Maass et al., 2017). Mini–Mental State Examination total score was used to drive the classification algorithm separately for both Aβ and tau data to obtain thresholds that (i) are consistent between both Aβ and tau PET data (i.e. to avoid an Aβ threshold defined on pathologically confirmed diagnoses versus a tau threshold that is not validated in this way) and (ii) use a global cognitive measure independent from neuropsychological outcome variables used in later group comparisons. Analyses for each Braak composite stage were conducted in a hierarchical manner: first, thresholds were determined for Braak V/VI; individuals positive for Braak V/VI were then removed from subsequent analyses prior to the determination of Braak III/IV thresholds; and finally, following the removal of individuals positive for Braak III/IV, thresholds for Braak I/II were determined. Analyses were conducted in a single algorithm for Aβ using the cortical summary measure, given the more spatially diffuse emergence of Aβ deposits relative to the consistent hierarchical spatiotemporal progression of tau pathology. Thresholds were taken at the lowest bifurcation in the tree, unless otherwise noted; if two bifurcations were made at the same level, the threshold was instead taken from the level above. Importantly, unlike prior studies that have thresholded tau conditionally on Aβ (Wang et al., 2016; Mishra et al., 2017; Jack et al., 2019), thresholds for Aβ and tau positivity were derived independently of the other to avoid bias in the prevalence estimates of A/T groups.
Diagnostic classification
Individuals with dementia were included only during derivation of the SUVR thresholds. Clinical diagnosis of dementia was made based on the following criteria utilized by ADNI (http://adni.loni.usc.edu/): (i) subjective memory complaint reported by the participant, study partner or clinician; (ii) objective memory impairment defined by a score below education-adjusted cut-offs on Logical Memory Delayed Recall, Story A of the Wechsler Memory Scale—Revised; (iii) score between 20 and 26 on the Mini–Mental State Examination; (iv) score 0.5 or 1.0 on the Clinical Dementia Rating Scale; and (v) met NINCDS/ADRDA criteria (McKhann et al., 1984) for probable Alzheimer’s disease.
Clinical diagnosis of MCI was determined using comprehensive neuropsychological criteria (Jak et al., 2009; Bondi et al., 2014). Regression-based z-scores adjusting for age, sex and education were derived for all participants for the following measures: Trail Making Test Parts A and B (attention/executive domain); confrontation naming (i.e. Boston Naming Test or Multilingual Naming Test) and Animal Fluency (language domain); and Rey Auditory Verbal Learning Recall and Recognition (memory domain). Participants’ observed scores were compared with the predicted scores for a group of ‘robust’ CN participants (i.e. maintained CN status throughout the duration of their participation in ADNI) and standardized to create z-scores. A diagnosis of MCI was made for any participant with z-scores >1 SD below the mean for either (i) both tests within a domain or (ii) at least one test across all three domains. All other participants with scores above these cut-points were considered CN.
Clinical and cognitive variables
Demographic variables including age, sex and education were measured for all participants and adjusted for within the cognitive z-scores. Apolipoprotein E (APOE) ε4 positivity was determined by the presence of at least one APOE ε4 allele. Cardiovascular risk was assessed using pulse pressure and calculated as systolic minus diastolic blood pressure, to ascertain the possible contribution of vascular pathology to group differences in cognition given that pulse pressure has been associated with post-mortem cerebrovascular disease (Nation et al., 2012). For a subset of individuals (n = 76) with Hachinski ischemic scale scores, an aggregate measure of vascular risk, group differences in this measure are also reported to further rule out vascular contributions to cognitive differences.
Neuropsychological composite scores were created by calculating z-scores for individual measures, as described in the previous section, and averaging within the following domains: memory recall (Logical Memory Immediate Recall, Logical Memory Delayed Recall and Rey Auditory Verbal Learning Delayed Recall), attention/executive function (Trail Making Test Parts A and B) and language [confrontation naming (Boston Naming Test or Multilingual Naming Test) and animal fluency]. For the creation of domain composites, all observations indicating discontinuation of the test due to failure were retained as meaningful values [i.e. maximum raw score of 300 s on Trails B (n = 7) or 0 on confrontation naming (n = 7)].
Statistical analyses
Individuals were first assigned to one of the four A/T groups based on Aβ and tau PET positivity (i.e. A−/T−, A−/T+, A+/T− or A+/T+). Group comparisons on demographic, clinical, and PET SUVR data were conducted using chi-square tests for independence for categorical variables and one-way ANOVAs for continuous variables. Analyses assessing group differences in neuropsychological composite scores were conducted using ANCOVAs adjusting for APOE ε4 positivity with four observations (one per group) deleted due to missing APOE ε4 data. To assess the effect of using a newly derived Aβ PET threshold, we additionally ran these analyses using the conventional 1.11 threshold for Aβ. Residuals for all three domain composite scores indicated a moderate-to-strong negative skew; therefore, prior to data analysis, these variables were shifted to a positive scale [1 + x − min(x)] and normalized using Box-Cox transformation [(xλ − 1)/λ where λ = 1.7 for memory, 4.2 for executive, and 3.4 for language]. Reported statistics of interest includes F-ratios and partial eta-squares (ηp2s) for omnibus tests and t-ratios and Cohen’s d statistics for all pairwise contrasts. P-values were also reported for all statistical tests with Tukey adjustment for pairwise contrasts within each domain. Effect sizes were interpreted as per convention: ηp2 = 0.01 (small effect), ηp2 = 0.09 (medium effect), ηp2 = 0.25 (large effect); Cohen’s d = 0.2 (small effect), Cohen’s d = 0.5 (medium effect), Cohen’s d = 0.8 (large effect). All 95% confidence intervals were included with effect size statistics. Although the reported statistics reflect the difference between estimated marginal means of Box-Cox transformed values, untransformed and unadjusted z-score means for each group are presented in tables and figures to facilitate interpretation. Finally, within-group assessment of associations between transformed neuropsychological domain scores and continuous cortical Aβ and Braak I/II SUVR levels were conducted using Pearson partial correlations controlling for APOE ε4 positivity, with partial rs and unadjusted P-values as the reported statistics. Effect sizes were again interpreted as per convention: r = 0.1 (small effect), r = 0.3 (moderate effect), r = 0.5 (large effect). Scatterplots include Box-Cox transformed and residualized values to depict tau PET and cognitive associations as modelled. Hypothesis tests were two-sided and considered statistically significant at α = 0.05 unless otherwise noted. All analyses and figures were generated in R version 3.5.0 (R Core Team, 2017), including the following extension packages: MASS, dplyr, car, emmeans, sjstats, compute.es, psych, and ggplot2. Raincloud plots were generated from code supplied in Allen et al. (2019).
Data availability
Data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public–private partnership. The primary goal of ADNI has been to test whether serial magnetic resonance imaging, PET, other biological markers and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early Alzheimer’s disease. This research was approved by the Institutional Review Boards of all participating sites, and written informed consent was obtained for all study participants.
Results
Amyloid and tau positivity
Thresholds were determined using a sample of 523 individuals (355 CN, 122 MCI, and 46 with dementia) with tau PET data and 350 individuals (219 CN, 95 MCI and 36 with dementia) who additionally had Aβ PET data. Supplementary Figs. 1 and 2 depict decision tree outputs from the conditional inference classification algorithm with Mini–Mental State Examination score as the response variable. A positivity threshold of >1.14 was derived for the intensity normalized Aβ summary SUVR (P < 0.001), resulting in 140 individuals [40% overall; 59 CN (26.9%), 47 MCI (49.5%), 34 Alzheimer’s disease (94.4%)] classified as A+. Although there was a lower-level bifurcation in the tree at a value of 0.95 (P = 0.03), the first bifurcation value of 1.14 was chosen to maintain consistency with prior conventional positivity thresholds (i.e. 1.11; Joshi et al., 2012; Landau and Jagust, 2015) and because selection of the lower threshold would have resulted in 90% of individuals without dementia categorized as A+, which does not comport with prior PET studies (10–30% in CN using k-means cluster methods in Cohen et al., 2013; 29% in CN and 43% in early MCI in Landau et al., 2012; 25% in CN and abnormal in Lewczuk et al., 2017) nor with neuropathological studies (e.g. Arriagada et al., 1992; Braak and Del Tredici, 2015).
Braak stage thresholding began with the intensity normalized Braak V/VI SUVR, for which a positivity threshold of >1.96 was derived (P < 0.001); 22 individuals [3 CN (0.8%), 7 MCI (5.7%), 12 with dementia (26.0%)] were classified as Braak V/VI+ and were removed from subsequent Braak staging classifications (Schöll et al., 2016). For the remaining 501 Braak V/VI− individuals, a positivity threshold of >1.51 was derived for the intensity normalized Braak III/IV SUVR (P < 0.001); 101 individuals [37 CN (10.5%), 42 MCI (41.7%), 22 with dementia (64.7%)] were classified as Braak III/IV+ and were removed from the final Braak staging classification. Finally, for the remaining 400 Braak III/IV− individuals, a positivity threshold of >1.18 was derived for the intensity normalized Braak I/II+ SUVR (P = 0.02); 278 individuals [210 CN (66.7%), 56 MCI (76.7%), 12 with dementia (100%)] were classified as Braak I/II+ and the remaining 122 individuals [105 CN (33.3%), 17 MCI (23.3%)] were classified as Braak I/II−. Braak III/IV+ and Braak V/VI+ individuals iteratively removed during the classification process were also assigned positivity for lower Braak stages. Any individual classified as Braak I/II+ was considered T+. Notably, there were no individuals classified as Braak III/IV+ who were not also Braak I/II+.
A/T group classification
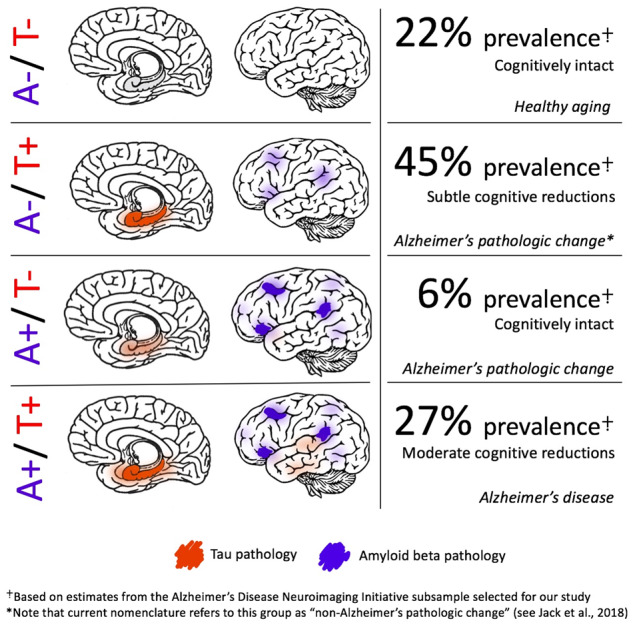
Three hundred one individuals without dementia (212 CN, 89 MCI) who had florbetapir and flortaucipir PET data obtained within 12 months of one another were assigned to one of the four groups to investigate the concurrence of Aβ and tau positivity: A−/T−, A−/T+, A+/T− or A+/T+. Table 1 depicts a cross-tabulation of Aβ and tau positivity. The largest group of individuals [135 total (45%); 103 CN, 32 MCI] had evidence of prominent tau in the absence of Aβ (A−/T+). In contrast, only 18 individuals (6%; 14 CN, 4 MCI) had evidence of prominent Aβ in the absence of tau (A+/T−); 81 individuals (27%; 42 CN, 39 MCI) were positive for both Aβ and tau (A+/T+); and 67 individuals (22%; 53 CN, 14 MCI) were negative for both pathologies (A−/T−).
Table 1.
Cross-tabulation of amyloid and tau positivity resulting in four group classifications
| T− | T+ | |
|---|---|---|
| A− | 67 (22%) (53 CN, 14 MCI) | 135 (45%) (103 CN, 32 MCI) |
| A+ | 18 (6%) (14 CN, 4 MCI) | 81 (27%) (42 CN, 39 MCI) |
Notably, when the conventional 1.11 threshold for Aβ was used to categorize groups, group proportions largely remained the same with the A−/T− group at 21%, A−/T+ at 40%, A+/T− at 7% and A+/T+ at 32% (see Supplementary Table 1). For completeness, classification of the 36 excluded individuals with dementia with the original 1.14 threshold for Aβ resulted in the following: 1 individual was A−/T−, 1 individual was A−/T+, 1 individual was A+/T−, and 33 individuals were A+/T+.
Florbetapir/flortaucipir PET summary statistics
The means and standard deviations for cortical Aβ, Braak I/II and Braak III/IV SUVR levels across the four A/T groups are reported in Supplementary Table 2, and distributions of these variables are displayed Fig. 1. One individual in the A−/T+ group had an SUVR value of 3.46 for Braak I/II that was suppressed in Fig. 1 to improve visualization of the rest of the data; given that this value is physiologically plausible and the Braak III/IV value for this individual was well within the range of the sample (1.89), this individual was not excluded from any analyses. Notably, as indicated by Fig. 1C, individuals in the A−/T+ group had a range of Aβ SUVR values and were not clustered just below the threshold for Aβ positivity. Furthermore, some individuals in the A−/T+ group exhibited notable flortaucipir binding in Braak III/IV regions typically considered free from tau pathology in the absence of significant cortical Aβ (see Fig. 1B; mean SUVR = 1.42; 18 individuals Braak III/IV+).
Figure 1.

PET SUVR distributions by A/T group. Raincloud plots depicting distributions of SUVR values for AV1451 Braak stage I/II (A), AV1451 Braak stage III/IV (B) and AV45 cortical summary index (C) across all A/T groups. A−/T− is denoted in blue, A−/T+ is denoted in green, A+/T− is denoted in orange and A+/T+ is denoted in red. One individual had an SUVR value of 3.46 for Braak I/II that was suppressed in all panels to improve the visualization of the rest of the data.
Demographic and clinical comparisons
Stratification of demographic and clinical characteristics by A/T group can be found in Table 2. Groups statistically differed in age (F = 13.6, ηp2 = 0.12, P < 0.001) such that the A−/T− group was younger than all other groups. The A+/T+ group had the largest proportion of individuals with MCI (48.1%), whereas all other groups consisted of 20–24% individuals with MCI (χ2 = 18.50, ηp2 = 0.10, P < 0.001). Groups also differed in genetic risk (χ2 = 29.60, ηp2 = 0.15, P < 0.001), with a higher proportion of APOE ε4 carriers in the A+/T+ groups relative to the A−/T− and A−/T+ groups. The A+/T− group did not statistically differ from any other groups, although it is notable that there were no ε4/ε4 homozygotes in this group. There was no difference in vascular risk between groups as indexed by pulse pressure, although the A+/T− group had a numerically lower pulse pressure by an average of ∼8 mmHg. Furthermore, for a subset of individuals with Hachinski ischemic scale scores, there were no group differences in this measure of vascular risk.
Table 2.
A/T group differences in demographic and clinical characteristics
| Group, mean (SD) |
Group differences |
||||||
|---|---|---|---|---|---|---|---|
| A−/T− | A−/T+ | A+/T− | A+/T+ | F or χ2 | η 2 | P-value | |
| n (%) | 67 (22.2) | 135 (44.9) | 18 (6.0) | 81 (26.9) | |||
| Age | 71.13 (6.28) | 76.50 (7.21) | 75.39 (4.85) | 78.06 (7.17) | 13.60 | 0.12 | <0.001 |
| Sex (% female) | 46.3 | 49.6 | 38.9 | 53.1 | 1.50 | 0.01 | 0.68 |
| Education (years) | 16.55 (2.43) | 16.95 (2.56) | 16.28 (2.45) | 16.48 (2.61) | 0.87 | 0.01 | 0.46 |
| Diagnosis (% MCI) | 20.9 | 23.7 | 22.2 | 48.1 | 18.50 | 0.10 | <0.001 |
| APOE risk (n 0/1/2 ε4 alleles) | 46/19/1 | 105/23/6 | 10/7/0 | 37/33/10 | 29.60 | 0.15 | <0.001 |
| Pulse pressure | 57.93 (14.01) | 59.18 (16.30) | 51.28 (11.06) | 62.19 (16.24) | 2.69 | 0.03 | 0.05 |
| Hachinski risk score | 0.58 (0.93) (n = 24) | 0.53 (0.73) (n = 30) | 0.60 (0.55) (n = 5) | 0.35 (0.61) (n = 17) | 0.34 | 0.01 | 0.79 |
Cognitive comparisons
Memory recall
ANCOVAs assessing differences in memory domain scores between A/T groups yielded a statistically significant omnibus effect of moderate size (F = 5.55, ηp2 = 0.06, P = 0.001; see Table 3 and Fig. 2; see also Supplementary Table 3 for all pairwise contrasts including Tukey-adjusted P-values). Follow-up pairwise contrasts revealed large effects for differences between the A+/T+ group and the A−/T− (Cohen’s d = 0.84, 95% CI = [0.33, 1.33]) and A+/T− (Cohen’s d = 1.32, 95% CI = [0.28, 2.34]) groups, with lower scores observed for the A+/T+ group. There was also a moderate effect for the difference between A+/T+ and A−/T+ groups (Cohen’s d = 0.55, 95% CI = [0.20, 0.89]) with lower scores again observed for the A+/T+ group. Lastly, there was a small effect for the difference between A−/T− and A−/T+ groups (Cohen’s d = 0.18, 95% CI = [−0.30, 0.66]), with lower scores observed for the A−/T+ group. Results remained largely the same when group comparisons were made using the 1.11 threshold.
Table 3.
A/T group differences in neuropsychological domain performance
| Group, mean (SD) |
Group differences |
||||||
|---|---|---|---|---|---|---|---|
| A−/T−, n = 67 | A−/T+, n = 135 | A+/T−, n = 18 | A+/T+, n = 81 | F-ratio | η p 2 | P-value | |
| Memory | −0.24 (0.85) | −0.35 (0.99) | −0.07 (0.73) | −0.96 (1.30) | 5.55 | 0.06 | 0.001 |
| Executive | −0.005 (0.64) | −0.21 (0.94) | −0.05 (0.77) | −0.78 (1.69) | 4.15 | 0.05 | 0.007 |
| Language | −0.14 (0.86) | −0.48 (1.62) | 0.07 (0.67) | −0.59 (1.25) | 2.51 | 0.03 | 0.06 |
Note: Reported values reflect untransformed and unadjusted z-score means to facilitate the interpretation of group differences; these values were not used in models from which statistics were derived.
Figure 2.

Cognitive z-score distributions by A/T group. Dot-boxplots depicting cognitive domain scores for memory (A), executive function (B) and language (C) composites across all A/T groups. A−/T− is denoted in blue, A−/T+ is denoted in green, A+/T− is denoted in orange and A+/T+ is denoted in red. Values reflect untransformed and unadjusted z-scores.
Attention/executive function
ANCOVAs assessing differences in executive domain scores between A/T groups yielded a statistically significant omnibus effect of moderate size (F = 4.15, ηp2 = 0.05, P = 0.007; see Table 3 and Fig. 2; see also Supplementary Table 4 for all pairwise contrasts including Tukey-adjusted P-values). Follow-up pairwise contrasts revealed large effects for differences between the A+/T+ group and the A−/T− (Cohen’s d = 0.80, 95% CI = [0.30, 1.29]) and A+/T− (Cohen’s d = 0.96, 95% CI = [−0.03, 1.93]) groups, with lower scores observed for the A+/T+ group. There was also a moderate effect for the difference between A+/T+ and A−/T+ groups (Cohen’s d = 0.44, 95% CI = [0.10, 0.78]) with lower scores again observed for the A+/T+ group. Lastly, there was a small-to-moderate effect for the difference between A−/T− and A−/T+ groups (Cohen’s d = 0.28, 95% CI = [−0.20, 0.76]), with lower scores observed for the A−/T+ group. Results remained largely the same when group comparisons were made using the 1.11 threshold.
Language
ANCOVAs assessing differences in language domain scores between A/T groups yielded an omnibus effect of small size (F = 2.51, ηp2 = 0.03, P = 0.06; see Table 3 and Fig. 2; see also Supplementary Table 5 for all pairwise contrasts including Tukey-adjusted P-values). Follow-up pairwise contrasts revealed a large effect for the difference between A+/T+ and A+/T− groups (Cohen’s d = 1.05, 95% CI = [0.04, 2.03]), with lower scores observed for the A+/T+ group. There was also a moderate effect for the difference between A+/T+ and A−/T− groups (Cohen’s d = 0.52, 95% CI = [0.03, 1.01]) with lower scores again observed for the A+/T+ group. No notable difference was observed between A+/T+ and A−/T+ groups. However, there were small-to-moderate effects for the difference between the A−/T+ group and the A−/T− (Cohen’s d = 0.29, 95% CI = [−0.19, 0.77]) and A+/T− (Cohen’s d = 0.27, 95% CI = [−0.07, 0.61]) groups, with lower scores observed for the A−/T+ group. Results remained largely the same when group comparisons were made using the 1.11 threshold.
Cognition and PET SUVR associations
Partial correlations controlling for APOE ε4 positivity revealed negative associations between Braak I/II SUVR levels and all neuropsychological domain scores for only the A−/T+ (small-to-moderate rs from −0.18 to −0.27) and A+/T+ (moderate-to-large rs from −0.29 to −0.48) groups (see Table 4). Effect sizes were considerably larger for the A+/T+ group across all domains and most notably within the memory domain. The A+/T− group exhibited a moderately strong association within the executive domain (r = −0.30), with an effect as large as that of the A+/T+ group. Notably, when one observation from the A−/T+ group was excluded due to an outlier of 3.46 in Braak I/II SUVR (6 SD above sample mean), all results were retained with similar effects. In contrast, there were no associations between cortical Aβ SUVR levels and any neuropsychological domain score, although a moderate effect was again observed for the A+/T− group within the executive domain (r = −0.37). Table 4 displays partial rs with confidence intervals for all correlations and P-values, and Fig. 3 displays scatterplots for these associations.
Table 4.
Correlations between cortical amyloid/tau Braak I/II SUVR levels and neuropsychological domain performance, stratified by A/T group
| Partial correlation coefficients with 95% confidence intervals [LB, r, UB] |
||||
|---|---|---|---|---|
| A−/T−, n = 67 | A−/T+, n = 135 | A+/T−, n = 18 | A+/T+, n = 81 | |
| Tau, memory | [−0.28, −0.04, 0.20] P = 0.75 | [−0.42, −0.27, −0.11]* P = 0.001 | [−0.63, −0.21, 0.30] P = 0.41 | [−0.63, −0.48, −0.29]* P < 0.001 |
| Tau, executive | [−0.19, 0.06, 0.29] P = 0.65 | [−0.34, −0.18, −0.01]* P = 0.04 | [−0.68, −0.30, 0.22] P = 0.25 | [−0.48, −0.29, −0.07]* P = 0.01 |
| Tau, language | [−0.06, 0.18, 0.41] P = 0.14 | [−0.38, −0.22, −0.05]* P = 0.01 | [−0.55, −0.10, 0.40] P = 0.71 | [−0.56, −0.38, −0.18]* P < 0.001 |
| Amyloid, memory | [−0.15, 0.09, 0.33] P = 0.45 | [−0.14, 0.03, 0.20] P = 0.76 | [−0.53, −0.07, 0.42] P = 0.79 | [−0.18, 0.04, 0.26] P = 0.74 |
| Amyloid, executive | [−0.35, −0.12, 0.13] P = 0.34 | [−0.04, 0.13, 0.30] P = 0.12 | [−0.72, −0.37, 0.13] P = 0.14 | [−0.32, −0.11, 0.11] P = 0.32 |
| Amyloid, language | [−0.13, 0.12, 0.35] P = 0.34 | [−0.11, 0.06, 0.22] P = 0.52 | [−0.64, −0.23, 0.28] P = 0.37 | [−0.20, 0.02, 0.24] P = 0.83 |
LB = 95% CI lower bound; UB = 95% CI upper bound. r values are given in bold.
Statistically significant association at P < 0.05.
Figure 3.

Tau PET and cognition associations for T+ groups. Scatterplots depicting the association between Braak I/II SUVR and memory (A), executive function (B) and language (C) composites. A−/T+ is denoted in gray and A+/T+ is denoted in purple. Values have been residualized with respect to APOE ε4 positivity and cognitive composites transformed to meet normality assumptions. Figures exclude one outlier with a residualized Braak I/II SUVR of 2.14 to improve visualization, but effects remained statistically and qualitatively similar.
Discussion
Despite the primacy of Aβ over tau postulated by the amyloid cascade model and AT(N) framework for AD pathological progression, our investigation of discrepancies in Aβ and tau PET positivity revealed that the largest proportion of our sample (45%) had MTL tau accumulation in the absence of Aβ (A−/T+). In contrast, only 6% of the sample had evidence of Aβ in the absence of tau (A+/T−), despite the use of equivalent methods for deriving positivity thresholds. The A−/T+ group tended to perform more poorly across memory, executive function, and language domains relative to the A−/T− and A+/T− groups, although not as poorly as the A+/T+ group. Only the A−/T+ and A+/T+ groups exhibited associations between tau SUVR levels and all cognitive domains, with stronger effects observed for the A+/T+ group. Importantly, our results provide preliminary evidence to suggest that tau pathology may emerge independent of beta-amyloidosis and that A−/T+ individuals, given their widespread early cognitive compromises, may be best represented on the Alzheimer’s continuum. These findings are critically important in the conceptualization and treatment of AD because they suggest a wider spectrum of individuals as part of the AD prodrome, as well as provide further evidence that tau represents a relevant and promising treatment target for Alzheimer’s disease.
Although there has been a recent surge of research utilizing tau PET imaging in the context of Alzheimer’s disease, many studies have focused on tau only in the presence of Aβ, rather than assessing them independently (Vemuri et al., 2016; Bejanin et al., 2017; Ossenkoppele et al., 2019a,b). In addition, positivity for tau PET is often determined conditionally based on Aβ (Wang et al., 2016; Mishra et al., 2017; Jack et al., 2019). Importantly, this strategy obviates the possibility of assessing discordance between Aβ and tau positivities and offers incomplete conclusions about the respective roles of these pathologies in the Alzheimer’s disease clinical continuum. It is for this reason that we derived thresholds for Aβ and tau independently of one another and independent of our clinical and cognitive outcome variables, by using Mini–Mental State Examination as a global index of cognition to drive the classification algorithm. This process yielded tau positivity thresholds that were similar to, albeit more conservative than, those previously reported using analogous methods in a more heterogeneous sample (Schöll et al., 2016; Maass et al., 2017), providing convergent validity for the values derived in this study. Notably, although the value we derived for the Aβ positivity threshold (>1.14) was slightly more liberal than the conventional threshold using pathologically confirmed data (>1.11; Landau and Jagust, 2015), a reanalysis of the data with the conventional 1.11 threshold revealed that group classifications remained largely the same (see Supplementary Table 1) and cognitive comparisons did not notably differ from the results reported above. Thus, we believe that the methods used for threshold derivation in this study provide valid and reliable biomarker categorizations.
Applying these newly derived positivity thresholds to examine discrepancies in Aβ and tau positivity, we found that the A−/T+ group represented the largest proportion of the sample (45%), whereas more than a 7-fold lower proportion (6%) were categorized as A+/T−. Notably, these proportions differ substantially from a recent study of AT(N) categorizations, which found only 11.5% to be classified as A−/T+/(N− or +) using PET methods (Jack et al., 2019). Importantly, this prior study derived a threshold for tau positivity that was contingent on Aβ status, with the tau SUVR threshold determined based on optimal discriminability of young CN A− individuals from older cognitively impaired A+ individuals. Using a model based on discrimination of A− and A+ groups necessarily yields a threshold value that will maximally group high tau SUVRs with the A+ category and low tau SUVRs with the A− category, thus increasing the likelihood of producing A+/T+ and A−/T− groups with fewer individuals falling into discrepant groups. We, in contrast, derived A+ and T+ threshold independently of one another by fitting our model to a global cognitive outcome regardless of PET status for the other biomarker to reduce bias.
Our resultant classifications of 45% A−/T+ and 6% A+/T− differ from the temporal sequence of pathological events anticipated by the amyloid cascade model and AT(N) framework, both of which purport that Aβ is the primary pathological substrate of Alzheimer’s disease that emerges some years prior to the tauopathy of Alzheimer’s disease. However, post-mortem histopathological studies of Alzheimer’s disease have indicated early pathological tau changes that occur in the brainstem, specifically the locus coeruleus, which subsequently propagate to medial temporal regions encompassing Braak stage I/II. These events largely occur before the evidence of Aβ accumulation (Braak et al., 2011; Braak and Del Tredici, 2015; Ehrenberg et al., 2017), in accord with the high proportion of A−/T+ observed in our study. Furthermore, our findings of a high prevalence of T+ in the context of A− comport with the post-mortem pathological findings of Braak and Del Tredici (2015). In their study, ∼75–80% of individuals aged 70–80 years had evidence of Braak stages I–II tau pathology, whereas only ∼10–35% over that same age range had evidence of Aβ phases 1–3 (note that the average age of our study participants was 75.6 years). Moreover, across all ages, 953 (61%) of the autopsied subjects with Braak stage I/II tau pathology had Aβ phase 0 pathology. In a separate study, Braak and Del Tredici (2014) further demonstrated that, of the 80% of individuals between the ages of 70–80 years with neuropathological evidence of Braak stages I–II tau pathology, only half also had evidence of Aβ (including phase 1), which again aligns with our findings of 45% A−/T+ prevalence.
Prior research has acknowledged this group of A−/T+ individuals, although historically they have not been considered part of the Alzheimer’s developmental continuum. Instead, individuals with this pathological presentation of MTL tau in the absence of Aβ have been relegated to alternative categorizations such as PART, which views medial temporal tauopathy in the absence of Aβ as a feature of aging separate from AD (Crary et al., 2014; Jellinger et al., 2015). Individuals under the PART designation have been characterized as older, having lower APOE ε4 allelic frequencies, and typically more cognitively intact with only localized amnestic changes observed in severe cases (Crary et al., 2014). However, other groups contend that PART itself is a developmental stage (i.e. Braak I/II) of Alzheimer’s disease pathogenesis (Braak and Del Tredici, 2014; Duyckaerts et al., 2015; Josephs et al., 2017). Most investigations into PART have been conducted using post-mortem histopathological data, which limits the ability to assess cognitive status in close temporal proximity to the pathological determinants due to the use of retrospective cognitive data that often varies on the order of years in its proximity to the time of autopsy. In contrast, our study was able to investigate these groupings using in vivo PET markers of Aβ and tau pathology that more closely coincided with the administration of a neuropsychological battery, allowing for an improved cognitive characterization of A−/T+ individuals.
Importantly, our results do not recapitulate the usual demographic and clinical features of the A−/T+ group proposed by proponents of PART (Crary et al., 2014). For example, our A−/T+ group did not differ from the non-pathological A−/T− group in terms of APOE ε4 allelic frequency. Furthermore, although the A−/T+ group was older than the A−/T− group, they did not differ from the other groups (A+/T− and A+/T+). Notably, vascular risk assessed through multiple indices did not differ between the groups and therefore any group differences in cognition are likely not attributable to vascular differences. The proportion of individuals with MCI did not differ among the A−/T−, A−/T+ and A+/T− groups (21-24%), but it was twice as high in the A+/T+ group (48%). Significantly, the A−/T+ group had an MCI rate of 24%, which conflicts with the claim that PART is a condition observed primarily in CN individuals (Crary et al., 2014). However, given that this rate did not statistically differ from the A−/T− group, we also examined differences in neuropsychological domain scores to further explore the pattern of cognitive performance across groups.
From the amyloid cascade and PART perspectives, one would predict similar cognitive performances between the A−/T− and A−/T+ groups, particularly in non-amnestic domains, followed by a lower-performing A+/T− group, followed by the lowest-performing A+/T+ group. This pattern was not observed across any of the three cognitive domains. Instead, the A−/T− and A+/T− groups performed most similarly, often with the A+/T− group demonstrating the best performances of all groups. In contrast, the A−/T+ group exhibited small-to-moderate decreases in performance relative to the A−/T− group across all domains. The A+/T+ group had the poorest performances in all domains with the exception of language, for which the A−/T+ group performed equally as poorly. Furthermore, examination of partial correlations between domain performance and Braak stage I/II SUVR levels revealed associations only for the A−/T+ and A+/T+ groups, with stronger effects for the latter. Despite its confinement to the MTL in Braak stage I/II, tau appears to have widespread effects on cognition across memory, executive, and language domains, even in the absence of Aβ positivity. The cognitive differences and associations in executive and language domains in the A−/T+ group are particularly noteworthy given that PART purports to impart only amnestic cognitive changes in the most severe cases. Thus, tau appears to be exerting deleterious effects on cognition within the MTL and beyond even in A− individuals, possibly through network changes to broader association cortices associated with language and executive functions (Ossenkoppele et al., 2019a,b). Notably, the reported effect sizes for group differences in cognition are relatively small and the conclusions drawn should be interpreted cautiously in the absence of further longitudinal research. Nevertheless, these preliminary findings suggest that MTL tau pathology in the absence of Aβ may not be an anticipated consequence of aging, as the PART hypothesis implies.
Given these findings, we propose that prominent MTL tau pathology itself is sufficient to be considered part of the Alzheimer’s continuum, rather than the distinct entity of PART, with subsequent beta-amyloidosis accelerating the disease stage. These data indicate that a revision to the AT(N) nomenclature (Jack et al., 2016, 2018a,b) is also warranted such that A−/T+ profiles should fall under the category of ‘Alzheimer’s pathologic change’ rather than ‘non-Alzheimer’s pathologic change,’ with the term ‘Alzheimer’s disease’ reserved for A+/T+. If an individual falls into one of these categories in the context of intact cognition, they would be considered in a preclinical stage (i.e. preclinical Alzheimer’s pathologic change or preclinical Alzheimer’s disease; see Fig. 4). Interestingly, a prior study investigating the prevalence of abnormal biomarkers without regard to their temporal ordering noted that neurodegeneration (possibly due to MTL tau pathology) was more likely to be the first abnormal biomarker than Aβ (Edmonds et al., 2015); perhaps the most appropriate staging model for Alzheimer’s pathology would follow this ‘tally’ system to maintain agnosticism with regard to the temporal emergence of these biomarkers, given the continued uncertainty over the developmental cascade leading to AD. Such a reframing in nosology would also have critical implications for drug discovery and clinical trials. As our findings suggest, waiting for Aβ positivity to become evident before attempting its clearance, particularly in those with MTL tau positivity, would negate the opportunity to clear pathological tau before it begins to affect cognition broadly across memory, language and executive functions.
Figure 4.

Suggested nomenclature for the Alzheimer’s pathological continuum. Theoretical model of Alzheimer’s nomenclature based on A/T group and cognitive status, including relative group proportions based on A/T positivity prevalence findings in the current sample.
Prevailing theory postulates that tau pathology migrates from the brainstem to the MTL as part of the aging process and remains in this region until sufficient Aβ pathology has accumulated to drive it out to adjacent neocortical regions (Bennett et al., 2017). Our data showed that within the A−/T+ group, all of whom were Braak stage I/II+ (i.e. confined to MTL), 18 individuals (13% of the A−/T+ group) were also Braak stage III/IV+ despite their Aβ negativity. These data suggest that, in some cases, tau pathology may extend beyond the MTL independent of the influence of Aβ. Nonetheless, it remains unclear whether Aβ is needed to further the spread of tau beyond the MTL and the literature offers conflicting evidence. On the one hand, several studies have demonstrated that tau accumulates at a faster rate in the presence of Aβ (Lockhart et al., 2017; Jack et al., 2018b; Leal et al., 2018; Schultz et al., 2018) and our results indicated a stronger association between tau and cognitive performance, as well as generally poorer performance, in the A+/T+ group relative to the A−/T+ group. On the other hand, other studies show that tau has the ability to spread beyond the MTL to adjacent cortical regions in the absence of Aβ, both in animal models (Kaufman et al., 2018) and human PET studies (Lowe et al., 2018). Further post-mortem evidence demonstrates the continued progression of tau pathology despite the enduring clearance of Aβ from anti-Aβ therapies administered prior to death, suggesting an Aβ-independent mechanism of tau propagation (Nicoll et al., 2019).
Notably, there are several other pathologies in addition to Aβ and tau that comprise the Alzheimer’s syndrome, and the majority of individuals with Alzheimer’s disease have multiple pathologies (Schneider et al., 2009; Boyle et al., 2018). Cerebrovascular pathology has proven to be a prominent and early contributor to Alzheimer’s processes, with neuroimaging markers of small-vessel disease emerging as early as 22 years prior to symptom onset in autosomal-dominant AD (Lee et al., 2016). Data further indicate synergistic interactions between cerebrovascular pathology and hallmark Alzheimer’s pathologies, such that cerebrovascular dysfunction may accelerate Aβ and tau pathological progression, and vice versa (Blair et al., 2015; Nation et al., 2015; Rabin et al., 2019). Another important pathological feature in older adults is TDP-43, which defines the new conceptualization of limbic-predominant age-related TDP-43 encephalopathy (i.e. LATE; Nelson et al., 2019) and is often comorbid with Aβ and tau pathologies (James et al., 2016). As research progresses, a polypathologic staging scheme for AD that extends beyond Aβ and tau may be warranted. As previously suggested, a ‘tally’ system for these multiple pathologies (which also includes the presence of subtle or mild cognitive declines) may be the best nosology for representing their cumulative contributions to the Alzheimer’s dementia syndrome (see Edmonds et al., 2015).
There are several notable strengths of the current study. First, the measurement of Aβ and tau positivity with PET imaging allowed for perhaps the first in vivo investigation of PART, with improved temporal proximity between the assessment of cognition and the measurement of pathology relative to previous post-mortem studies. The independent derivation of positivity thresholds for Aβ and tau is also a unique feature of our study relative to prior studies and ensured any potential A−/T+ individuals could accurately be detected, given that Aβ positivity was not considered in the determination of T− and T+ groups. Furthermore, the use of neuropsychological measures sensitive to early AD provided a multi-domain assessment of cognition, rather than relying on brief cognitive screening or rating measures insensitive to the AD prodrome and not validated outside of clinic settings (see De Roeck et al., 2019 for a review). These methodological strengths allowed for an improved examination of the concept of PART, leading to the conclusion that this A−/T+ pathological profile has deleterious effects on cognition and may be better considered as a stage of the Alzheimer’s continuum.
Despite its strengths, we also acknowledge that this study is not without its limitations. Importantly, the homogeneity of the ADNI sample may limit generalizability to more representative community-based samples and further studies sampling from a diverse population are needed to ensure that these effects persist among individuals of underrepresented races/ethnicities, socioeconomic strata and comorbid medical or mental health conditions. Furthermore, our study’s cross-sectional analyses limited our ability to directly examine within-subject temporal emergence of Aβ and tau pathologies as well as cognitive change over time across the A/T groups, and longitudinal investigations of such phenomena will be an important next step to corroborate the preliminary findings presented in this study and further our understanding of pathological progression along the Alzheimer’s continuum within individuals over time. It is also noteworthy that the florbetapir PET tracer for Aβ has a high affinity for neuritic plaques, but it may not detect diffuse plaques or soluble oligomers (Beach et al., 2014); thus, we cannot rule out that these other forms of Aβ pathology are present in A− individuals and contribute to neurofibrillary tangle formation (Koss et al., 2016; Abner et al., 2018). That said, proponents of PART allow for the presence of diffuse plaques in addition to MTL tau (Crary et al., 2014) and, therefore, the detection of these diffuse plaques would not change the categorization of this group.
One may also question the decision to restrict the definition of tau positivity based on spatial location (i.e. MTL) while instead using a broader cortical summary measure for Aβ positivity. Importantly, the use of a composite across diffuse cortical regions may dilute sensitivity of the detection of pathology in any single region. However, given that Aβ pathology presents diffusely and variably throughout cortex (Braak and Braak, 1995; Lockhart et al., 2017), a composite region may better capture the presence of this accumulated pathology given that measurement of a single region may produce false negatives if Aβ has instead accumulated in other unsampled cortical regions. Conversely, tau pathology emerges in a specific spatiotemporal sequence across the Alzheimer’s continuum (Braak and Braak, 1991), which increases our sensitivity for the detection of tau pathology if we select these regions predicted by neuropathological staging (e.g. MTL regions for early detection). For this reason, we and other representative PET studies (see Maass et al., 2017 ) have determined tau positivity based on SUVR level within the MTL (i.e. Braak stage I/II). Notably, selection of this approach likely contributed to our relatively high prevalence of T+ within this study (Braak et al., 2011), whereas a composite of Braak stages I–IV would be more conservative in detecting T+. However, given that we observed subtle cognitive deficits among A−/T+ individuals (with tau largely confined to the MTL), this finding suggests that the observed highly prevalent MTL tau in the absence of amyloid is an important pathological grouping that may be considered part of the Alzheimer’s continuum rather than a feature of normal aging.
That said, other approaches to thresholding or staging of tau PET are worth considering and may yield different prevalence rates of A−/T+. For example, one study derived a composite summary measure, which included four regions that maximally separated low and high levels of flortaucipir binding, and proceeded to define a positivity threshold based on discrimination of Aβ− and Aβ+ older adults (Mishra et al., 2017). Another approach taken by Ossenkoppele et al. (2018) selected regions of interest across Braak stages I–IV and defined a positivity threshold that was 2 SD above the mean for cognitively healthy older adults. We recognize that selection of a thresholding approach is highly influential to the resultant positivity groupings and, therefore, future studies investigating alternative methods for the determination of PET positivity and resultant A/T groupings are critically needed to inform our understanding of pathological changes along the Alzheimer’s disease continuum. However, regardless of the selection of regions or thresholding method, we believe that it important to determine tau positivity independently of Aβ, given the emerging early and independent role of tau within the Alzheimer’s disease prodrome (Jefferson-George et al., 2017; Aschenbrenner et al., 2018; Maass et al., 2018; Nicoll et al., 2019).
In summary, our analyses of PET data within the ADNI cohort suggest that tau pathology is common among individuals without significant beta-amyloidosis, given that 45% of the sample was categorized as A−/T+ relative to 6% as A+/T−, contrary to prevailing amyloid cascade and AT(N) frameworks. Furthermore, the relatively poor cognitive performances of the A−/T+ group, as well as the associations between Braak I/II tau levels and cognition in this group, support the notion that tau pathology confined to the MTL and, in the absence of Aβ, may be part of the Alzheimer’s developmental cascade rather than a feature of aging with few-to-no circumscribed cognitive deficits (i.e. PART). Future studies using longitudinal PET and cognitive data will provide an improved characterization of cognitive change within these A/T groups and better profile how they may transition over time. If realized, this work will lead to a better understanding of the respective spatiotemporal relationships between Aβ and tau in the AD prodrome and offer important implications for person-specific interventions in future anti-Aβ and anti-tau treatment efforts.
Supplementary Material
Acknowledgements
The authors acknowledge the valuable consultation from Dr. Susan Landau and Dr. Anne Maass regarding methods for SUVR threshold derivation. The authors are also grateful to ADNI study investigators and participants for providing the data used in this article and especially thank all individuals at the University of California, Berkeley, who processed and made available the PET data used in this article.
Funding
This work was supported by National Science Foundation Graduate Research Fellowship DGE-1650112 (A.J.W.) and National Institutes of Health grants R01 AG049810 and R01 AG054049 (M.W.B.). Data collection and sharing for this project were funded by the ADNI (National Institutes of Health grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging and the National Institute of Biomedical Imaging and Bioengineering and through generous contributions from the following: AbbVie; Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer, Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Competing interests
A.J.W., K.J.B., K.R.T., L.D.-W., P.E.G., and A.M.B. report no competing interests. M.W.B. receives royalties from Oxford University Press and serves as a consultant for Eisai, Novartis and Roche Pharmaceutical companies.
Glossary
- ADNI
Alzheimer’s Disease Neuroimaging Initiative
- AT(N)
amyloid-tau-neurodegeneration
- Aβ
amyloid beta
- APOE
apolipoprotein E
- CN
cognitively normal
- MTL
medial temporal lobe
- MCI
mild cognitive impairment
- PART
primary age-related tauopathy
- SUVR
standardized uptake variable ratio
Appendix I: ADNI study investigators
I. ADNI I, GO, II and III Part A: Leadership and Infrastructure Principal Investigator Michael W. Weiner, MD UC San Francisco ATRI PI and Director of Coordinating Center Clinical Core Paul Aisen, MD University of Southern California Executive Committee Michael Weiner, MD UC San Francisco Paul Aisen, MD University of Southern California Ronald Petersen, MD, PhD Mayo Clinic, Rochester Clifford R. Jack, Jr., MD Mayo Clinic, Rochester William Jagust, MD UC Berkeley John Q. Trojanowki, MD, PhD U Pennsylvania Arthur W. Toga, PhD USC Laurel Beckett, PhD UC Davis Robert C. Green, MD, MPH Brigham and Women’s Hospital/Harvard Medical School Andrew J. Saykin, PsyD Indiana University John Morris, MD Washington University St. Louis Leslie M. Shaw University of Pennsylvania ADNI External Advisory Board (ESAB) Zaven Khachaturian, PhD Prevent Alzheimer’s Disease 2020 (Chair) Greg Sorensen, MD Siemens Maria Carrillo, PhD Alzheimer’s Association Lew Kuller, MD University of Pittsburgh Marc Raichle, MD Washington University St. Louis Steven Paul, MD Cornell University Peter Davies, MD Albert Einstein College of Medicine of Yeshiva University Howard Fillit, MD AD Drug Discovery Foundation Franz Hefti, PhD Acumen Pharmaceuticals David Holtzman, MD Washington University St. Louis M. Marcel Mesulam, MD Northwestern University William Potter, MD National Institute of Mental Health Peter Snyder, PhD Brown University ADNI 3 Private Partner Scientific Board (PPSB) Rev Sept 20, 2017 Veronika Logovinsky, MD, PhD Eli Lilly (Chair) Data and Publications Committee Robert C. Green, MD, MPH BWH/HMS (Chair) Resource Allocation Review Committee Tom Montine, MD, PhD University of Washington (Chair) Clinical Core Leaders Ronald Petersen, MD, PhD Mayo Clinic, Rochester (Core PI) Paul Aisen, MD University of Southern California Clinical Informatics and Operations Gustavo Jimenez, MBS USC Michael Donohue, PhD USC Devon Gessert, BS USC Kelly Harless, BA USC Jennifer Salazar, MBS USC Yuliana Cabrera, BS USC Sarah Walter, MSc USC Lindsey Hergesheimer, BS USC Biostatistics Core Leaders and Key Personnel Laurel Beckett, PhD UC Davis (Core PI) Danielle Harvey, PhD UC Davis Michael Donohue, PhD UC San Diego MRI Core Leaders and Key Personnel Clifford R. Jack, Jr., MD Mayo Clinic, Rochester (Core PI) Matthew Bernstein, PhD Mayo Clinic, Rochester Nick Fox, MD University of London Paul Thompson, PhD UCLA School of Medicine Norbert Schuff, PhD UCSF MRI Charles DeCArli, MD UC Davis Bret Borowski, RT Mayo Clinic Jeff Gunter, PhD Mayo Clinic Matt Senjem, MS Mayo Clinic Prashanthi Vemuri, PhD Mayo Clinic David Jones, MD Mayo Clinic Kejal Kantarci Mayo Clinic Chad Ward Mayo Clinic PET Core Leaders and Key Personnel William Jagust, MD UC Berkeley (Core PI) Robert A. Koeppe, PhD University of Michigan Norm Foster, MD University of Utah Eric M. Reiman, MD Banner Alzheimer’s Institute Kewei Chen, PhD Banner Alzheimer’s Institute Chet Mathis, MD University of Pittsburgh Rev Sept 20, 2017 Susan Landau, PhD UC Berkeley Neuropathology Core Leaders John C. Morris, MD Washington University St. Louis Nigel J. Cairns, PhD, FRCPath Washington University St. Louis Erin Franklin, MS, CCRP Washington University St. Louis Lisa Taylor-Reinwald, BA, HTL Washington University St. Louis (ASCP)—Past Investigator Biomarkers Core Leaders and Key Personnel Leslie M. Shaw, PhD UPenn School of Medicine John Q. Trojanowki, MD, PhD UPenn School of Medicine Virginia Lee, PhD, MBA UPenn School of Medicine Magdalena Korecka, PhD UPenn School of Medicine Michal Figurski, PhD UPenn School of Medicine Informatics Core Leaders and Key Personnel Arthur W. Toga, PhD USC (Core PI) Karen Crawford USC Scott Neu, PhD USC Genetics Core Leaders and Key Personnel Andrew J. Saykin, PsyD Indiana University Tatiana M. Foroud, PhD Indiana University Steven Potkin, MD UC UC Irvine Li Shen, PhD Indiana University Kelley Faber, MS, CCRC Indiana University Sungeun Kim, PhD Indiana University Kwangsik Nho, PhD Indiana University Initial Concept Planning & Development Michael W. Weiner, MD UC San Francisco Lean Thal, MD UC San Diego Zaven Khachaturian, PhD Prevent Alzheimer’s Disease 2020 Early Project Proposal Development Leon Thal, MD UC San Diego Neil Buckholtz National Institute on Aging Michael W. Weiner, MD UC San Francisco Peter J. Snyder, PhD Brown University William Potter, MD National Institute of Mental Health Steven Paul, MD Cornell University Marilyn Albert, PhD Johns Hopkins University Richard Frank, MD, PhD Richard Frank Consulting Zaven Khachaturian, PhD Prevent Alzheimer’s Disease 2020 NIA John Hsiao, MD National Institute on Aging Rev Sept 20, 2017 Part B: Investigators By Site Oregon Health & Science University: Joseph Quinn, MD Lisa C. Silbert, MD Betty Lind, BS Jeffrey A. Kaye, MD, A.—Past Investigator Raina Carter, BA—Past Investigator Sara Dolen, BS—Past Investigator University of Southern California: Lon S. Schneider, MD Sonia Pawluczyk, MD Mauricio Becerra, BS Liberty Teodoro, RN Bryan M. Spann, DO, PhD—Past Investigator University of California—San Diego: James Brewer, MD, PhD Helen Vanderswag, RN Adam Fleisher, MD—Past Investigator University of Michigan: Jaimie Ziolkowski, MA, BS, TLLP Judith L. Heidebrink, MD, MS Joanne L. Lord, LPN, BA, CCRC—Past Investigator Mayo Clinic, Rochester: Ronald Petersen, MD, PhD Sara S. Mason, RN Colleen S. Albers, RN David Knopman, MD Kris Johnson, RN—Past Investigator Baylor College of Medicine: Javier Villanueva-Meyer, MD Valory Pavlik, PhD Nathaniel Pacini, MA Ashley Lamb, MA Joseph S. Kass, MD, LD, FAAN Rachelle S. Doody, MD, PhD—Past Investigator Victoria Shibley, MS—Past Investigator Munir Chowdhury, MBBS, MS—Past Investigator Susan Rountree, MD—Past Investigator Mimi Dang, MD—Past Investigator Columbia University Medical Center: Yaakov Stern, PhD Lawrence S. Honig, MD, PhD Karen L. Bell, MD Randy Yeh, MD Washington University, St. Louis: Beau Ances, MD, PhD, MSc John C. Morris, MD David Winkfield, BS Maria Carroll, RN, MSN, GCNS-BC Angela Oliver, RN, BSN, MSG Mary L. Creech, RN, MSW—Past Investigator Mark A. Mintun, MD—Past Investigator Stacy Schneider, APRN, BC, GNP—Past Investigator University of Alabama—Birmingham: Daniel Marson, JD, PhD David Geldmacher, MD Marissa Natelson Love, MD Randall Griffith, PhD, ABPP—Past Investigator David Clark, MD—Past Investigator John Brockington, MD—Past Investigator Mount Sinai School of Medicine: Hillel Grossman, MD Effie Mitsis, PhD—Past Investigator Rush University Medical Center: Raj C. Shah, MD Melissa Lamar, PhD Patricia Samuels Wien Center: Ranjan Duara, MD Maria T. Greig-Custo, MD Rosemarie Rodriguez, PhD Johns Hopkins University: Marilyn Albert, PhD Chiadi Onyike, MD Daniel D’Agostino II, BS Stephanie Kielb, BS—Past Investigator Rev Sept 20, 2017 New York University: Martin Sadowski, MD, PhD Mohammed O. Sheikh, MD Jamika Singleton-Garvin, CCRP Anaztasia Ulysse Mrunalini Gaikwad Duke University Medical Center: P. Murali Doraiswamy, MBBS, FRCP Jeffrey R. Petrella, MD Olga James, MD Salvador Borges-Neto, MD Terence Z. Wong, MD—Past Investigator Edward Coleman—Past Investigator University of Pennsylvania: Jason H. Karlawish, MD David A. Wolk, MD Sanjeev Vaishnavi, MD Christopher M. Clark, MD—Past Investigator Steven E. Arnold, MD—Past Investigator University of Kentucky: Charles D. Smith, MD Greg Jicha, MD Peter Hardy, PhD Riham El Khouli, MD Elizabeth Oates, MD Gary Conrad, MD University of Pittsburgh: Oscar L. Lopez, MD MaryAnn Oakley, MA Donna M. Simpson, CRNP, MPH University of Rochester Medical Center: Anton P. Porsteinsson, MD Kim Martin, RN Nancy Kowalksi, MS, RNC Melanie Keltz, RN Bonnie S. Goldstein, MS, NP—Past Investigator Kelly M. Makino, BS—Past Investigator M. Saleem Ismail, MD—Past Investigator Connie Brand, RN—Past Investigator University of California Irvine IMIND: Gaby Thai, MD Aimee Pierce, MD Beatriz Yanez, RN Elizabeth Sosa, PhD Megan Witbracht, PhD University of Texas Southwestern Medical School: Kyle Womack, MD Dana Mathews, MD, PhD Mary Quiceno, MD Emory University: Allan I. Levey, MD, PhD James J. Lah, MD, PhD Janet S. Cellar, DNP, PMHCNS-BC University of Kansas, Medical Center: Jeffrey M. Burns, MD Russell H. Swerdlow, MD William M. Brooks, PhD University of California, Los Angeles: Ellen Woo, PhD Daniel H.S. Silverman, MD, PhD Edmond Teng, MD, PhD Sarah Kremen, MD Liana Apostolova, MD—Past Investigator Kathleen Tingus, PhD—Past Investigator Po H. Lu, PsyD—Past Investigator George Bartzokis, MD—Past Investigator Mayo Clinic, Jacksonville: Neill R Graff-Radford, MBBCH, FRCP (London) Francine Parfitt, MSH, CCRC Kim Poki-Walker, BA Indiana University: Martin R. Farlow, MD Ann Marie Hake, MD Brandy R. Matthews, MD—Past Investigator Jared R. Brosch, MD Scott Herring, RN, CCRC Yale University School of Medicine: Christopher H. van Dyck, MD Richard E. Carson, PhD Pradeep Varma, MD McGill Univ., Montreal-Jewish General Hospital: Howard Chertkow, MD Howard Bergman, MD Chris Hosein, MEd Rev Sept 20, 2017 Sunnybrook Health Sciences, Ontario: Sandra Black, MD, FRCPC Bojana Stefanovic, PhD Chris (Chinthaka) Heyn, BSC, PhD, MD, FRCPC U.B.C. Clinic for AD & Related Disorders: Ging-Yuek Robin Hsiung, MD, MHSc, FRCPC Benita Mudge, BS Vesna Sossi, PhD Howard Feldman, MD, FRCPC—Past Investigator Michele Assaly, MA—Past Investigator Cognitive Neurology—St. Joseph's, Ontario: Elizabeth Finger, MD Stephen Pasternack, MD, PhD William Pavlosky, MD Irina Rachinsky, MD—Past Investigator Dick Drost, PhD—Past Investigator Andrew Kertesz, MD—Past Investigator Cleveland Clinic Lou Ruvo Center for Brain Health: Charles Bernick, MD, MPH Donna Munic, PhD Northwestern University: Marek-Marsel Mesulam, MD Emily Rogalski, PhD Kristine Lipowski, MA Sandra Weintraub, PhD Borna Bonakdarpour, MD Diana Kerwin, MD—Past Investigator Chuang-Kuo Wu, MD, PhD—Past Investigator Nancy Johnson, PhD—Past Investigator Premiere Research Inst (Palm Beach Neurology): Carl Sadowsky, MD Teresa Villena, MD Georgetown University Medical Center: Raymond Scott Turner, MD, PhD Kathleen Johnson, NP Brigid Reynolds, NP Brigham and Women's Hospital: Reisa A. Sperling, MD Keith A. Johnson, MD Gad A. Marshall, MD Stanford University: Jerome Yesavage, MD Joy L. Taylor, PhD Steven Chao, MD, PhD Barton Lane, MD—Past Investigator Allyson Rosen, PhD—Past Investigator Jared Tinklenberg, MD—Past Investigator Banner Sun Health Research Institute: Edward Zamrini, MD Christine M. Belden, PsyD Sherye A. Sirrel, CCRC Boston University: Neil Kowall, MD Ronald Killiany, PhD Andrew E. Budson, MD Alexander Norbash, MD—Past Investigator Patricia Lynn Johnson, BA—Past Investigator Howard University: Thomas O. Obisesan, MD, MPH Ntekim E. Oyonumo, MD, PhD Joanne Allard, PhD Olu Ogunlana, BPharm Case Western Reserve University: Alan Lerner, MD Paula Ogrocki, PhD Curtis Tatsuoka, PhD Parianne Fatica, BA, CCRC University of California, Davis—Sacramento: Evan Fletcher, PhD Pauline Maillard, PhD John Olichney, MD Charles DeCarli, MD Owen Carmichael, PhD—Past Investigator Neurological Care of CNY: Smita Kittur, MD—Past Investigator Parkwood Institute: Michael Borrie, MB ChB T-Y Lee, PhD Dr Rob Bartha, PhD University of Wisconsin: Sterling Johnson, PhD Sanjay Asthana, MD Rev Sept 20, 2017 Cynthia M. Carlsson, MD, MS Banner Alzheimer's Institute: Pierre Tariot, MD Anna Burke, MD Joel Hetelle, BS Kathryn DeMarco, BS Nadira Trncic, MD, PhD, CCRC—Past Investigator Adam Fleisher, MD—Past Investigator Stephanie Reeder, BA—Past Investigator Dent Neurologic Institute: Vernice Bates, MD Horacio Capote, MD Michelle Rainka, PharmD, CCRP Ohio State University: Douglas W. Scharre, MD Maria Kataki, MD, PhD Rawan Tarawneh, MD Albany Medical College: Earl A. Zimmerman, MD Dzintra Celmins, MD David Hart, MD Hartford Hospital, Olin Neuropsychiatry Research Center: Godfrey D. Pearlson, MD Karen Blank, MD Karen Anderson, RN Dartmouth-Hitchcock Medical Center: Laura A. Flashman, PhD Marc Seltzer, MD Mary L. Hynes, RN, MPH Robert B. Santulli, MD—Past Investigator Wake Forest University Health Sciences: Kaycee M. Sink, MD, MAS Mia Yang, MD Akiva Mintz, MD, PhD Rhode Island Hospital: Brian R. Ott, MD Geoffrey Tremont, PhD Lori A. Daiello, Pharm.D, ScM Butler Hospital: Stephen Salloway, MD, MS Paul Malloy, PhD Stephen Correia, PhD Athena Lee, PhD UC San Francisco: Howard J. Rosen, MD Bruce L. Miller, MD David Perry, MD Medical University South Carolina: Jacobo Mintzer, MD, MBA Kenneth Spicer, MD, PhD David Bachman, MD St. Joseph’s Health Care: Elizabeth Finger, MD Stephen Pasternak, MD Irina Rachinsky, MD John Rogers, MD Andrew Kertesz, MD—Past Investigator Dick Drost, MD—Past Investigator Nathan Kline Institute Nunzio Pomara, MD Raymundo Hernando, MD Antero Sarrael, MD University of Iowa College of Medicine Delwyn D. Miller, PharmD, MD Karen Ekstam Smith, RN Hristina Koleva, MD Ki Won Nam, MD Hyungsub Shim, MD Susan K. Schultz, MD—Past Investigator Cornell University Norman Relkin, MD, PhD Gloria Chiang, MD Michael Lin, MD Lisa Ravdin, PhD University of South Florida: USF Health Byrd Alzheimer’s Institute Amanda Smith, MD Christi Leach, MD Balebail Ashok Raj, MD—Past Investigator Kristin Fargher, MD—Past Investigator Courtney Bodge, PhD
References
- Abner EL, Neltner JH, Jicha GA, Patel E, Anderson SL, Wilcock DM, et al. Diffuse amyloid-β plaques, neurofibrillary tangles, and the impact of APOE in elderly persons' brains lacking neuritic amyloid plaques. J Alzheimers Dis 2018; 64: 1307–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, Poggiali D, Whitaker K, Marshall TR, Kievit RA.. Raincloud plots: a multi-platform tool for robust data visualization. Wellcome Open Res 2019; 4: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Allg Zeitschr Psychiatr Psychiatr Gerichtl Med 1907; 146–8. [Google Scholar]
- Alzheimer’s Disease Neuroimaging Initiative; 2019. http://adni.loni.usc.edu/ (15 July 2019, date last accessed).
- Arriagada PV, Marzloff K, Hyman BT.. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology 1992; 42: 1681–8. [DOI] [PubMed] [Google Scholar]
- Aschenbrenner AJ, Gordon BA, Benzinger TLS, Morris JC, Hassenstab JJ.. Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology 2018; 91: e859–e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SL, Maass A, Jagust WJ.. Considerations and code for partial volume correcting [18F]-AV-1451 tau PET data. Data Brief 2017; 15: 648–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, Schneider JA, Sue LI, Serrano G, Dugger BN, Monsell SE, et al. Theoretical impact of florbetapir (18F) amyloid imaging on diagnosis of Alzheimer dementia and detection of preclinical cortical amyloid. J Neuropathol Exp Neurol 2014; 73: 948–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejanin A, Schonhaut DR, La Joie R, Kramer JH, Baker SL, Sosa N, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer's disease. Brain 2017; 140: 3286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell WR, An Y, Kageyama Y, et al. Neuropathologic, genetic, and longitudinal cognitive profiles in primary age-related tauopathy (PART) and Alzheimer’s disease. Alzheimers Dement 2019; 15: 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RE, DeVos SL, Dujardin S, Corjuc B, Gor R, Gonzalez J, et al. Enhanced tau aggregation in the presence of amyloid β. Am J Pathol 2017; 187: 1601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacker DB, Tanzi RE.. The genetics of Alzheimer’s disease: current status and future prospects. Arch Neurol 1998; 55: 294–6. [DOI] [PubMed] [Google Scholar]
- Blair LJ, Frauen HD, Zhang B, Nordhues BA, Bijan S, Lin Y-C, et al. Tau depletion prevents progressive blood–brain barrier damage in a mouse model of tauopathy. Acta Neuropathol Commun 2015; 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondi MW, Edmonds EC, Jak AJ, Clark LR, Delano-Wood L, McDonald CR, et al. Neuropsychological criteria for mild cognitive impairment improves diagnostic precision, biomarker associations, and progression rates. J Alzheimers Dis 2014; 42: 275–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA.. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 2018; 83: 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E.. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239–59. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E.. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 1995; 16: 271–8. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K.. Are cases with tau pathology occurring in the absence of Aβ deposits part of the AD-related pathological process? Acta Neuropathol 2014; 128: 767–72. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K.. The preclinical phase of the pathologic process underlying sporadic Alzheimer’s disease. Brain 2015; 138: 2814–33. [DOI] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K.. Stages of the pathologic process in Alzheimer’s disease: age categories from 1-100 years. J Neuropathol Exp Neurol 2011; 70: 960–9. [DOI] [PubMed] [Google Scholar]
- Burnham SC, Bourgeat P, Doré V, Savage G, Brown B, Laws S, et al. Clinical and cognitive trajectories in cognitively health elderly individuals with suspected non-Alzheimer’s disease pathology (SNAP) or Alzheimer’s disease pathology: a longitudinal study. Lancet Neurol 2016; 15: 1044–53. [DOI] [PubMed] [Google Scholar]
- Cohen AD, Mowrey W, Weissfeld LA, Aizenstein HJ, McDade E, Mountz JM, et al. Classification of amyloid-positivity in controls: comparison of visual read and quantitative approaches. Neuroimage 2013; 71: 207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crary JF. Primary age-related tauopathy and the amyloid cascade hypothesis: the exception that proves the rule? J Neurol Neuromed 2016; 1: 53–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014; 128: 755–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Roeck EE, De Deyn PP, Dierckx E, Engelborghs S.. Brief cognitive screening instruments for early detection of Alzheimer’s disease: a systematic review. Alzheimers Res Ther 2019; 11: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Braak H, Brion J-P, Buée L, Del Tredici K, Goedert M, et al. PART is part of Alzheimer disease. Acta Neuropathol 2015; 129: 749–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonds EC, Delano-Wood L, Galasko DR, Salmon DP, Bondi MW; Alzheimer’s Disease Neuroimaging Initiative. Subtle cognitive decline and biomarker staging in preclinical Alzheimer's disease. J Alzheimers Dis 2015; 47: 231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Kost J, Tariot PN, Aisen PS, Cummings JL, Vellas B, et al. Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N Engl J Med 2018; 378: 1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg AJ, Nguy AK, Theofilas P, Dunlop S, Suemoto CK, Di Lorenzo Alho AT, et al. Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: the pathological building blocks of early Alzheimer's disease. Neuropathol Appl Neurobiol 2017; 43: 393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW.. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 1984; 122: 1131–5. [DOI] [PubMed] [Google Scholar]
- Gordon BA, Blazey T, Su Y, Fagan AM, Holtzman DM, Morris JC, et al. Longitudinal β-amyloid deposition and hippocampal volume in preclinical Alzheimer disease and suspected non-Alzheimer disease pathophysiology. JAMA Neurol 2016; 73: 1192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. The discovery of Alzheimer-causing mutations in the APP gene and the formulation of the “amyloid cascade hypothesis”. FEBS J 2017; 284: 1040–4. [DOI] [PubMed] [Google Scholar]
- Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 2018; 378: 321–30. [DOI] [PubMed] [Google Scholar]
- Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018a; 14: 535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016; 87: 539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Schwarz CG, Lowe VJ, Senjem ML, Vemuri P, et al. Longitudinal tau PET in ageing and Alzheimer's disease. Brain 2018b; 141: 1517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Therneau TM, Weigand SD, Knopman DS, Mielke MM, et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA 2019; 321: 2316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W. Imaging the evolution and pathophysiology of Alzheimer’s disease. Nat Rev Neurosci 2018; 19: 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jak AJ, Bondi MW, Delano-Wood L, Wierenga C, Corey-Bloom J, Salmon DP, et al. Quantification of five neuropsychological approaches to defining mild cognitive impairment. Am J Geriatr Psychiatry 2009; 17: 368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA.. TDP-43 stage, mixed pathologies, and clinical Alzheimer's-type dementia. Brain 2016; 139: 2983–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson-George KS, Wolk DA, Lee EB, McMillan CT.. Cognitive decline associated with pathological burden in primary age-related tauopathy. Alzheimers Dement 2017; 13: 1048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, Alafuzoff I, Attems J, Beach TG, Cairns NJ, Crary JF, et al. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol 2015; 129: 757–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Tosakulwong N, Whitwell JL, Knopman DS, Machulda MM, et al. Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: a clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol 2017; 133: 705–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AD, Pontecorvo MJ, Clark CM, Carpenter AP, Jennings DL, Sadowsky CH, et al. ; Florbetapir F 18 Study Investigators. Performance characteristics of amyloid PET with florbetapir F 18 in patients with Alzheimer’s disease and cognitively normal subjects. J Nucl Med 2012; 53: 378–84. [DOI] [PubMed] [Google Scholar]
- Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI.. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer's disease and PART. Acta Neuropathol 2018; 136: 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, Jack CR, Wiste HJ, Weigand SD, Vemuri P, Lowe VJ, et al. Brain injury biomarkers are not dependent on β-amyloid in normal elderly. Ann Neurol 2013; 73: 472–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss DJ, Jones G, Cranston A, Gardner H, Kanaan NM, Platt B.. Soluble pre-fibrillar tau and β-amyloid species emerge in early human Alzheimer's disease and track disease progression and cognitive decline. Acta Neuropathol 2016; 132: 875–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau S, Jagust W. Florbetapir Processing Methods; 2015. http://adni.loni.usc.edu/ (15 July 2019, date last accessed).
- Landau S, Jagust W. Flortaucipir (AV-1451) Processing Methods; 2016. http://adni.loni.usc.edu/ (15 July 2019, date last accessed).
- Landau SM, Breault C, Joshi AD, Pontecorvo M, Mathis CA, Jagust WJ, et al. Amyloid-beta imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med 2013; 54: 70–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Horng A, Fero A, Jagust WJ; Alzheimer's Disease Neuroimaging Initiative. Amyloid negativity in patients with clinically diagnosed Alzheimer disease and MCI. Neurology 2016; 86: 1377–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, et al. ; Alzheimer's Disease Neuroimaging Initiative. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol 2012; 72: 578–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal SL, Lockhart SN, Maass A, Bell RK, Jagust WJ.. Subthreshold amyloid predicts tau deposition in aging. J Neurosci 2018; 38: 4482–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Viqar F, Zimmerman ME, Narkhede A, Tosto G, Benzinger TLS, et al. ; Dominantly Inherited Alzheimer Network. White matter hyperintensities are a core feature of Alzheimer's disease: Evidence from the dominantly inherited Alzheimer network. Ann Neurol 2016; 79: 929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewczuk P, Matzen A, Blennow K, Parnetti L, Molinuevo JL, Eusebi P, et al. Cerebrospinal fluid Aβ42/40 corresponds better than Aβ42 to amyloid PET in Alzheimer's disease. J Alzheimers Dis 2017; 55: 813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart SN, Schöll M, Baker SL, Ayakta N, Swinnerton KN, Bell RK, et al. Amyloid and tau PET demonstrate region-specific associations in normal older people. Neuroimage 2017; 150: 191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe VJ, Wiste HJ, Senjem ML, Weigand SD, Therneau TM, Boeve BF, et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer's dementia. Brain 2018; 141: 271–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass A, Lockhart SN, Harrison TM, Bell RK, Mellinger T, Swinnerton K, . et al. Entorhinal tau pathology, episodic memory decline, and neurodegeneration in aging. J Neurosci 2018; 38: 530–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass A, Landau S, Baker SL, Horng A, Lockhart SN, La Joie R, et al. Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer's disease. Neuroimage 2017; 157: 448–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984; 34: 939. [DOI] [PubMed] [Google Scholar]
- Mishra S, Gordon BA, Su Y, Christensen J, Friedrichsen K, Jackson K, et al. AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: defining a summary measure. Neuroimage 2017; 161: 171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Papp KV, Rentz DM, Schultz AP, LaPoint M, Amariglio R, et al. Heterogeneity in suspected non-Alzheimer disease pathophysiology among clinically normal older individuals. JAMA Neurol 2016; 73: 1185–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GP, Clark IA, Vissel B.. Questions concerning the role of amyloid-β in the definition, aetiology, and diagnosis of Alzheimer’s disease. Acta Neuropathol 2018; 136: 663–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nation DA, Delano-Wood L, Bangen KJ, Wierenga CE, Jak AJ, Hansen LA, et al. Antemortem pulse pressure elevation predicts cerebrovascular disease in autopsy-confirmed Alzheimer's disease. J Alzheimers Dis 2012; 30: 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nation DA, Edmonds EC, Bangen KJ, Delano-Wood L, Scanlon BK, Han SD, et al. Pulse pressure in relation to tau-mediated neurodegeneration, cerebral amyloidosis, and progression to dementia in very old adults. JAMA Neurol 2015; 72: 546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019; 142: 1503–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JAR, Buckland GR, Harrison CH, Page A, Harris S, Love S, et al. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer’s disease. Brain 2019; 142: 2113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Iaccarino L, Schonhaut DR, Brown JA, La Joie R, O'Neil JP, et al. Tau covariance patterns in Alzheimer's disease patients match intrinsic connectivity networks in the healthy brain. Neuroimage Clin 2019a; 23:101848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Rabinovici GD, Smith R, Cho H, Schöll M, Strandberg O, et al. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA 2018; 320: 1151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Smith R, Ohlsson T, Strandberg O, Mattsson N, Insel PS, et al. Associations between tau, Aβ, and cortical thickness with cognition in Alzheimer disease. Neurology 2019b; 92: e601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxtoby NP, Young AL, Cash DM, Benzinger TLS, Fagan AM, Morris JC, et al. Data-driven models of dominantly-inherited Alzheimer's disease progression. Brain 2018; 141: 1529–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017) R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. https://www.R-project.org (15 July 2019, date last accessed).
- Rabin JS, Yang H-S, Schultz AP, Hanseeuw BJ, Hedden T, Viswanathan A, et al. Vascular risk and β-amyloid are synergistically associated with cortical tau. Ann Neurol 2019; 85: 272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciarelli R, Fedele E.. The amyloid cascade hypothesis in Alzheimer’s disease: it’s time to change our mind. Curr Neuropharmacol 2017; 15: 926–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA.. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 2009; 66: 200–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöll M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossenkoppele R, et al. PET imaging of tau deposition in the aging human brain. Neuron 2016; 89: 971–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz SA, Gordon BA, Mishra S, Su Y, Perrin RJ, Cairns NJ, et al. Widespread distribution of tauopathy in preclinical Alzheimer's disease. Neurobiol Aging 2018; 72: 177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. Alzheimer disease and aducanumab: adjusting our approach. Nat Rev Neurol 2019; 15: 365–6. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Lowe VJ, Knopman DS, et al. Tau-PET uptake: regional variation in average SUVR and impact of amyloid deposition. Alzheimers Dement (Amst) 2016; 6: 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, et al. Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between β-amyloid and tauopathy. JAMA Neurol 2016; 73: 1070–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public–private partnership. The primary goal of ADNI has been to test whether serial magnetic resonance imaging, PET, other biological markers and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early Alzheimer’s disease. This research was approved by the Institutional Review Boards of all participating sites, and written informed consent was obtained for all study participants.